

Abstract
Steroidal bivalent ligands for the estrogen receptor (ER) were designed using crystal structures of ERα dimers as a template. The syntheses of several 17α-ethynylestradiol-based bivalent ligands with varying linker compositions and lengths are described. The binding affinities of these bivalent ligands for ERα and ERβ were determined. In the two series of bivalent ligands that we synthesized, there is a clear correlation between linker length and binding affinity, both of which reach a maximum at the same tether length. Further studies are underway to explore aspects of bivalent ligand and control compound binding to the ERs and their effects on ER dimer formation; these results will be reported in a subsequent publication.
Keywords: Estrogen Receptor, Bivalent Ligand, Multivalent Ligand, Dimer
Introduction
A multivalent ligand is a single molecule comprised of multiple individual ligands linked by a tether. Such ligands have often been used to increase binding affinity, to probe dimer or cluster formation or to explore subsite binding on a receptor.1-7 In medicinal chemistry, multivalent ligands have been used as inhibitors of several biological targets, including, among others, HIV protease, glycosidase and the opioid and muscarinic receptors.8
The estrogen receptor has a brief history involving multivalent, specifically bivalent, ligands (Figure 1). In 1994, our research group was the first to investigate the effects of bivalent ligands on ER by tethering two hexestrol (a non-steroidal ER agonist) ligands with varying lengths of polymethylene and polyethylene glycol (PEG) spacers (1).9 Another early bivalent ER ligand involved dimer formation through an iron carbonyl system built from ethynylestradiol (2); this molecule gave poor results in binding assays, presumably because its tether length was too short.10 In 1999, Bérubé et al.11 reported the synthesis of tethered triphenylethylene molecules (3); ER binding affinities were not reported, and these compounds showed low potencies in cell-based assays, with no evidence that effects were ER mediated. The most recent reports of bivalent ligands for ER involved the synthesis of two estradiol molecules linked at the 17α position by varying lengths of PEG chains (4), but again the binding affinities for these compounds were not reported.12,13 By using the crystal structure of ER to rationally design bivalent ligands, we hoped to prepare higher affinity ligands with varying tether lengths and compositions which could offer insight into ER dimer formation and stability.
Figure 1.

Some previously reported ER bivalent ligands and their relative binding affinity (RBA) values, estradiol = 100%
Results and Discussion
Steroidal Bivalent Ligand Design
Our bivalent ER ligands were designed based on the crystal structure of the ERα ligand binding domain bound to estradiol (E2) and literature reports of estrogen ligand conjugates. The bivalent ligands prepared by Bergman et al.9 and Osella et al.10 were published before the crystal structure of the ER ligand binding domain was released,14 so it could not have been used as a guide for their ligand design. Although Bérubé and coworkers reported their first bivalent ligands in 1999,11 two years after the publication of the first ER crystal structure, they do not mention using it as a basis for bivalent ligand design in this work,11 or in their later papers.12,13 We hoped to gain a better understanding of bivalent ligand binding to ER by considering not only the structure of the receptor but also by carefully selecting how the ligands are tethered, relying as well on published reports of functionalized estradiol.15
The crystal structure of the ERα dimer with E2 bound16 was modeled using the Sybyl modeling software package (Figure 2). It is worth noting that the ER dimer possesses C2 symmetry, which simplifies bivalent ligand design, because bivalent ligands built from the same enantiomer of E2 are also C2 symmetric. The closest distance between the E2 ligands is between the carbons at the 17-position of the steroids and was found to be 26 Å, seemingly close enough to be linked by a molecular tether. It is fortunate that the point of closest distance between ligands in the crystal structure, the 17α-position, is known to tolerate substitution very well;15 in fact, 17α-ethynylestradiol (EE2) binds with a nearly three-fold higher affinity to ER than E2 itself.17
Figure 2.

Figure showing the distance between the 17α carbons of two estradiol molecules (space filling: grey = carbon, red = oxygen) within the ERα homodimer (teal ribbon). Figures were generated with Sybyl 7.3 from the corresponding research collaboratory for structural bioinformatics protein data bank (RCSB-PDB file names: 1ERE for E2).
Our design was also supported by the numerous reports of 17α-phenylethynylestradiol derivatives tethered to other molecules such as dendrimers,18 fluorophores,19 radioactive and luminescent organometallic complexes,20-22 Pt(II) fragments,23 and photo affinity tags,24 all of which show reasonably high binding affinity for ER. While modeling of a phenyl group onto the end of the ethynyl substituent causes severe interactions with the ERα-estradiol protein structure, X-ray analysis of closely related 17α-phenylvinyl estrogens with ERα shows that when presented with this larger substituent at 17α, the ER unwinds a short helix (helix 8), thereby generating a large pocket that accommodates the phenyl group and provides access to the exterior of the protein.25 This suggests that ER will respond in the same way to the phenylethynyl group, which is nearly isostructural with the phenylvinyl group.
Due to the synthetic accessibility of 17α-phenylethynylestradiols and the well established and structurally confirmed tolerance of ER for this substitution, we chose to prepare bivalent ligands with varying tether lengths and composition based on 17α-phenylethynylestradiol (5, Scheme 1). The synthesis of these estrogens is relatively straightforward and involves palladium-catalyzed Sonogashira coupling of EE2 with an aryl halide.26
Scheme 1.

Synthesis of Bivalent Ligands
Initially, bivalent ligands with hexaethylene glycol tethers were prepared because the linker is commercially available and seemed long enough to bridge the distance between the ligand binding pockets in the ER dimer complex (26 Å). With this commercial tether, the distance between the 17-carbons in the hexaethylene glycol bivalent ligand in a fully extended conformation was found to be 35.7 Å using Chem3D Pro 7.0 (see below). Because it is possible that the hexaethylene glycol linker could adopt a non-staggered, coiled conformation (see below),27-30 we felt that the additional 10 Å in the tether length might help the two steroids to span the 17α-17α distance of 26 Å.
The synthesis of a hexaethylene glycol tethered bivalent ligand with amine linkages (10) is shown in Scheme 2. Hexaethylene glycol was refluxed with p-toluenesulfonyl chloride in CH2Cl2 in the presence of excess triethylamine and DMAP to afford di-tosylated hexaethylene glycol (6) in good yield (Scheme 2). Diamino hexaethylene glycol (8) was prepared from the ditosylate 6 by forming the diazide (7), followed by reduction in the presence of palladium on carbon in methanol under a hydrogen atmosphere. Bivalent ligand 10 was prepared by stirring the benzaldehyde derivative of 17α-ethynylestradiol (9)18 and diamine 8 in CH2Cl2 in the presence of excess anhydrous K2CO3 for 24–48 hours. Formation of the imine was confirmed by proton NMR by the disappearance of the aldehyde peak around 10 ppm and the appearance of the imine peaks (cis and trans) near 8 ppm.18 The crude imine was dissolved in MeOH and excess NaBH4 was added and stirred at room temperature for 5-10 minutes to give the bivalent estrogen 10 in good yield over two steps.
Scheme 2.

A second hexaethylene glycol bivalent ligand, with phenyl ether linkages, was also synthesized (12, Scheme 3). Bis-aryl iodide (11) was prepared by stirring the PEG ditosylate 6 with 4-iodophenol and excess K2CO3 in 2-butanone for 24 hours. Palladium-catalyzed Sonogashira coupling31,32 was used to make phenylethynyl derivatives of 17α-ethynylestradiol (EE2) from aryl iodides 11. By stirring 11 with EE2 in presence of CuI and Pd catalyst in Et3N and CH3CN at room temperature, bivalent ligand 12 was isolated in low yield. The low yield for the Sonogashira coupling can be attributed to homo coupling of the EE2, giving a diacetylene dimer, and incomplete reaction to give the mono-coupled product, both of which were isolated as side products. Homo coupling of alkynes is a common side reaction in Sonogashira couplings.33
Scheme 3.

Because ethylene glycol polymers are reported to form coiled conformations27-30 and are relatively polar, we chose to explore tethers constituted of less polar glycol ethers, namely the polyethylene and polybutylene glycol ethers. In these systems, the helical or coiled conformation enforced by the inherent gauche preference of the ethylene glycol unit should be relaxed. Polyethylene glycols are commercially available in discrete lengths, but polypropylene and polybutylene glycols are not. Therefore, we prepared discrete polypropylene and polybutylene glycols using modifications of previously described syntheses,34 the details of which can be found in the supporting information. Bivalent ligands with these polypropylene and polybutylene linkers were synthesized as shown in Scheme 4. The phenol linkage was chosen over the amine because it can be prepared in fewer synthetic steps. The linker diol was converted to the methanesulfonate, which was found to be more reactive and easier to prepare than the corresponding tosylate. The mesylates were then alkylated with p-iodophenol and subjected to Sonogashira coupling to give the bivalent ligands.
Scheme 4.

Bis-mesylates were prepared from the polypropylene and polybutylene glycol linkers in good to excellent yield by stirring with methanesulfonyl chloride in Et3N and CH2Cl2 at 0 °C for 2 hours (Scheme 4). The yields for this reaction ranged from 76-100%, with the differences being attributed to different workup and purification procedures. Some isolated products were quite pure by 1H NMR and could be used in subsequent reactions without further purification (see below), while others needed to be purified by column chromatography. In these latter cases, the yields were lower because these ether methanesulfonates lack a chromophore; so, it was difficult to determine by TLC when they were eluting from the column. The mesylates were then converted to the corresponding bis-aryliodides in good yield by stirring with p-iodophenol and K2CO3 in 2-butanone at reflux. Once chromophores were present, purification of the alkylated products using silica gel flash chromatography was straightforward, and any impurities remaining in the mesylates could be easily removed in this step.
Bivalent ligands were prepared in moderate yields by coupling the bis-aryliodides with EE2 under improved Sonogashira conditions. By changing solvents from CH3CN to THF, using piperidine in place of Et3N as the base, and palladium tetrakis(triphenylphosphine) as the catalyst, yields were improved substantially. Generally, purification on multiple (2-3) silica gel columns using different solvent systems was necessary to obtain the products in pure form, usually as white foamy solids. All of the bivalent ligands were completely characterized by 1H and 13C NMR and high resolution mass spectrometry. Compound purity was determined using 1H NMR and HPLC, with all bivalent ligands being at least 95% pure. The presence of even a small amount of residual EE2 in the bivalent ligand product would result in higher than actual RBA values. The difference in HPLC retention times of EE2 and the bivalent ligands is substantial, making HPLC an excellent tool for the detection of residual EE2. All bivalent ligands were free of EE2 by this analysis method.
Biological Results for Steroidal Bivalent Ligands
The eleven bivalent ligands were tested in the competitive radiometric relative binding affinity (RBA) assay for each subtype of ER, using [3H]estradiol as a tracer and estradiol as a standard.35,36 Binding affinities expressed as RBA values (i.e., relative to the binding affinity of estradiol = 100%) are shown in Table 1. All of the bivalent ligands had lower affinity for ER than estradiol, with the best compounds (10 and 15) having RBA values just below 7%. In all cases, the RBAs were higher for ERα than for ERβ, which may be attributed to the generally larger ligand binding pocket in ERα.37
Table 1.
Structures, ERα and ERβ binding affinities, and maximum tether lengths for bivalent estrogens
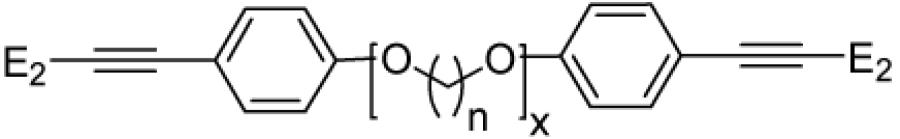 | |||||
|---|---|---|---|---|---|
| Bivalent Ligand |
n | x | C17-C17 Distance (Å)† |
ERα RBA‡ (%) |
ERβ RBA‡ (%) |
| EE2-(eg)6-EE2 (Amine, 10) |
2 | 6 | 39.8 | 6.8 ± 1.0 |
3.6 ± 0.3 |
| EE2-(eg)6-EE2 (Phenol, 12) |
2 | 6 | 35.7 | 1.2 ± 0.4 |
0.68 ± 0.22 |
| EE2-(pg)2-EE2 (13) |
3 | 2 | 24.2 | 0.90 ± 0.04 |
0.38 ± 0.21 |
| EE2-(pg)3-EE2 (14) |
3 | 3 | 29.4 | 2.1 ± 0.5 |
0.53 ± 0.23 |
| EE2-(pg)4-EE2 (15) |
3 | 4 | 34.1 | 6.9 ± 1.8 |
0.39 ± 0.003 |
| EE2-(pg)5-EE2 (16) |
3 | 5 | 39.1 | 2.3 ± 0.6 |
0.69 ± 0.56 |
| EE2-(pg)6-EE2 (17) |
3 | 6 | 43.9 | 1.5 ± 0.5 |
0.38 ± 0.34 |
| EE2-(bg)2-EE2 (18) |
4 | 2 | 26.4 | 1.8 ± 0.4 |
0.82 ± 0.25 |
| EE2-(bg)3-EE2 (19) |
4 | 3 | 33.5 | 4.5 ± 0.4 |
1.5 ± 0.6 |
| EE2-(bg)4-EE2 (20) |
4 | 4 | 38.9 | 2.6 ± 0.7 |
0.76 ± 0.28 |
| EE2-(bg)5-EE2 (21) |
4 | 5 | 45.3 | 1.6 ± 0.3 |
0.22 ± 0.04 |
The maximum C17-C17 distance was determined using Chem 3D Pro to generate structures with fully extended chains (all staggered conformations), and then minimizing the energy to 0.05 RMS gradient and measuring the distance between the 17-carbons of the steroids.
RBA is relative binding affinity, i.e., binding relative to estradiol (E2); the Kd of E2 for ERα is 0.2 nM and for ERβ is 0.5 nM.
We examined the relationship between the tether length and binding affinity in the propylene glycol ethers and butylene glycol ethers— two series in which bivalent ligands linked with multiple tether lengths were prepared. To estimate tether lengths, the bivalent ligands were drawn using ChemDraw 7.0 and then transferred to Chem3D Pro 7.0 and minimized to a RMS gradient of 0.05 with staggered chain conformations. The distance between the carbons at the 17 position (C17) of the steroid were then measured; this gives what are considered maximum tether lengths, which are recorded in Table 1. By plotting the C17-C17 distance against the RBA, clear affinity-length relationships can be seen (Figure 1).
Figure 3 is a plot of the RBA versus the maximum tether length in Angstroms (Å). For both the propylene and butylene glycol ether-linked bivalent ligands (black lines), an optimum ERα binding affinity is observed for ligands with C17-C17 maximum tether lengths of 34-35 Å. Notably, this maximum tether length of ~35 Å is considerably greater than the 26 Å C17-C17 distance measured in the estradiol-liganded ERα crystal structure (Figure 2). The straight-line distance measured directly between the ligands in the crystal structure, however, actually passes through some portions of the ER. Realistically, when the ligands bind with their phenylethynyl linkages projecting outward from the monomer units of the receptor dimer, the tether would need to find a minimum energy path—probably more tortuous than direct—to connect to the ends of the two phenyl groups. It is also possible that the tether chain adopts a conformation different from the all-extended one used to estimate the maximum tether length; conformations other than a fully staggered one would result in contraction of the tether, giving a somewhat shorter distance between the two steroids. Thus, a combination of conformational effects shortening the tether end-to-end distance and a non-linear low-energy route connecting the two steroids result in bivalent ligands with the ~35-Å maximum tether length giving the highest ERα binding affinities.
Figure 3.

A plot of relative binding affinity vs. maximum tether length between the C17 carbons for propylene glycol and butylenes glycol ether-tethered bivalent ligands. The ethyleneglycol ether-tethered (12) and amine-tethered (10) ligands are shown as single points at 35.7Å and 39.8Å, respectively.
The binding affinities of the two ethylene glycol-linked bivalent ligands appear relevant to this point. Compound 12 has the same ether linkage to the phenylethynyl group as do the bivalent ligands in the other two series, and its maximum tether length (35.7 Å) is close to that which gives the peak of (1.2%) than that of the comparable tether-length members of the other two series (6.9% and 4.5%). In water, polyethylene glycols are known to adopt a helical or coiled-like conformation, due to the preferred gauche conformation around the C-C bond bearing two electronegative substituents.29,30 Because of this tendency towards helicity, ethylene glycol ether tethers are more likely to span a considerably shorter distance than do the propylene glycol and butylene glycol ether tether chains, even though their maximum, fully extended (fully staggered) lengths might be the same. It was this conformational uncertainty that led us to focus on ligands linked by the butylene and propylene glycol ether tethers in which this preferred gauche effect-enforced helicity no longer applies. We believe as well that the higher lipophilicity of the three and four-carbon ether monomer units might be contributing to their higher binding affinities. It is notable that the other bivalent ligand having an ethylene glycol tether (10) has an ERα binding affinity comparable to the best of those in the other two series. However, it has a tether that is 4 Å longer than that in compound 12, which might compensate for the greater tendency of the polyethylene glycol chain to coil. Also, the tether in this compound is attached to the phenylethynyl group via a benzyl amine rather than a phenyl ether. Thus, the structure of the tether-to-steroid linkage, as well as the tether length and composition likely all contribute to the binding affinity of these bivalent ligands.
All of these compounds have lower binding affinities for ERβ. Although the linear scale for RBA values compresses the curve, it is clear that the butylene glycol bivalent ligands also reach maximum ERβ binding affinity at 35 Å. The ERβ binding of the propylene glycol linked bivalent ligands, however, does not show a clear relationship to tether length.
Conclusions
Bivalent ligands for the estrogen receptor (ER) were prepared with glycol ether tethers of different lengths and compositions, and their binding affinities for ERα and ERβ were tested in a radiometric competitive binding assay. Members of the propylene glycol and butylene glycol ether bivalent ligand series show a pronounced peak in ERα binding affinity with tether lengths of ~35 Å. Affinities for ERβ are lower, but a similar maximum binding with the butylene glycol-linked ligands was evident. Further biological studies of these bivalent ligands and control compounds are underway to examine the effect of bivalent ligand tether length and composition on the binding affinity, agonist/antagonist function and stability and conformation of ER dimers. The results from these studies will be the subject of a separate publication.
Experimental
All reagents were purchased from commercial suppliers and were used without further purification. Anhydrous solvents (with the exception of DMF) were obtained from an anhydrous solvent dispensing system, and anhydrous DMF was obtained by distillation over molecular sieves. For all reactions employing anhydrous solvents, glassware was oven-dried overnight and cooled under vacuum, then purged with argon; all such reactions were conducted under argon.
1H NMR spectra were recorded on either a 400 or 500 MHz Varian Oxford instrument and 13C NMR were recorded at either 100 or 125 MHz on the same instruments. NMR spectra are reported in ppm and were referenced to the solvent peak and processed using ACD Labs 5.0 software. EI mass spectra were recorded at 70 eV using the 70-VSE mass spectrometer, and ESI mass spectra were recorded using the Quattro mass spectrometer. Melting points are uncorrected and were obtained using a Thomas Hoover Uni-Melt capillary melting point apparatus.
Hexaethylene Glycol 1,20-Ditosylate (6)
Hexaethylene glycol (1.41 g, 5 mmol) and DMAP (120 mg, 1 mmol) were dissolved in Et3N (7 mL, 50 mmol) and CH2Cl2 (100 mL) and cooled to 0 °C. Tosyl chloride (3.21 g, 20 mmol) was added and the reaction warmed to room temperature and stirred for 24 h until complete by TLC (EtOAc). The mixture was diluted with CH2Cl2 (200 mL), washed with saturated NaHCO3 (3 × 60 mL), dried over MgSO4, filtered and concentrated. Purification by silica gel column (2:1 EtOAc/hexanes) gave the product as a clear oil (2.1 g, 72%). 1H NMR (500 MHz, chloroform-d) δ: 2.42 (s, 6 H) 3.55 (s, 8 H) 3.56 - 3.62 (m, 8 H) 3.63 - 3.68 (m, 4 H) 4.08 - 4.16 (m, 4 H) 7.28 - 7.36 (m, 4 H) 7.73 - 7.81 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 21.56, 68.58, 69.19, 70.42, 70.47, 70.52, 70.65, 127.91, 129.78, 132.89, 144.77. HRMS: Calc’d for C26H38O11S2 [M]+: 590.1856. Found: 590.1871.
Hexaethylene glycol 1,20-diazide (7)
Hexaethylene glycol ditosylate (6, 1 g, 1.7 mmol) was dissolved in DMF (10 mL) and sodium azide (663 mg, 10.2 mmol) was added and the mixture was heated to 80°C and stirred for 24 h. The reaction mixture was cooled to room temperature and added to water (50 mL) and extracted with EtOAc (2 × 75 mL). The combined organic layers were washed with saturated LiCl (2 × 30 mL), dried over MgSO4, filtered and concentrated. The product was isolated as a clear oil (493 mg, 87%). 1H NMR (500 MHz, chloroform-d) δ: 3.61 (m, 20H), 3.34 (m, 3H). 13C NMR (125 MHz, chloroform-d) δ: 70.6. 70.5, 70.4, 70.3, 69.1, 50.1. ESI MS: [M+H]+ = 333.0.
Hexaethylene glycol 1,20-diamine (8)
Hexaethylene glycol 1,20-diazide (7, 480 mg, 1.44 mmol) was dissolved in CH3OH (20 mL) and 10% Pd/C (100 mg) was added and the mixture was stirred at room temperature for 24 h under a H2 atmosphere. The mixture was filtered through Celite and concentrated, affording the product as a yellow oil (445 mg, 100%). 1H NMR (500 MHz, chloroform-d) δ: 3.59 (m, 10H), 3.47 (t, J = 5.35 Hz, 4H), 2.82 (t, J = 5.25 Hz, 4H), 1.47 (s, 4H). 13C NMR (125 MHz, chloroform-d) δ: 73.4, 70.53, 70.52, 70.49, 70.2, 41.73. HRMS: Calc’d for C12H28N2O5 [M]+: 281.2076. Found: 281.2064.
Hexaethylene Glyocol Bivalent EE2 Ligand with Amine Linker (10)
The benzaldehyde derivative of EE2 (9, 31.0 mg, 0.078 mmol) was dissolved in CH2Cl2 (5 mL) and anhydrous K2CO3 (54 mg, 0.39 mmol) and hexaethyleneglycol diamine (8, 10.8 mg, 0.039 mmol) were added. The mixture was stirred at room temperature for 48 h and concentrated. 1H NMR of the crude product indicated complete conversion to the imine. The product was redissolved in CH3OH (5 mL) and NaBH4 (43 mg, 0.12 mmol) was added and stirred for 5 min at which point no imine was observed by TLC (EtOAc, silica gel). The mixture was concentrated and redissolved in EtOAc (50 mL) and washed with water (2 × 15 mL), dried over MgSO4, filtered and concentrated. The product was a clear oil (27 mg, 79%). 1H NMR (500 MHz, chloroform-d) δ: 1H NMR (500 MHz, chloroform-d) δ: 3.50 - 3.67 (m, 24 H) 3.76 - 3.85 (m, 4 H) 6.42 - 6.69 (m, 4 H) 7.06 - 7.17 (m, 2 H) 7.23 - 7.32 (m, 4 H) 7.37 (t, J=8.15 Hz, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 154.3, 154.0, 138.1, 128.3, 121.8, 115.4, 113.2, 112.9, 80.2, 70.5, 70.4, 70.2, 70.1, 69.9, 53.3, 49.7, 48.4, 43.7, 39.5, 33.1, 29.7, 27.2, 26.5, 22.9, 12.9, 1.0. HRMS: Calc’d for C66H85N2O9 [M]+: 1049.6255. Found: 1049.6296.
Hexaethylene glycol 1,20-di(4-iodophenyl)ether (11)
Hexaethylene glycol ditosylate (6, 300 mg, 0.5 mmol) was dissolved in acetone (20 mL) and K2CO3 (474 mg, 3 mmol) and p-iodophenol (330 mg, 1.5 mmol) were added and the mixture was refluxed for 24 h. The reaction mixture was concentrated and redissolved in CHCl3 (100 mL) and washed with 1 M NaOH (2 × 25 mL) and dried over MgSO4, filtered and concentrated. The product was a clear oil (630 mg, 92%). 1H NMR (500 MHz, chloroform-d) δ: 7.51 (m, 4H), 6.66 (m, 4H), 4.05 (dd, J = 5.25, 4.27 Hz, 4H), 3.80 (m, 4H), 3.68 (m, 4H), 3.63 (m, 12H). 13C NMR (125 MHz, chloroform-d) δ: 158.5, 138.0, 118.0, 116.9, 82.8, 70.7, 70.5, 70.4, 69.4, 67.3. HRMS: Calc’d for C24H32I2O7 [M]+: 686.0237. Found: 686.0230.
Hexaethylene Glycol Bivalent EE2 Ligand with Phenol Linker (12)
Hexaethylene glycol 1,20-di(4-iodophenyl)ether (11, 172 mg, 0.25 mmol), 17α-ethynylestradiol (133 mg, 0.45 mmol), PdCl2(Ph3P)2 (7 mg, 0.01 mmol) and CuI (1 mg, 0.005 mmol) were added to an oven-dried flask under argon and dissolved in anhydrous, degassed CH3CN (15 mL) and Et3N (5 mL). The reaction was stirred for 24 h at room temperature, until complete by TLC (EtOAc). The mixture was concentrated and purified by silica gel column chromatography (column 1: 5% CH3OH/CHCl3, column 2: 3:1 CHCl3/acetone) to give the product as a white solid (54 mg, 21%). mp 83-84 °C. 1H NMR (500 MHz, chloroform-d) δ: 0.90 (s, 6 H) 1.21 - 1.55 (m, 10 H) 1.66 - 1.90 (m, 8 H) 1.91 - 2.00 (m, 2 H) 2.02 - 2.12 (m, 2 H) 2.14 - 2.24 (m, 4 H) 2.27 - 2.42 (m, 4 H) 2.68 - 2.86 (m, 4 H) 3.58 - 3.66 (m, 12 H) 3.66 - 3.72 (m, 4 H) 3.76 - 3.84 (m, 4 H) 4.04 - 4.12 (m, 4 H) 6.54 (d, J=2.69 Hz, 2 H) 6.62 (dd, J=8.48, 2.75 Hz, 2 H) 6.78 - 6.87 (m, 4 H) 7.11 (d, J=8.42 Hz, 2 H) 7.31 - 7.37 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.89, 22.88, 26.45, 27.16, 29.62, 33.02, 38.99, 39.44, 43.58, 47.56, 49.65, 67.36, 69.57, 70.48, 70.52, 70.54, 70.74, 80.36, 85.77, 91.36, 112.74, 114.53, 115.19, 115.27, 126.46, 132.30, 133.04, 138.15, 153.56, 158.72. ESI MS [M+H2O]+ = 1040.5.
General Procedure for Formation of Linker bis-Mesylate
Linker diol (1 equivalent) was added to an oven-dried flask under argon, dissolved in Et3N (4 equivalents) and CH2Cl2 (10 mL/mmol linker diol) and cooled to −78 °C. Mesyl chloride (3 equivalents) was added and the reaction was warmed to 0 °C and stirred for 2 h until starting material was consumed by TLC (10% CH3OH/EtOAc). The mixture was diluted with CHCl3, washed with saturated NaHCO3 and brine, dried over MgSO4, filtered, concentrated and purified by silica gel column chromatography. Details for individual products including yield, NMR and mass spec data can be found in the supporting information.
General Procedure for Formation of Linker bis-Aryl Iodide
Linker bis-mesylate (1 equivalent), p-iodophenol (4 equivalents) and K2CO3 (8 equivalents) were dissolved in 2-butanone (15 mL) and refluxed for 20 h. The reaction mixture was diluted with EtOAc (100 mL), washed with 3 M KOH (4 × 25 mL) and brine (25 mL), dried over MgSO4, filtered, concentrated and purified by silica gel column chromatography. Details for individual products including yield, NMR and mass spec data can be found in the supporting information.
General Procedure for Sonogashira Coupling for Bivalent Ligands
17α-Ethynylestradiol (2.5 equivalents), Pd(Ph3P)4 (10 mol %), CuI (20 mol %) and piperidine (20 equivalents) were added to an oven-dried flask under argon. A solution of the linker bis-aryl iodide (1 equivalent) in anhydrous THF (15 mL) was added, and the reaction was refluxed for 18 h until complete by TLC (7:1 CHCl3/acetone). The mixture was diluted with EtOAc (100 mL), washed with water (3 × 35 mL) and brine (2 × 25 mL), dried over MgSO4, filtered, concentrated and purified by silica gel chromatography.
Dipropylene Glycol Bivalent Ligand (13)
Following the general Sonogashira coupling procedure, dipropylene glycol bis-aryl iodide (89 mg, 0.17 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (26 mg, 20%), after purification by silica gel column chromatography (column 1: 1:1 hexanes/EtOAc, column 2: 10% acetone/CHCl3, column 3: 3:2 hexanes/acetone). mp 110-112 °C. 1H NMR (400 MHz, chloroform-d) δ: 0.90 - 0.98 (m, 6 H) 1.18 - 1.92 (m, 18 H) 1.91 - 2.28 (m, 12 H) 2.29 - 2.50 (m, 4 H) 2.72 - 2.94 (m, 4 H) 4.01 (t, J=6.23 Hz, 4 H) 4.88 (br. s., 2 H) 6.57 (d, J=2.69 Hz, 2 H) 6.64 (dd, J=8.55, 2.69 Hz, 2 H) 6.72 - 6.82 (m, 4 H) 7.17 (d, J=8.30 Hz, 2 H) 7.31 - 7.40 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.94, 22.92, 26.49, 27.17, 29.43, 29.65, 33.06, 39.05, 39.44, 43.62, 47.57, 49.67, 64.76, 67.09, 77.79, 80.36, 85.85, 91.30, 112.65, 114.35, 115.22, 126.55, 133.06, 138.27, 153.26, 158.95, 177.21. HRMS: Calc’d for C58H66O7Na [M+Na]+: 897.4706. Found: 897.4714.
Tripropylene Glycol Bivalent Ligand (14)
Following the general Sonogashira coupling procedure, tripropylene glycol bis-aryl iodide (106 mg, 0.18 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (63 mg, 38%), after purification by silica gel column chromatography (column 1: 3:1 CHCl3/acetone, column 2: 3:2 hexanes/acetone). mp 110 °C. 1H NMR (400 MHz, chloroform-d) δ: 0.93 (s, 6 H) 1.24 - 1.59 (m, 8 H) 1.63 - 1.92 (m, 10 H) 1.92 - 2.15 (m, 8 H) 2.16 - 2.30 (m, 4 H) 2.29 - 2.49 (m, 4 H) 2.75 - 2.91 (m, 4 H) 3.50 (t, J=6.35 Hz, 4 H) 3.56 (t, J=6.10 Hz, 4 H) 4.03 (t, J=6.23 Hz, 4 H) 5.25 (br. s., 2 H) 6.57 (d, J=2.44 Hz, 2 H) 6.64 (dd, J=8.30, 2.69 Hz, 2 H) 6.78 - 6.86 (m, 4 H) 7.10 - 7.19 (m, 2 H) 7.32 - 7.42 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.92, 22.89, 26.46, 27.16, 29.46, 29.63, 29.88, 33.02, 39.01, 39.42, 43.60, 47.57, 49.65, 64.82, 67.13, 67.75, 80.40, 85.85, 91.20, 112.67, 114.38, 114.85, 115.24, 126.51, 132.43, 133.04, 138.21, 153.36, 158.94. HRMS: Calc’d for C61H72O8Na [M+Na]+: 955.5125. Found: 955.5106.
Tetrapropylene Glycol Bivalent Ligand (15)
Following the general Sonogashira coupling procedure, tetrapropylene glycol bis-aryl iodide (115 mg, 0.18 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (79 mg, 45%), after purification by silica gel column chromatography (column 1: 4:1 CHCl3/acetone, column 2: 3:2 hexanes/acetone). mp 100-102 °C. 1H NMR (400 MHz, chloroform-d) δ: 0.93 (s, 6 H) 1.23 - 1.60 (m, 8 H) 1.68 - 1.92 (m, 12 H) 1.91 - 2.15 (m, 8 H) 2.15 - 2.30 (m, 4 H) 2.30 - 2.50 (m, 4 H) 2.75 - 2.89 (m, 4 H) 3.45 (t, J=6.35 Hz, 4 H) 3.50 (t, J=6.47 Hz, 4 H) 3.58 (t, J=6.10 Hz, 4 H) 4.04 (t, J=6.23 Hz, 4 H) 5.43 (br. s., 2 H) 6.57 (d, J=2.69 Hz, 2 H) 6.64 (dd, J=8.55, 2.69 Hz, 2 H) 6.77 - 6.87 (m, 4 H) 7.15 (d, J=8.30 Hz, 2 H) 7.32 - 7.42 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.92, 22.89, 26.46, 27.17, 29.49, 29.64, 29.89, 33.03, 39.01, 39.43, 43.59, 47.56, 49.65, 64.86, 67.11, 67.72, 67.86, 80.37, 85.83, 91.22, 112.68, 114.39, 114.86, 115.24, 126.49, 132.36, 133.04, 138.19, 153.44, 158.95. HRMS: Calc’d for C64H78O9Na [M+Na]+: 1013.5544. Found: 1013.5530.
Pentapropylene Glycol Bivalent Ligand (16)
Following the general Sonogashira coupling procedure, pentapropylene glycol bis-aryl iodide (65 mg, 0.09 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (48 mg, 50%), after purification by silica gel column chromatography (column 1: 3:1 CHCl3/acetone, column 2: 3:2 hexanes/acetone). mp 80-82 °C. 1H NMR (500 MHz, chloroform-d) δ: 0.93 (s, 6 H) 1.31 - 1.58 (m, 8 H) 1.69 - 1.93 (m, 14 H) 1.93 - 2.15 (m, 8 H) 2.16 - 2.27 (m, 4 H) 2.32 - 2.47 (m, 4 H) 2.75 - 2.91 (m, 4 H) 3.41 - 3.48 (m, 8 H) 3.51 (t, J=6.43 Hz, 4 H) 3.58 (t, J=6.11 Hz, 4 H) 4.05 (t, J=6.32 Hz, 4 H) 5.14 (br. s., 2 H) 6.57 (d, J=2.57 Hz, 2 H) 6.64 (dd, J=8.36, 2.79 Hz, 2 H) 6.80 - 6.87 (m, 4 H) 7.16 (d, J=8.36 Hz, 2 H) 7.33 - 7.41 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.90, 22.88, 26.46, 27.15, 29.50, 29.63, 29.91, 29.93, 33.02, 39.01, 39.42, 43.59, 47.56, 49.64, 64.87, 67.11, 67.71, 67.82, 67.90, 80.34, 85.83, 89.34, 91.25, 112.67, 114.42, 114.89, 115.24, 126.54, 132.48, 133.07, 138.25, 159.00. HRMS: Calc’d for C67H84O10Na [M+Na]+: 1071.5692. Found: 1071.5966.
Hexapropylene Glycol Bivalent Ligand (17)
Following the general Sonogashira coupling procedure, hexapropylene glycol bis-aryl iodide (69 mg, 0.09 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (42 mg, 42%), after purification by silica gel column chromatography (column 1: 3:1 CHCl3/acetone, column 2: 3:2 hexanes/acetone). mp 76 °C. 1H NMR (400 MHz, chloroform-d) δ: 0.93 (s, 6 H) 1.29 - 1.60 (m, 8 H) 1.72 - 1.92 (m, 16 H) 1.93 - 2.15 (m, 8 H) 2.16 - 2.30 (m, 4 H) 2.32 - 2.51 (m, 4 H) 2.81 (d, J=4.88 Hz, 4 H) 3.41 - 3.54 (m, 16 H) 3.59 (t, J=6.10 Hz, 4 H) 4.05 (t, J=6.23 Hz, 4 H) 5.17 (br. s., 2 H) 6.57 (d, J=2.69 Hz, 2 H) 6.64 (dd, J=8.30, 2.69 Hz, 2 H) 6.79 - 6.88 (m, 4 H) 7.16 (d, J=8.55 Hz, 2 H) 7.33 - 7.42 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 13.24, 23.22, 26.80, 27.49, 29.84, 29.98, 30.27, 33.35, 39.34, 39.76, 43.93, 47.89, 49.97, 65.19, 67.43, 68.04, 68.12, 68.18, 68.22, 78.10, 80.64, 86.12, 91.57, 112.97, 114.72, 115.20, 115.54, 126.84, 132.76, 133.36, 138.55, 153.70, 159.29. HRMS: Calc’d for C70H90O11Na [M+Na]+: 1129.6381. Found: 1129.6376.
Dibutylene Glycol Bivalent Ligand (18)
Following the general Sonogashira coupling procedure, dibutylene glycol bis-aryl iodide (43 mg, 0.08 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (37 mg, 54%), after purification by silica gel column chromatography (column 1: 7:1 CHCl3/acetone, column 2: 3:2 hexanes/acetone). mp 84-86 °C. 1H NMR (400 MHz, chloroform-d) δ: 0.90 - 1.00 (m, 6 H) 1.22 - 1.58 (m, 12 H) 1.69 - 1.92 (m, 12 H) 1.91 - 2.27 (m, 8 H) 2.30 - 2.49 (m, 4 H) 2.75 - 2.91 (m, 4 H) 3.50 (t, J=6.23 Hz, 4 H) 3.97 (t, J=6.23 Hz, 4 H) 5.37 (br. s., 2 H) 6.57 (d, J=2.69 Hz, 2 H) 6.64 (dd, J=8.30, 2.69 Hz, 2 H) 6.76 - 6.87 (m, 4 H) 7.16 (d, J=8.55 Hz, 2 H) 7.32 - 7.42 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 26.03, 26.20, 26.47, 27.17, 29.17, 29.63, 31.71, 33.02, 39.00, 39.43, 43.59, 47.56, 49.64, 53.64, 67.67, 70.39, 80.38, 85.84, 112.67, 114.38, 115.23, 126.49, 132.38, 133.03, 138.19, 153.42, 158.96. HRMS: Calc’d for C60H70O7Na [M+Na]+: 925.5109. Found: 925.5034.
Tributylene Glycol Bivalent Ligand (19)
Following the general Sonogashira coupling procedure, tributylene glycol bis-aryl iodide (49 mg, 0.08 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (39 mg, 52%), after purification by silica gel column chromatography (1: 7:1 CHCl3/acetone, 2: 3:2 hexanes/acetone). mp 64-66 °C. 1H NMR (400 MHz, chloroform-d) δ: 0.91 - 0.97 (m, 6 H) 1.14 - 2.28 (m, 36 H) 2.29 - 2.50 (m, 6 H) 2.75 - 2.89 (m, 4 H) 3.38 - 3.53 (m, 8 H) 3.97 (t, J=6.35 Hz, 4 H) 6.57 (d, J=2.44 Hz, 2 H) 6.64 (dd, J=8.30, 2.44 Hz, 2 H) 6.77 - 6.87 (m, 4 H) 7.16 (d, J=8.30 Hz, 2 H) 7.33 - 7.43 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.91, 22.91, 26.03, 26.22, 26.44, 27.17, 29.20, 29.64, 33.03, 39.01, 39.45, 43.60, 47.57, 49.65, 53.66, 67.70, 70.31, 70.65, 80.37, 85.86, 91.17, 112.66, 114.39, 115.22, 126.52, 132.45, 133.03, 138.22, 153.38, 158.99. HRMS: Calc’d for C64H78O8Na [M+Na]+: 997.5594. Found: 997.5565.
Tetrabutylene Glycol Bivalent Ligand (20)
Following the general Sonogashira coupling procedure, tetrabutylene glycol bis-aryl iodide (55 mg, 0.08 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (43 mg, 53%), after purification by silica gel column chromatography (column 1: 15% acetone/CHCl3, column 2: 3:2 hexanes/acetone). mp 80 °C. 1H NMR (400 MHz, chloroform-d) δ: 1.23 - 1.31 (m, 6 H) 1.31 - 1.54 (m, 8 H) 1.56 - 1.68 (m, 6 H) 1.68 - 1.92 (m, 12 H) 1.91 - 2.26 (m, 16 H) 2.38 (d, J=9.28 Hz, 4 H) 2.81 (d, J=4.88 Hz, 4 H) 3.38 - 3.52 (m, 10 H) 3.97 (t, J=6.35 Hz, 2 H) 5.35 (br. s., 4 H) 6.57 (d, J=2.69 Hz, 2 H) 6.64 (dd, J=8.42, 2.81 Hz, 2 H) 6.78 - 6.86 (m, 4 H) 7.16 (d, J=8.06 Hz, 2 H) 7.33 - 7.41 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.89, 22.86, 25.98, 26.17, 26.37, 26.44, 27.15, 29.15, 29.61, 33.00, 38.98, 39.42, 43.57, 47.55, 49.62, 67.68, 70.30, 70.57, 70.64, 80.36, 85.85, 91.20, 112.69, 114.39, 114.80, 115.25, 126.49, 132.32, 133.04, 138.18, 153.53, 159.00. HRMS: Calc’d for C68H86O9Na [M+Na]+: 1069.6170. Found: 1069.6149.
Pentabutylene Glycol Bivalent Ligand (21)
Following the general Sonogashira coupling procedure, pentabutylene glycol bis-aryl iodide (61 mg, 0.08 mmol) was coupled with 17α-ethynylestradiol to give the product as a white solid (42 mg, 49%), after purification by silica gel column chromatography (column 1: 15% acetone/CHCl3, column 2: 3:2 hexanes/acetone). mp 60-62 °C. 1H NMR (400 MHz, chloroform-d) δ: 1.22 - 2.28 (m, 48 H) 2.29 - 2.52 (m, 4 H) 2.73 - 2.89 (m, 4 H) 3.36 - 3.52 (m, 16 H) 3.97 (t, J=6.35 Hz, 4 H) 5.66 (br. s., 4 H) 6.56 (d, J=2.69 Hz, 2 H) 6.63 (dd, J=8.42, 2.81 Hz, 2 H) 6.78 - 6.87 (m, 4 H) 7.15 (d, J=8.30 Hz, 2 H) 7.32 - 7.41 (m, 4 H). 13C NMR (125 MHz, chloroform-d) δ: 12.89, 22.87, 26.01, 26.19, 26.39, 26.41, 27.16, 29.63, 33.01, 39.00, 39.44, 43.59, 47.56, 49.64, 67.03, 67.23, 67.69, 70.31, 70.56, 70.59, 70.66, 80.33, 85.83, 91.22, 112.68, 114.41, 114.82, 115.24, 126.51, 133.05, 137.10, 138.21, 153.53, 160.99. HRMS: Calc’d for C72H44O10Na [M+Na]+: 1141.6745. Found: 1141.6732.
Estrogen receptor binding assays
Relative binding affinities were determined by competitive radiometric binding assays with 2 nM [3H]E2 as tracer ([2,4,6,7-3H]estra-1,3,5,(10)-triene-3,17β-diol, 70-120 Ci/mmol, GE Healthcare, Piscataway, NJ), as a modification of methods previously described.35,36 The source of ER was purified full-length human ERαand ERβ purchased from Pan Vera/Invitrogen (Carlsbad, CA). Incubations were done at 0 °C for 18-24 h, and hydroxyapatite (Bio-Rad, Hercules, CA) was used to absorb the purified receptor-ligand complexes.35 The binding affinities are expressed as relative binding affinity (RBA) values, where the RBA of estradiol is 100%; under these conditions, the Kd of estradiol for ERα is ca. 0.2 nM, and for ERβ0.5 nM. The determination of these RBA values is reproducible in separate experiments with a CV of 0.3, and the values shown represent the average ± range or SD of 2 or more separate determinations.
Supplementary Material
Acknowledgments
We are grateful for support of this research from the National Institutes of Health (PHS R37 DK15556). Mass spectra were obtained on a 70-VSE mass spectrometer, purchased in part with a grant from the Division of Research Resources, National Institutes of Health (RR 04648)on a Quattro mass spectrometer, purchased in part with a grant from the Division of Research Resources, National Institutes of Health (RR 07141).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material Supplementary material is available which includes details on the synthesis and characterization of propylene and butylenes glycol linkers, linker mesylates and linker aryl iodides.
Supplementary Data Procedural details for the synthesis of the propylene and butylenes glycol tethers, with spectroscopic characterization data, are given in the supplementary data section.
References
- 1.Ballet S, Pietsch M, Abell AD. Protein Pept. Lett. 2008;15:668. doi: 10.2174/092986608785133672. [DOI] [PubMed] [Google Scholar]
- 2.Huskens J. Curr. Opin. Chem. Biol. 2006;10:537. doi: 10.1016/j.cbpa.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Kiessling LL, Gestwicki JE, Strong LE. Angew. Chem., Int. Ed. 2006;45:2348. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. J. Am. Chem. Soc. 2007;129:1312. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Messer WS., Jr. Curr. Pharm. Des. 2004;10:2015. doi: 10.2174/1381612043384213. [DOI] [PubMed] [Google Scholar]
- 6.Schamel WWA, Reth M. Adv Exp Med Biol. 2008;640:64. doi: 10.1007/978-0-387-09789-3_6. [DOI] [PubMed] [Google Scholar]
- 7.Zhang A, Liu Z, Kan Y. Curr. Top. Med. Chem. 2007;7:343. doi: 10.2174/156802607779941279. [DOI] [PubMed] [Google Scholar]
- 8.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. J Am Chem Soc. 2002;124:14922. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann KE, Wooge CH, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. J Steroid Biochem Mol Biol. 1994;49:139. doi: 10.1016/0960-0760(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 10.Osella D, Dutto G, Nervi C, McGlinchey MJ, Vessieres A, Jaouen G. J. Organomet. Chem. 1997;533:97. [Google Scholar]
- 11.Groleau S, Nault J, Lepage M, Couture M, Dallaire N, Berube G, C.-Gaudreault R. Bioorg. Chem. 1999;27:383. [Google Scholar]
- 12.Berube G, Rabouin D, Perron V, N’Zemba B, Gaudreault R-C, Parent S, Asselin E. Steroids. 2006;71:911. doi: 10.1016/j.steroids.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Rabouin D, Perron V, N’Zemba B, C.-Gaudreault R, Berube G. Bioorg. Med. Chem. Lett. 2003;13:557. doi: 10.1016/s0960-894x(02)00987-3. [DOI] [PubMed] [Google Scholar]
- 14.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson J-A, Carlquist M. Nature. 1997;389:753. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 15.Anstead GM, Carlson KE, Katzenellenbogen JA. Steroids. 1997;62:268. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 16.Figures were generated with Sybyl 7.3 from the corresponding research collaboratory for structural bioinformatics protein data bank (RCSB)-PDB file names: 1 ERE for E2.
- 17.Pomper MG, VanBrocklin H, Thieme AM, Thomas RD, Kiesewetter DO, Carlson KE, Mathias CJ, Welch MJ, Katzenellenbogen JA. J Med Chem. 1990;33:3143. doi: 10.1021/jm00174a009. [DOI] [PubMed] [Google Scholar]
- 18.Kim SH, Katzenellenbogen JA. Angew. Chem., Int. Ed. 2006;45:7243. doi: 10.1002/anie.200601923. [DOI] [PubMed] [Google Scholar]
- 19.Salman M, Reddy BR, Delgado P, Stotter PL, Fulcher LC, Chamness GC. Steroids. 1991;56:375. doi: 10.1016/0039-128x(91)90070-c. [DOI] [PubMed] [Google Scholar]
- 20.El Amouri H, Vessieres A, Vichard D, Top S, Gruselle M, Jaouen G. J. Med. Chem. 1992;35:3130. doi: 10.1021/jm00095a006. [DOI] [PubMed] [Google Scholar]
- 21.Lo KK-W, Zhang KY, Chung C-K, Kwok KY. Chem. Eur. J. 2007;13:7110. doi: 10.1002/chem.200700530. [DOI] [PubMed] [Google Scholar]
- 22.Top S, El Hafa H, Vessieres A, Quivy J, Vaissermann J, Hughes DW, McGlinchey MJ, Mornon J-P, Thoreau E, Jaouen G. J. Am. Chem. Soc. 1995;117:8372. [Google Scholar]
- 23.Gabano E, Cassino C, Bonetti S, Prandi C, Colangelo D, Ghiglia A, Osella D. Org. Biomol. Chem. 2005;3:3531. doi: 10.1039/b507716h. [DOI] [PubMed] [Google Scholar]
- 24.Purohit A, Wyatt J, Hynd G, Wright J, El-Shafey A, Swamy N, Ray R, Jones GB. Tetrahedron Lett. 2001;42:8579. [Google Scholar]
- 25.Nettles KW, Bruning JB, Gil G, O’Neill EE, Nowak J, Hughs A, Kim Y, DeSombre ER, Dilis R, Hanson RN, Joachimiak A, Greene GL. EMBO Rep. 2007;8:563. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arterburn JB, Rao KV, Perry MC. Tetrahedron Lett. 2000;41:839. [Google Scholar]
- 27.Kreuzer HJ, Wang RLC, Grunze M. New J. Phys. 1999;1:21–1. [Google Scholar]
- 28.Sundaralingam M. Nature. 1968;217:35. doi: 10.1038/217035a0. [DOI] [PubMed] [Google Scholar]
- 29.Wiberg KB, Murcko MA, Laidig KE, MacDougall PJ. J. Phys. Chem. 1990;94:6956. [Google Scholar]
- 30.Wolfe S. Accounts Chem. Res. 1972;5:102. [Google Scholar]
- 31.Chinchilla R, Najera C. Chem. Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 32.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467. [Google Scholar]
- 33.Elangovan A, Wang Y-H, Ho T-I. Org. Lett. 2003;5:1841. doi: 10.1021/ol034320+. [DOI] [PubMed] [Google Scholar]
- 34.Knuf EC, Jiang J-K, Gin MS. J. Org. Chem. 2003;68:9166. doi: 10.1021/jo034728k. [DOI] [PubMed] [Google Scholar]
- 35.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Biochemistry. 1997;36:14897. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 36.Katzenellenbogen JA, Johnson HJ, Jr., Myers HN. Biochemistry. 1973;12:4085. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 37.Pike ACW, Brzozowski AM, Hubbard RE, Bonn T, Thorsell A-G, Engstrom O, Ljunggren J, Gustafsson J-A, Carlquist M. Embo J. 1999;18:4608. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.