

Abstract
Deoxyhemoglobin reduces nitrite to nitric oxide (NO). In order to study the effect of the hemoglobin quaternary conformation on the nitrite reaction, we compared T-state deoxyhemoglobin with R-state deoxyhemoglobin produced by reacting hemoglobin with carboxypeptidase-A prior to deoxygenation. The nitrite reaction with deoxyhemoglobin was followed by chemiluminescence, electron paramagnetic resonance and visible spectroscopy. The initial steps in this reaction involve the binding of nitrite to deoxyhemoglobin followed by the formation of an electron delocalized metastable intermediate that retains potential NO bioactivity. This reaction is shown by visible spectroscopy to occur 5.6 times faster in the R-state than in the T-state. However, the dissociation of NO from the delocalized intermediate is shown to be facilitated by the T-quaternary conformation with a 9.6 fold increase in the rate constant. The preferred NO-release in the T-state, which has a higher affinity for the membrane, can result in the NO diffusing out of the RBC and being released to the vasculature at low partial pressures of oxygen.
Keywords: nitrite reduction, nitric oxide, hemoglobin quaternary conformation
Introduction
Nitric oxide as a vasodilator plays a major role in regulating blood flow and vascular tone [1]. The primary source for the synthesis of NO in the circulatory system involves endothelial nitric oxide synthase [2]. Since nitric oxide has a life time in plasma of <0.1ms [3], effective delivery of NO to the vasculature would require that the NO is synthesized at the site where it is needed. The reported [4] reduced activity of nitric oxide synthase at reduced partial pressures of oxygen may limit the ability of endothelial nitric oxide synthase to supply NO to the microcirculation. To resolve this dilemma mechanisms for the transport of NO activity by RBCs have been proposed. A mechanism involving the transfer of NO to a thiol group producing S-nitrosylated hemoglobin (SNOHb) was originally proposed in 1996 by the seminal paper of Stamler and collaborators [5], with alternative hypotheses proposed by other investigators involving the hypoxic release of ATP [6] and reduction of nitrite to NO by deoxygenated hemoglobin in the RBC [7,8].
Any role for RBCs in transporting nitric oxide must, however, be able to avoid the very efficient scavenging of nitric oxide in RBCs by both oxyhemoglobin (oxyHb) and deoxyhemoglobin (deoxyHb). The hypothesis suggesting hypoxic release of ATP does not involve a pool of RBC NO that can react with hemoglobin [6,9]. The Stamler hypothesis bypassed this difficulty by transferring the NO to the β-93 thiol group with the formation of SNOHb [5]. This NO can then be transferred to endothelial cells through transnitrosation of membrane thiol groups [10]. The 2003 studies by Rifkind and Gladwin and their collaborators [7,8] proposed a mechanism that involved the reduction of nitrite back to NO by a reaction with deoxyHb. The attraction of this mechanism is that it reuses the NO produced by the endothelium, without requiring the transport of unstable nitric oxide. The feasibility of this mechanism, however, requires that the nitrite reduced back to NO is not quenched by hemoglobin.
Gladwin and collaborators originally suggested that the nitrite is reduced to NO by a metabolon (a complex of band 3, carbonic anhydrase, and deoxyhemoglobin reductase) localized on the membrane at the point where it is needed [11]. More recently [12] they have proposed that nitrite complexes with oxidized hemoglobin (metHb) formed during the reduction of nitrite by deoxyHb and that the reaction of the NO formed with this complex produces N2O3 that can diffuse out of the cell and/or react with thiols to form S-nitrosothiols. The difficulty with this mechanism is that it uses the final products (metHb and NO) formed by the reaction and requires that the NO produced react with the low levels of nitrite reacted metHb instead of the readily available oxyHb and deoxyHb.
To explain how NO can be formed in RBCs without reacting with oxyHb or deoxyHb, Rifkind and collaborators have been able to demonstrate the formation of an intermediate with the electron shared between the NO, the heme iron and perhaps the β-93 thiol of hemoglobin [8,3–15]. This electron delocalization provides for a metastable NO that remains associated with hemoglobin, but which can under the proper conditions be released to the vasculature. These studies provide the basis for the accumulation, within RBCs, of a pool of potentially bioactive NO. It is, however, still necessary to explain how this NO can be released from the RBC.
Recent studies have shown that the nitrite reaction depends on the hemoglobin quaternary conformation [16–18]. We have previously developed a kinetic method to distinguish between the initial nitrite reaction and the dissociation of NO from the electron-delocalized metastable nitrite reacted hemoglobin [14]. Since the dissociation step and not the initial nitrite reaction would be involved in the release of NO from RBCs, we wanted to analyze the effect of the quaternary conformation on the two steps involved in the nitrite reaction. To investigate the effect of the quaternary conformation on the two distinct steps involved in the nitrite reaction, we have in this paper used carboxypeptidase A (CPA) modified hemoglobin, which retains the R-state even when fully deoxygenated. We were then able to perform a detailed analysis of the nitrite reaction for fully deoxygenated T and R state hemoglobins utilizing a combination of chemiluminescence, visible spectroscopy, electron paramagnetic resonance and filtration methods. Based on this analysis, we have shown that while the nitrite reaction is enhanced by R state hemoglobin as previously reported [16] the dissociation of NO from the delocalized intermediate is actually enhanced in T-state hemoglobin. These results can explain the allosteric release of NO to the vasculature by combining the accumulation of potentially bioactive NO in the R quaternary conformation with the release from hemoglobin in the T quaternary conformation when the hemoglobin is also bound to the membrane.
Material and methods
Preparation of Hemoglobin
Hemoglobin was prepared from fresh RBCs as described earlier [8].
Preparation of R-state deoxyhemoglobin
Hemoglobin was incubated with CPA obtained from Sigma Chemical Co., St. Louis, Missouri for 2 hours at 37°C in 0.1M Tris-HCl buffer, pH 8.0. and then passed through a Sephadex G-100 column to remove the enzyme [19]. This reaction cleaved the terminal histidine and tyrosine in the β-chains stabilizing the R–state even when hemoglobin is fully deoxygenated.
Preparation of methemoglobin
Methemoglobin was prepared by oxidizing oxyhemoglobin with a 1.2 molar excess of potassium ferricyanide. The oxidized hemoglobin was then passed down a G-25 Sephadex column to remove the excess ferricyanide and the ferrocyanide formed during the reaction.
T-state methemoglobin was prepared by adjusting the pH to 6.5 and adding one molar equivalent of inositol hexaphosphate (IHP) [20]
Deoxygenation of Hemoglobin
Hemoglobin in 50 mM NaCl and 4 mM phosphate buffer, pH 7.4 (PBS) was deoxygenated in an anaerobic Coy glove box. The glove box uses hydrogen and a palladium catalyst to remove any residual oxygen resulting in <1ppm oxygen. Multicomponent fitting of the deoxyHb sample obtained by this procedure did not detect any oxyHb and no oxyHb accumulation was detected during the course of the experiment.
Determination of Nitrite Consumption
1mM deoxyHb was reacted with ~0.1mM nitrite in PBS, pH 7.4 for 60 min at room temperature (22 °C) in the anaerobic Coy glove box. To quantitate the consumption of nitrite, the reaction was carried out in a petri dish containing 5 ml of R or T state deoxyHb. The reaction solution was placed on a shaker and, at specific time intervals, 0.5 ml aliquots were transferred to microfilterfuge tubes (Rainin Instrument Co. Inc. filters with a 10,000 mol. wt. cutoff) and centrifuged for 2 min at 6,500 RPM using a microfuge (LK Scientific) inside the anaerobic glove box. After centrifugation, 5μl of the filtrate was injected into the Nitric Oxide Analyzer (NOA) purge vessel to determine nitrite by chemiluminescence (see below). At zero time the free nitrite indicates the actual concentration of nitrite added.
Chemiluminescence Method to Determine Nitrite
The Model 280 NOA from Sievers Instruments was used. The sample was injected into the NOA purge vessel containing 7 ml of glacial acetic acid and 1ml of 0.5M ascorbic acid in order to determine the concentration of nitrite as previously described [21].
Chemiluminescence Method to Determine Total Heme-NO
The Model 280 NOA from Sievers Instruments was used in accordance with previously published procedures [8]. With the purge vessel containing 5.5 ml of 100 mM sulfanilamide dissolved in 87.5% glacial acetic acid, 1.0 ml of 1.0 M potassium ferricyanide in water and 0.1 ml antifoam reagent at 37°C, the chemiluminescence measures the concentration of total heme–NO (Hb(II)NO, Hb(III)NO and any heme based intermediates) in a hemoglobin sample [15].
Electron Paramagnetic Resonance (EPR) Determination of Hb(II)NO
Samples were transferred anaerobically to 4 mm clear fused Quartz EPR tubes (707 SQ 250M – WILMAD) and frozen immediately by submerging the EPR tubes in liquid nitrogen and stored at 77 K until EPR measurements were performed. EPR spectra were measured using a Bruker EMX spectrometer with 100 KHz modulation as previously described [8].
The Reaction of Nitrite with deoxyhemoglobin followed by Visible Spectroscopy
After hemoglobin (100 μM) or CPA reacted hemoglobin (57.3 μM) were deoxygenated, the nitrite reaction was initiated by using a gas tight syringe to add nitrite in a septum sealed cuvette with a 1:1 final molar ratio of nitrite to heme. Spectra of hemoglobin from 490 to 640 nm were continuously recorded on a Perkin Elmer Lambda 35 spectrophotometer for 35 min. The resultant spectra obtained in any experiment were analyzed using a least squares multicomponent fitting program (Perkin Elmer Spectrum QuantC v 4.51) to obtain concentrations of deoxyHb, metHb, Hb(II)NO, oxyHb and nitrite bound metHb. Our spectral analysis does not include spectra for the intermediates that we have shown [15] to be formed during the nitrite reaction. As discussed in our early spectral analysis of the nitrite reaction [14], these intermediates are thought to have spectra similar to one of the metHb components and result in higher levels of total metHb than Hb(II)NO. Since no oxyHb was detected, the spectra were also fit excluding oxyHb, without any changes in the concentrations of the other components. To compare the affect of quaternary conformation, the different concentrations of R-state and T-state deoxyhemoglobin (see above) required that the spectral changes determined by visible spectroscopy be plotted as percent changes.
The Reaction of Nitrite with deoxyhemoglobin followed by Chemiluminescence and Electron Paramagnetic Resonance
1 mM deoxyHb or CPA treated deoxyHb was reacted with 250 μM nitrite. For EPR measurements the reaction mixture was prepared in the glove box and at various time intervals aliquots were transferred to EPR tubes and frozen. For the chemiluminescence measurements, the reaction was initiated by using a gas tight syringe to add deoxygenated nitrite to the hemoglobin. 20 to 50 μl samples were injected into the NOA purge vessel at different time intervals using a gas tight syringe, to determine total heme-NO.
The reaction of R and T state methemoglobin with NO
An NO saturated solution of PBS was prepared by bubbling NO through deoxygenated PBS in a sealed vial. The NO used was first bubbled through saturated NaOH to remove impurities. The concentration of this NO saturated solution was determined by chemiluminescence. R or T state metHb was deoxygenated in a septum sealed cuvette. Using a gas tight syringe a volume of the NO saturated solution was injected into the cuvette that contained an amount of NO that was equivalent to the concentration of metHb. Starting immediately spectra from 490 nm to 640 nm were continuously recorded on a Perkin Elmer Lambda 6 spectrophotometer. The resultant spectra obtained in any experiment were analyzed using a least squares multicomponent fitting program to obtain concentrations of metHb, metNO and Hb(II)NO.
Statistical Analysis
Origin 6.1 (Microcal Software, Northampton, MA) was used for analysis of the data. The paired student’s t test was used for comparing samples. Two tailed values of p<0.05 were considered statistically significant.
Results
The Initial Reaction of Nitrite with fully deoxygenated R and T state Hemoglobin
Other investigators [16,17] have used increased oxygenation of hemoglobin to study the effect of the quaternary conformation on the nitrite reaction. However, nitrite reacts with both deoxygenated and oxygenated chains by completely different mechanisms. Thus, the reaction of nitrite with oxygenated chains does not involve the reduction of nitrite, but nevertheless results in the formation of metHb. The interpretation of the nitrite reaction for partially oxygenated hemoglobin is, therefore, complex. To avoid these difficulties, we used CPA to cleave the two carboxy-terminal residues of the β-chains (histidine-146 and tyrosine-145) [19]. This modification disrupts the salt bridges that stabilize the T-state. The oxygen affinity increases and the binding is non-cooperative indicative of the retention of the R-state even when the hemoglobin is fully deoxygenated [22]. We then were able to compare the T and R quaternary conformation of fully deoxygenated hemoglobin without any oxygenation.
The initial process when nitrite reacts with deoxyHb involves the consumption of nitrite together with the utilization of deoxyHb.
| (1) |
To investigate this initial reaction, the consumption of both reactants in Eq 1 (nitrite and deoxyHb) were measured. The consumption of nitrite was determined (Fig. 1) by chemiluminescent analysis of the filtrate (see Methods Section). The consumption of R and T state deoxyHb (Fig. 2) was determined by multicomponent fitting of the visible spectra (see Methods Section).
Figure 1.
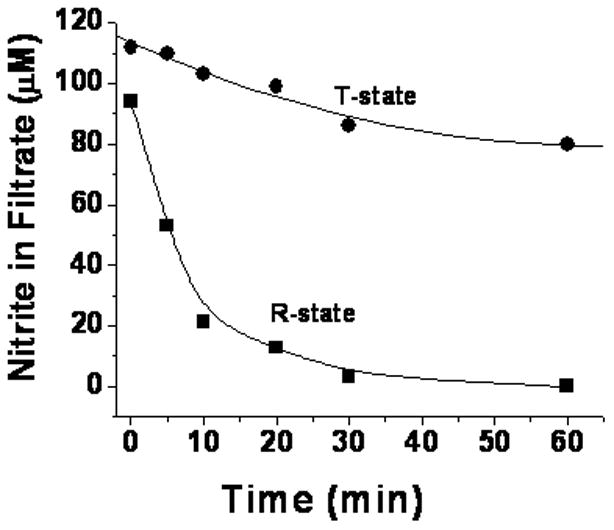
Consumption of nitrite during the reaction with deoxyhemoglobin as a function of time. Nitrite was reacted with 1 mM deoxyHb at a 1:1 molar ratio. At each time point an aliquot of the reaction mixture of T-state deoxygenated hemoglobin (●) and deoxygenated CPA treated hemoglobin in the R-state (■) was filtered under anaerobic conditions and the nitrite in the filtrate was determined by chemiluminescence.
Figure 2.
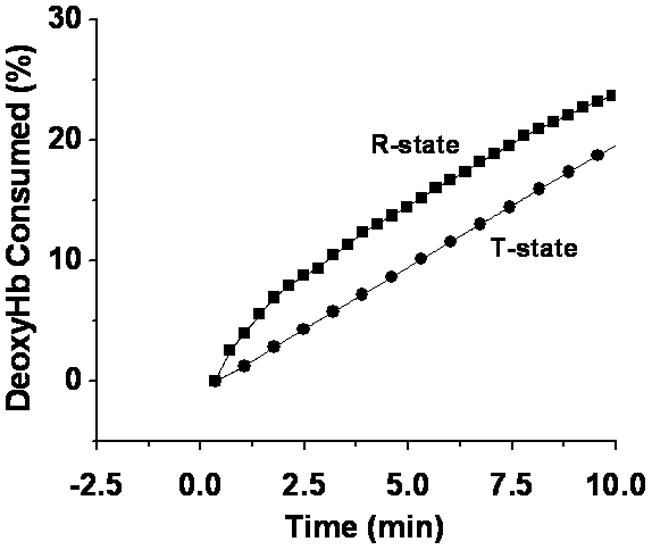
The Percent deoxygenated hemoglobin consumed as a function of time. Nitrite was reacted with deoxyHb at a molar ratio of 1:1 and changes in the visible spectrum were monitored as a function of time for deoxygenated hemoglobin (100 μM) in the T-state (●) and deoxygenated CPA reacted hemoglobin (57.3 μM) in the R-state (■). The percent decrease in deoxyHb at each time point was determined by multicomponent fitting of the spectra (see methods section).
In both experiments R-state hemoglobin reacted faster than T-state hemoglobin. This finding is consistent with the increase in the rate for the reaction when the sample was partially oxygenated [17]. An analogous increase for the reaction was reported when the R-state was trapped by encapsulating oxyHb into a sol-gel prior to deoxygenating the sample [18].
To determine the rate constant (k1) for the initial nitrite reaction we used the initial slope for the consumption of deoxyHb (Fig. 2) obtained from the visible spectroscopy data together with the initial concentrations of deoxyHb and nitrite.
| (2) |
This rate for the initial reaction could have been obtained from the initial slope of either the consumption of deoxyHb or nitrite. We, however, calculated the rate constants using the visible spectral data, which provides multiple values over a narrow time range on the same sample, which is not possible for the filtration determination. The visible spectroscopy data, therefore, provides a more reliable determination of the initial slope and the rate constant. The higher rate constant for the initial reaction with R–state than with T-state deoxyHb obtained from Eq 2 is shown in Fig. 3A. The 5.6 fold higher rate constant for R state hemoglobin is consistent with kinetic data reported by Salhany [23].
Figure 3.
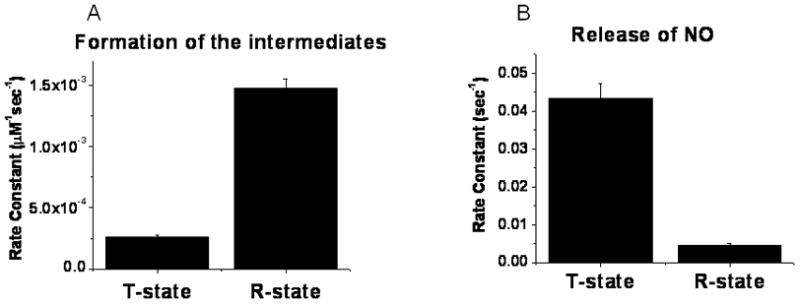
Comparison of rate constants ±SE deteremined from visible spectroscopy data for (A) formation of intermediates and (B) dissociation of NO from the intermediates during the reaction of T-state deoxyHb and CPA reacted R-state deoxyHb with nitrite (details of the procedures given in methods section).
The Formation of the Electron Delocalized Species in the R and T Quaternary Conformation
The initial intermediate formed when nitrite reacts with deoxyHb is the nitrite bound to deoxyHb {Hb(II)--ONO− }, which then goes on to form the electron delocalized species {Hb(II)NO+ ↔Hb(III)NO} where the electron is shared between the NO and the iron [15].
| (3) |
We have shown [15] that the chemiluminescence signal obtained in acetic acid without added ascorbate to reduce any bound nitrite detects the delocalized intermediate as well as any Hb(II)NO, but not the nitrite bound to deoxyHb, which accounts for ~50% of the reacted nitrite. In figure 4 we show the time dependent increase in this chemiluminescence signal for the R and T quaternary conformation. The chemiluminescence signal, which does not include Hb(II)--ONO−, levels of well below the concentration of reacted nitrite. However, the faster time dependent formation of the chemiluminescent signal for the R state than the T-state implies that the formation of the delocalized intermediate is also accelerated in the R quaternary conformation. This increase, however, cannot be used to determine the rate constants for the formation of the delocalized intermediate for two reasons. (1) Since the initial nitrite complex needs to form prior to the formation of the intermediate (Eq. 3), the slower formation of the chemiluminescence signal in the T-state can at least in part be attributed to a smaller steady state level of the nitrite bound deoxyHb and not a slower rate for the formation of the delocalized intermediate. (2) The chemiluminescence detects Hb(II)NO in addition to the delocalized intermediate. Therefore, the increase in the rate for the formation of the chemiluminescent signal in the R conformation includes a contribution of the Hb(II)NO formed by the binding of NO, released from the intermediate, to unreacted deoxyHb.
Figure 4.
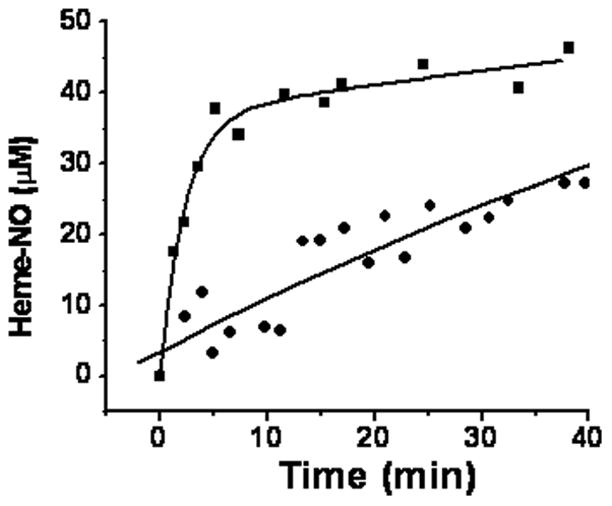
Formation of heme-NO detected by chemiluminescence (see methods for chemiluminescence method) as a function of time. 120μM nitrite was reacted with 480μM deoxygenated hemoglobin in the T-state (●) or deoxygenated CPA treated hemoglobin in the R-state (■).
The Dissociation of NO from the Delocalized Intermediate and the Formation of Hb(II)NO in R-state and T-state deoxyhemoglobin
To compare the release of NO in the R and T state it is necessary to determine the rate constant for the dissociation of NO from the intermediate. We have previously shown [14] that the delocalized intermediate is in equilibrium with an Hb(III)NO complex that has been shown to rapidly dissociate NO [24]. The rate limiting step for the release of NO is, therefore, the conversion of the delocalized species to the non-delocalized Hb(III)NO complex, which rapidly dissociates the NO.
| (4) |
| (5) |
The released NO immediately (kon= 1.4 × 107) reacts with any available deoxyHb in both the R-state and T-state forming Hb(II)NO [ref methods in nitric oxide research VG Kharitonov, j boneventutre and VJ Sharmapp39–45)].
| (6) |
The formation of Hb(II)NO determined by multicomponent analysis of the changes in visible spectroscopy (Fig. 5), thus, reflects the dissociation of NO from the intermediate. This formation of Hb(II)NO, however, requires the prior formation of the delocalized intermediate {Hb(II)NO+↔Hb(III)NO} and would not be expected to produce the initial increase observed for R-state deoxyHb. This initial increase can, however, be related to the formation of a paramagnetic intermediate that is being investigated {reference abstract biophysics meeting 2011}.
Figure 5.
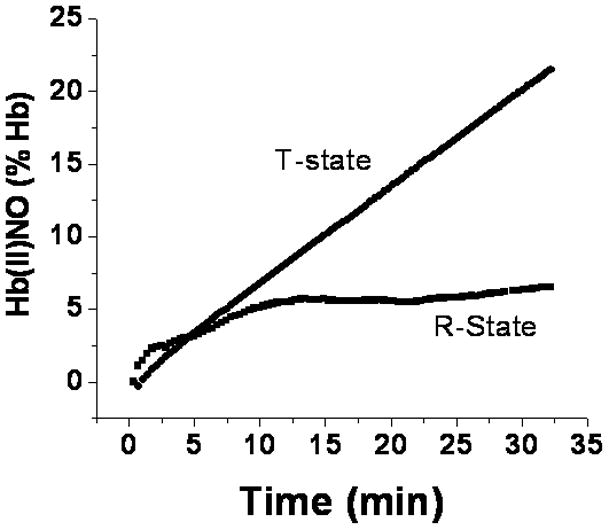
Determination of the percent of hemoglobin converted to Hb(II)NO by visible spectroscopy as a function of time for a 1:1 molar ratio of nitrite and deoxyHb. (●), deoxygenated hemoglobin in the T-state and (■), deoxygenated CPA reacted hemoglobin in the R-state.
The rate constant for the release of NO (k2) is given by the expression,
| (7) |
and depends on the formation of Hb(II)NO (d[Hb(II)NO]/dt) as well as the concentration of the intermediates. Since there are no intermediates present at zero time, we have in our previous study determined the rate constant for the release of NO from the data obtained after 35 min. At this time the non-CPA reacted Hb is still in the T-state because of the limited consumption of nitrite (Fig. 1) and formation of metHb and Hb(II)NO, which can shift the conformation into the R-state. For this purpose, it is necessary to determine the final concentration of intermediates as well as the final values of d[Hb(II)NO]final/dt.
Since intermediates have visible spectra similar to the metHb components [14], they result in higher levels of metHb and have a negligible effect on the levels of the better defined deoxyHb and Hb(II)NO. The overall nitrite reaction, without the formation of intermediates, produces equal concentrations of metHb and Hb(II)NO and the decrease in deoxyHb should be equivalent to twice the concentration of Hb(II)NO. Therefore the concentration of intermediates can be directly obtained reliably from the multicomponent analysis of the visible spectroscopy data, which provides concentrations of deoxyHb and Hb(II)NO for each time point (Fig. 6).
Figure 6.
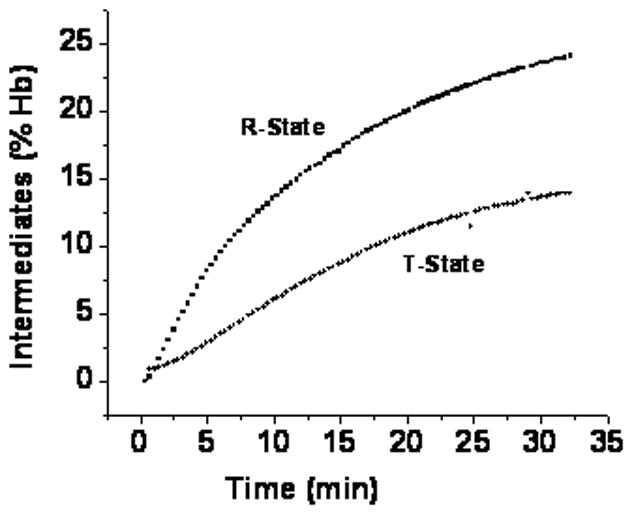
Time course for the formation of intermediates detected by visible spectroscopy. The Percent hemoglobin in the intermediate species was determined by substracting deoxyHb consumed from 2[Hb(II)NO] for deoxygenated T-state hemoglobin (●) and deoxygenated CPA reacted hemoglobin.
| (8) |
Although EPR can be used to determine Hb(II)NO, the limited number of points with different samples for each time point make it difficult to accurately determine d[Hb(II)NO]final/dt. Furthermore, a determination of the concentration of intermediates requires a comparison of chemiluminescence or filtration data with EPR data [8], which is difficult to reliably quantitate. We have, therefore, used the visible spectroscopy data (Figs. 5 & 6) to compare the rate for the dissociation of NO from the delocalized intermediate in the R and T quaternary conformation (Fig. 3B). The lower level of intermediates in the T-state than the R-state (Fig. 6) coupled with a faster final rate for the formation of Hb(II)NO (Fig. 5 ) results in a significantly higher rate for the dissociation of NO from the delocalized intermediate in the T-state than the R-state (Fig. 3B).
Although we are using the visible spectroscopy data to determine the rate constants in Fig. 3B, these results are consistent with the results obtained from EPR, filtration and chemiluminescence. We, thus, find a more pronounced R state increase in chemiluminescence (Fig. 4) and nitrite consumption (Fig. 2) that is not paralleled by an increase in the EPR determination of Hb(II)NO indicating lower levels of the intermediate in the T-state. Furthermore, determinations of d[Hb(II)NO]final from the EPR data (Table 1) also indicate that Hb(II)NO is forming more rapidly with T-state deoxyHb.
Table 1.
Final slope for the Formation of Hb(II)NO (d[Hb(II)NO]/dt) from the EPR data
Standard error
The reaction of NO with methemoglobin in the R and T Quaternary Conformation
Equations 3–5 involve the reaction of nitrite with deoxyHb forming the electron delocalized intermediate which releases NO producing metHb. This same reaction pathway going through the same intermediate with the eventual formation of deoxyHb instead of metHb takes place when NO is added to metHb [25].
| (9) |
Analogous to the initial formation of a nitrite complex with deoxyHb prior to the formation of the delocalized intermediate (Eq. 3), we have previously shown [14] that a complex of metHb with NO that has the characteristic spectrum of Hb(III)NO forms within seconds of the addition of NO. This complex decays within 10 min resulting in the formation of the electron delocalized intermediate. The non-delocalized Hb(III)NO formed in Eq. 9 is the same complex that forms when NO is released from the delocalized intermediate (Eqs. 4 & 5).
To complement our results on the role of the quaternary conformation on the nitrite reaction, we studied the effect of the quaternary conformation on the reaction of NO with metHb. This was accomplished by using low pH and IHP to shift the neutral pH metHb, which exist in the R-quaternary conformation, into the T-quaternary conformation [20]. As shown in Fig. 7 there is a rapid binding of NO to metHb in both quaternary conformations. The extent of this reaction was more pronounced in the R-state than the T-state, which can be attributed to the slower decay of the Hb(III)NO (Fig. 7) with a T1/2=155 sec in the R-state compared to a T1/2=56 sec in the T-state.
Figure 7. The reaction of NO with metHb.
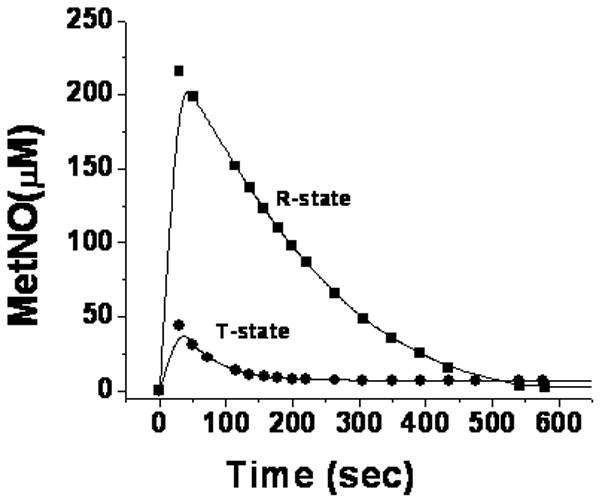
The reaction of 0.724mM metHb with a stoichiometric equivalent of NO. R-state metHb at neutral pH was converted to T-state metHb by adjusting the pH to 6.5 and adding a molar equivalent of IHP. The formation and decay of HbIII(NO) is monitored spectrophotometrically for T-state (●) and R-state (■) metHb.
The decay of the Hb(III)NO, is faster than reductive nitrosylation [25] and as indicated in Eq. 9, coincides with the formation of the electron delocalized complex [14]. This formation of the delocalized intermediate from Hb(III)NO is the reversal of the rate limiting step for the dissociation of NO during the nitrite reaction (eq. 4). These results, thus, indicate that both the forward and reverse rate constants for this reaction are slower in the R state than the T state. These kinetic results imply that the transition state involved in the dissociation reaction NO is favored by the T quaternary conformation.
In addition, by combining the kinetics for the forward and reverse transition between non-delocalized Hb(III)NO and the delocalized intermediate, the effect of the quaternary conformation on the equilibrium and the relative stability of the ground state for each complex can be obtained. Thus, the half times for the decay of the Hb(III)NO species in figure 7 indicate that the relative first order rate constants in the T-state is 2.8 fold greater than in the R state. This increase in the T-state rate for formation of the delocalized intermediate is about 3 fold less than the increase in the rate for the release of NO from the intermediate (Fig.2B). Therefore, the equilibrium constant for the formation of the metNO structure that does not involve electron delocalization between Hb(II)NO+ and Hb(III)NO (Eq. 4) is higher in the T-state than the R-state and the non-delocalized Hb(III)NO complex is more stable in the T-state.
The reaction of NO with metHb also produces Hb(II)NO. The kinetics for the formation of Hb(II)NO does not, however, provide information about the conversion of the intermediate back to Fe(II) hemoglobin, because of side reaction involving free NO. Free NO is the result of the relatively low affinity of metHb for NO. The excess NO, not present during the nitrite reduction process, affects the pathway for reductive nitrosylation [26]. In addition NO very rapidly reacts with the thiyl radical species that is in equilibrium with the electron delocalized intermediate [13] resulting in the formation of hemoglobin with NO on both the thiol and the heme iron (NOS---Hb(II)NO). Since the spectral properties of hemoglobin are dominated by the heme, the spectral properties of this complex correspond to that of Hb(II)NO.
Discussion
Previous studies have reported [16–18] that the nitrite reaction with deoxyHb is faster in the liganded R-quaternary conformation than in the T-quaternary conformation. The original studies by [17] Huang et al. made this important observation by comparing the nitrite reaction at different partial pressures of oxygen and observed an increase in the reaction rate at intermediate oxygen pressures where the quaternary conformations shifts from T to R and unliganded chains that can react with nitrite are still present. At these partial pressures of oxygen a mixture of R and T state hemoglobin exists and they were not able to directly compare the nitrite reaction of R and T state hemoglobin. In the study of Roche et al. [18] this issue was resolved by using encapsulated hemoglobin to retain hemoglobin in the T and R state. In our study we were able to compare T and R state hemoglobin by using CPA modified hemoglobin. CPA cleaves the two terminal residues of the β-chains resulting in hemoglobin that retains the R-state in solution even when fully deoxygenated. Results with this modified hemoglobin were then compared with fully deoxygenated HbA, which retains the T state until a major fraction of the hemes react with nitrite.
Our studies also indicate an increase in the initial steps of the nitrite reaction for R state hemoglobin (Scheme 1) as seen by the consumption of nitrite (Fig. 1), the decrease in deoxyHb (Fig. 2) and the formation of a chemiluminescence signal (Fig. 4).
Our earlier studies [8,14,15] have established the accumulation of an electron delocalized intermediate during the reaction of nitrite with deoxyHb. The release of NO from hemoglobin, therefore, does not depend on the rate for this initial nitrite reaction with deoxyHb, but on the rate for the dissociation of NO from the metastable delocalized intermediate. This question was not addressed by the other studies, which were for the most part performed with a large excess of nitrite, which reacts with the intermediate [14] destabilizing it.
At the low molar ratios of nitrite:heme used in our study, the formation of Hb(II)NO which monitors the final dissociation of NO is actually faster in the T-state than the R-state (Fig. 5, Table 1 & Scheme 1). Under these conditions, a detailed kinetic analysis of the consumption of deoxyHb and the formation of Hb(II)NO was able to distinguish between the formation of the intermediate and the release of NO from the intermediate (Fig. 3). Our kinetic analysis, thus, indicates that the R-state not only favors the initial reaction of nitrite with deoxyHb, which is consistent with a lower affinity in the T-state for all ligands, but also favors the formation of the electron delocalized intermediate (Scheme 1), which reflects the accumulation of the pool of potentially bioactive NO. Although the formation of the electron delocalized intermediate is favored by the R-state, the dissociation of NO from the intermediate and, therefore, the potential release of NO bioactivity is actually favored by the T-state (Scheme 1).
The same intermediate formed during the reaction of nitrite with deoxyhemoglobin is also formed when NO reacts with metHb {reference biochemistry paper visible spectroscopy}. The delocalized intermediate is formed after the initial binding of NO to metHb that produces the known metHbNO visible spectrum.
Some investigators {reference kim Shapiro papers }have argued that Hb(III)NO cannot exist as both an electron delocalized complex {Hb(II)NO+↔Hb(III)NO} and non delocalized Hb(III)NO (Eq.9). A recent paper (navati and friedman, JACS) was primarily concerned with the reaction of nitrite reacted MetHb with NO. They, however, also observed a new spectral species formed when NO was reacted with glass embedded metHb under dry conditions that appeared after the usual metHbNO species that was not the reduced Hb(II)NO. This spectral species can be the delocalized Hb(III)NO species that we have reported.
By combining kinetic data on the dissociation of NO during the nitrite reaction (Fig. 3B) with kinetic data for the formation of the delocalized intermediate when NO reacts with metHb (Fig. 7), we are able to show that the delocalized intermediate is more stable in the R- quaternary conformation than is the non-delocalized Hb(III)NO (Scheme 1). Furthermore the T-quaternary conformation facilitates the transition between the two complexes involving Hb(III)NO. Since non-delocalized Hb(III)NO is the complex with an off rate for NO of ~1 sec [27], the potential release of NO from hemoglobin is facilitated by the T quaternary conformation.
The opposite effects of the quaternary conformation on the formation of the intermediate and the dissociation of NO can be explained in terms of a conformational effect on the distal ligand pocket (Scheme 1), which influences the interactions involving the distal histidine. The proton facilitated binding of nitrite to deoxyHb (Scheme 1) would be expected to involve at neutral pH hydrogen bonding between the distal histidine and the bound nitrite [28]. Hydrogen bonding in the R-state to ligands of both Fe(II) and Fe(III) hemoglobins have been reported [29,30]. Hydrogen bonding of nitrite to the distal histidine would be particularly important for the binding of anionic nitrite to deoxyHb. The enhanced formation of the electron delocalized intermediate in the R-state suggests that the distal histidine interaction also stabilizes the electron delocalized intermediate, perhaps through an interaction of the oxygen of the nitrosonium ion with the Nε of the distal histidine (Scheme 1).
Our studies on the autoxidation of oxyHb and the release of superoxide [31] have been interpreted in terms of a nucleophilic interaction of the Nε of the distal histidine with the heme iron [32]. The autoxidation reaction is very slow in R-state fully oxygenated hemoglobin and is facilitated with partially oxygenated hemoglobin with greater T-state properties. (The autoxidation cannot be studied for fully deoxygenated T-state hemoglobin, since bound oxygen is required for the oxidation) These results suggest that the T-quaternary conformation alters the distal heme pocket configuration facilitating the approach of the histidine to the iron instead of the ligand (Scheme 1). This same ligand pocket rearrangement can facilitate the dissociation of NO from the delocalized intermediate by the nucleophilic interaction of the Nε of the distal histidine with the heme iron of the delocalized intermediate. This process will transfer an electron to the Hb(II)NO+ component of the intermediate facilitating the formation of the non-delocalized Hb(III)NO configuration (Scheme 1) and the subsequent displacement of NO from this complex.
The stabilization of the transition state between both forms of Hb(III)NO (the delocalized complex and the non-delocalized complex) by the T-quaternary conformation implies that bonding of the histidine with the heme-iron facilitates the required conformational change (Scheme 1). Although the non-delocalized Hb(III)NO complex is also stabilized by H-bonding of the histidine with the bound NO (Scheme 1), the greater stability of the delocalized intermediate in the R-state implies that H-bonding stabilizes the delocalized intermediate to a greater extent explaining the relatively rapid dissociation of NO from non-delocalized Hb(III)NO.
The increased rate for the formation of the intermediate in the R-state coupled with a slower dissociation of NO explains the accumulation of a pool of potentially bioactive NO as the hemoglobin begins to be deoxygenated, but retains the R-quaternary conformation (when 2–3 oxygens are still bound). At the same time the increased rate for the dissociation of NO in the T-state when hemoglobin becomes further deoxygenated is consistent with the release of NO to the vasculature by RBCs under hypoxic conditions [5].
Conclusion
The reduction of nitrite by deoxygenated hemoglobin chains to produce NO is well established. It is, however, necessary to explain how NO can be released from the RBC without being quenched by the rapid reactions of oxyHb and deoxyHb with NO. A potential explanation for this dilemma can be explained by the properties of the delocalized intermediate. As hemoglobin begins to be deoxygenated, while still in the R-state the intermediate accumulates retaining the potential NO bioactivity, while preventing its quenching by hemoglobin. Further deoxygenation that stabilizes the T-state enhances membrane binding and also facilitates the dissociation of NO from the delocalized intermediate. A significant fraction of the NO released from hemoglobin associated with the membrane can potentially diffuse out of the cell to the vasculature without being trapped by hemoglobin.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging.
Abbreviations
- DeoxyHb
deoxyhemoglobin
- metHb
methemoglobin
- oxyHb
oxyhemoglobin
- CPA
carboxypeptidase A
- IHP
Inositol hexaphosphate
- PBS
phosphate buffered saline
- NO
nitric oxide
- RBC
red blood cell
- SNOHb
S-nitrosohemoglobin
- NOA
nitric oxide analyzer
References
- 1.Ignarro LJ. Nitric oxide: a unique endogenous signaling molecule in vascular biology. Biosci Rep. 1999;19:51–71. doi: 10.1023/a:1020150124721. [DOI] [PubMed] [Google Scholar]
- 2.Gautier C, van FE, Mikula I, Martasek P, Slama-Schwok A. Endothelial nitric oxide synthase reduces nitrite anions to NO under anoxia. Biochem Biophys Res Commun. 2006;341:816–821. doi: 10.1016/j.bbrc.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR., Jr Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 4.Ostergaard L, Stankevicius E, Andersen MR, Eskildsen-Helmond Y, Ledet T, Mulvany MJ, Simonsen U. Diminished NO release in chronic hypoxic human endothelial cells. Am J Physiol Heart Circ Physiol. 2007;293:H2894–H2903. doi: 10.1152/ajpheart.01230.2006. [DOI] [PubMed] [Google Scholar]
- 5.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 6.Dietrich HH, Ellsworth ML, Sprague RS, Dacey RG., Jr Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol. 2000;278:H1294–H1298. doi: 10.1152/ajpheart.2000.278.4.H1294. [DOI] [PubMed] [Google Scholar]
- 7.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, III, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 8.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J Biol Chem. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 9.Cao Z, Bell JB, Mohanty JG, Nagababu E, Rifkind JM. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: a new mechanism for nitrite-induced vasodilation. Am J Physiol Heart Circ Physiol. 2009;297:H1494–H1503. doi: 10.1152/ajpheart.01233.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 11.Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006;291:H2026–H2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 12.Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A, Jiang A, He X, Azarov I, Seibert R, Mehta A, Patel R, King SB, Hogg N, Ghosh A, Gladwin MT, Kim-Shapiro DB. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol. 2007;3:785–794. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- 13.Nagababu E, Ramasamy S, Rifkind JM. S-nitrosohemoglobin: a mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide. 2006;15:20–29. doi: 10.1016/j.niox.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Nagababu E, Ramasamy S, Rifkind JM. Intermediates detected by visible spectroscopy during the reaction of nitrite with deoxyhemoglobin: the effect of nitrite concentration and diphosphoglycerate. Biochemistry. 2007;46:11650–11659. doi: 10.1021/bi700364e. [DOI] [PubMed] [Google Scholar]
- 15.Salgado MT, Nagababu E, Rifkind JM. Quantification of intermediates formed during the reduction of nitrite by deoxyhemoglobin. J Biol Chem. 2009;284:12710–12718. doi: 10.1074/jbc.M808647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, Hogg N. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem. 2005;280:31126–31131. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 17.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roche CJ, Dantsker D, Samuni U, Friedman JM. Nitrite reductase activity of sol-gel-encapsulated deoxyhemoglobin. Influence of quaternary and tertiary structure. J Biol Chem. 2006;281:36874–36882. doi: 10.1074/jbc.M603914200. [DOI] [PubMed] [Google Scholar]
- 19.Antonini E, Wyman J, ZITO R, Rossi-Fanelli A, CAPUTO A. Studies on carboxypeptidase digests of human hemoglobin. J Biol Chem. 1961;236:C60–C63. [PubMed] [Google Scholar]
- 20.Perutz MF, Fersht AR, Simon SR, Roberts GC. Influence of globin structure on the state of the heme. II. Allosteric transitions in methemoglobin. Biochemistry. 1974;13:2174–2186. doi: 10.1021/bi00707a027. [DOI] [PubMed] [Google Scholar]
- 21.Nagababu E, Rifkind JM. Measurement of plasma nitrite by chemiluminescence without interference of S-, N-nitroso and nitrated species. Free Radic Biol Med. 2007;42:1146–1154. doi: 10.1016/j.freeradbiomed.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kilmartin JV. Removal of specific C-terminal residues from human hemoglobin using carboxypeptidases A and B. Methods Enzymol. 1981;76:167–171. doi: 10.1016/0076-6879(81)76125-1. [DOI] [PubMed] [Google Scholar]
- 23.Salhany JM. Kinetics of reaction of nitrite with deoxy hemoglobin after rapid deoxygenation or predeoxygenation by dithionite measured in solution and bound to the cytoplasmic domain of band 3 (SLC4A1) Biochemistry. 2008;47:6059–6072. doi: 10.1021/bi8000819. [DOI] [PubMed] [Google Scholar]
- 24.Sharma VS, Traylor TG, Gardiner R, Mizukami H. Reaction of nitric oxide with heme proteins and model compounds of hemoglobin. Biochemistry. 1987;26:3837–3843. doi: 10.1021/bi00387a015. [DOI] [PubMed] [Google Scholar]
- 25.Ford PC, Fernandez BO, Lim MD. Mechanisms of reductive nitrosylation in iron and copper models relevant to biological systems. Chem Rev. 2005;105:2439–2455. doi: 10.1021/cr0307289. [DOI] [PubMed] [Google Scholar]
- 26.Addison AW, Stephanos JJ. Nitrosyliron(III) hemoglobin: autoreduction and spectroscopy. Biochemistry. 1986;25:4104–4113. doi: 10.1021/bi00362a018. [DOI] [PubMed] [Google Scholar]
- 27.Sharma VS, Isaacson RA, John ME, Waterman MR, Chevion M. Reaction of nitric oxide with heme proteins: studies on metmyoglobin, opossum methemoglobin, and microperoxidase. Biochemistry. 1983;22:3897–3902. doi: 10.1021/bi00285a026. [DOI] [PubMed] [Google Scholar]
- 28.Lukin JA, Simplaceanu V, Zou M, Ho NT, Ho C. NMR reveals hydrogen bonds between oxygen and distal histidines in oxyhemoglobin. Proc Natl Acad Sci U S A. 2000;97:10354–10358. doi: 10.1073/pnas.190254697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaanan B. Structure of human oxyhaemoglobin at 2.1 A resolution. J Mol Biol. 1983;171:31–59. doi: 10.1016/s0022-2836(83)80313-1. [DOI] [PubMed] [Google Scholar]
- 30.Asher SA, Adams ML, Schuster TM. Resonance Raman and absorption spectroscopic detection of distal histidine--fluoride interactions in human methemoglobin fluoride and sperm whale metmyoglobin fluoride: measurements of distal histidine ionization constants. Biochemistry. 1981;20:3339–3346. doi: 10.1021/bi00515a004. [DOI] [PubMed] [Google Scholar]
- 31.Balagopalakrishna C, Manoharan PT, Abugo OO, Rifkind JM. Production of superoxide from hemoglobin-bound oxygen under hypoxic conditions. Biochemistry. 1996;35:6393–6398. doi: 10.1021/bi952875+. [DOI] [PubMed] [Google Scholar]
- 32.Levy A, Rifkind JM. Low-temperature formation of a distal histidine complex in hemoglobin: a probe for heme pocket flexibility. Biochemistry. 1985;24:6050–6054. doi: 10.1021/bi00343a005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.