

Abstract
MRL/faslpr mice, which undergo a systemic autoimmune disease with similarities to systemic lupus erythematosus (SLE), display reduced pathology and prolonged survival if rendered deficient in ICAM-1. However, it remains unclear whether this is a result of the ability of ICAM-1 to promote the immune response or mediate leukocyte recruitment. Therefore, the aim of these studies was to compare the role of ICAM-1 in the elevated leukocyte-endothelial interactions, which affect MRL/faslpr mice. Intravital microscopy was used to compare leukocyte rolling and adhesion in postcapillary venules in the dermal and cerebral (pial) microcirculations of wild-type (ICAM+/+) and ICAM-1-deficient (ICAM-1−/−) MRL/faslpr mice. In the dermal microcirculation of 16-week MRL/faslpr mice, leukocyte adhesion was increased relative to nondiseased MRL+/+ mice. However, this increase was abolished in ICAM-1−/− MRL/faslpr mice. ICAM-1 deficiency was also associated with reduced dermal pathology. In contrast, in the pial microcirculation, the elevation in leukocyte adhesion observed in ICAM+/+ MRL/faslpr mice also occurred in ICAM-1−/− MRL/faslpr mice. VCAM-1 expression was detectable in both vascular beds, but higher levels were detected in the pial vasculature. Furthermore, VCAM-1 blockade significantly reduced leukocyte adhesion and rolling in the cerebral microcirculation of ICAM-1−/− MRL/faslpr mice. Therefore, ICAM-1 was critical for leukocyte adhesion in the skin but not the brain, where VCAM-1 assumed the major function. Given the ongoing development of anti-adhesion molecule therapies and their potential in inflammatory diseases such as SLE, these data indicate that implementation of these therapies in SLE should take into account the potential for tissue-specific functions of adhesion molecules.
Keywords: recruitment, adhesion, lupus, SLE, intravital microscopy
INTRODUCTION
Systemic lupus erythematosus (SLE) is a debilitating autoimmune disease in which multiple tissues are injured by aberrant inflammatory responses [1]. As part of these inflammatory responses, large numbers of leukocytes are recruited to affected tissues. Multiple lines of evidence suggest that to enter sites of inflammation, leukocytes in the circulation must undergo a sequence of interactions with the endothelial surface—first tethering and rolling on the endothelial surface prior to undergoing arrest on the endothelium, and subsequently, migrating into the affected site [2]. Examination of tissue biopsies from SLE patients has demonstrated that the endothelial adhesion molecules, which mediate these interactions, are expressed at elevated levels in SLE [3, 4]. Similarly, expression and function of leukocyte integrins are elevated significantly in SLE, most prominently, in patients with severe disease [5]. These correlations between adhesion molecule expression and disease severity suggest that increased leukocyte-endothelial cell interactions may be critical to disease expression in SLE and raise the possibility that anti-adhesion molecule therapies could be therapeutically effective in this intractable disease.
Support for this contention is derived from studies examining adhesion molecule expression in “lupus-prone” MRL/faslpr mice, demonstrating increased expression of adhesion molecules such as ICAM-1 and VCAM-1 in disease-affected tissues [6,7,8]. Furthermore, MRL/faslpr mice lacking ICAM-1 or one of the main ICAM-1 ligands, LFA-1, have an extended lifespan and decreased tissue inflammation relative to ICAM-1+/+/LFA-1+/+ littermates, providing further evidence that leukocyte recruitment is critical in progression of the disease [9,10,11]. However, ICAM-1 also contributes to lymphocyte costimulation, raising the possibility that the protection provided by ICAM-1 deficiency in this T cell-dependent model also relates to effects on lymphocyte activation [12,13,14,15]. To examine a role for ICAM-1 in directing leukocyte recruitment into tissue, Lloyd et al. [9] performed passive leukocyte transfer studies and demonstrated that the ability of transferred leukocytes to enter the lung was significantly reduced in ICAM-deficient (ICAM-1−/−) MRL/faslpr mice. However, these experiments did not examine the role of ICAM-1 in trafficking in other lupus-affected organs such as the skin and brain.
To investigate mechanisms of leukocyte recruitment in the MRL/faslpr model, we have previously used intravital microscopy to examine the microvasculature in disease-affected tissues in these animals [16, 17]. These studies demonstrated that in the skin and brain, two tissues commonly affected in human SLE, leukocyte-endothelial cell interactions in postcapillary venules were increased significantly at ages when disease pathology is accelerating [16, 17]. Moreover, we observed that the molecular basis for these increased interactions differed between tissues, with P- and E-selectin being of central importance in the skin versus the critical role of the α4-integrin/VCAM-1 pathway in the brain. These observations were important in that they demonstrated that despite the systemic nature of the inflammatory stimulus in this model, the adhesion molecules responsible for mediating local leukocyte recruitment differ between tissues. Therefore, the aim of this study was to directly examine the affected microvasculature in MRL/faslpr mice via intravital microscopy, using ICAM-1−/−mice as a way of determining the role of this molecule in mediating the critical interactions required to exit the vasculature. We examined two microvascular beds which we have previously observed to support increased rolling and adhesion in these mice: the dermal and cerebral (pial) microvasculatures [16, 17]. These experiments revealed that ICAM-1 is critical for leukocyte adhesion in the dermal vasculature but is not required for interactions within the cerebral microvasculature.
MATERIALS AND METHODS
Animals
MRL/MpJ-faslpr (MRL/faslpr) mice and MRL/MpJ (MRL+/+) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). ICAM-1−/− MRL/faslpr mice were generated by backcrossing a gene-targeted mutation onto the MRL/faslpr strain background, as described previously [10], and generously supplied by Dr. Daniel Bullard (University of Alabama at Birmingham, AL, USA). Mice were used at 16 weeks of age and weighed 40–50 g.
Antibodies
The antibodies used in this study were 6C7.1, a mAb against murine VCAM-1 (hybridoma provided by Dr. Dietmar Vestweber, Max Planck Institut, Meunster, Germany, and Dr. Britta Engelhardt, Theodor-Kocher Institut, Bern, Switzerland). This antibody has been shown previously to inhibit VCAM-1-dependent interactions in vivo and has also been used for detection of vascular VCAM-1 expression [18,19,20,21]. A110-2 (IgG2), a rat anti-keyhole limpet hemocyanin (KLH; BD Biosciences, San Jose, CA, USA), was used as an isotype-matched control in in vivo experiments. MEC 13.3 (BD Biosciences) was used for labeling mouse CD31/PECAM-1 [22].
Intravital microscopy
Animals were anesthetized by i.p. injection of a cocktail of 10 mg/kg xylazine (Bayer Pharmaceuticals, Pymble, NSW, Australia) and 150 mg/kg ketamine hydrochloride (Caringbah, NSW, Australia). A catheter was inserted in the tail vein or jugular vein to administer anesthetic, fluorescent dyes, and antibodies. The animal was placed on a thermo-controlled heating pad, regulating the core temperature to 37°C.
Cerebral microcirculation
The cerebral microcirculation was then prepared for microscopy as described previously [17, 23]. Briefly, a craniotomy was performed on the right parietal bone using a high-speed drill (Fine Science Tools Inc., North Vancouver, BC, Canada). A superfusion chamber was held in place over the craniotomy by securing the incised scalp around the lower lip of the chamber with dental cement and Loctite 401 rapid adhesive (Loctite Australia, Caringbah, NSW, Australia). Prior to removal of the dura and exposure of the pial microvasculature, artificial cerebrospinal fluid (CSF; ionic composition in mmol/L: NaCl 132, KCl 2.95, CaCl2 1.71, MgSO4 1.4, NaHCO3 24.6, glucose 3.71, urea 6.7, pH 7.4) was pumped continuously through the superfusion chamber at 37°C to maintain the exposed brain. A pH and gas tension similar to that of normal CSF was replicated in the artificial equivalent by constant bubbling with 12% O2, 5% CO2, and 83% N2. Animals were examined for a maximum of 1 h.
To visualize leukocytes, animals were injected (i.v.) with 50 μL 0.05% rhodamine 6G (Sigma Chemical Co., St. Louis, MO, USA) immediately prior to microscopy. Rhodamine 6G-associated fluorescence was visualized by epi-illumination at 510–560 nm, using a 590-nm emission filter [24, 25]. The pial microvasculature was visualized using an intravital microscope (Axioplan 2 Imaging; Carl Zeiss, Carnegie, Victoria, Australia) with a ×40 water immersion objective lens (Achroplan 40X/0.80 NA, Carl Zeiss) and a ×10 eyepiece. A SIT video camera (Dage-MTI VE-1000, Sci Tech Pty. Ltd., Preston South, Victoria, Australia) was used to project the images onto a monitor (Sony PVM-20N5E, Carl Zeiss), and the images were recorded for playback analysis using a videocassette recorder (Panasonic NV-HS950, Klapp Electronics, Prahran, Victoria, Australia). Multiple (three to five per mouse) postcapillary venules (25–50 μm in diameter) were selected in each experiment, and to minimize variability, the same section of venule was observed throughout the experiment. Venular diameter and the number of rolling and adherent leukocytes were determined off-line during video-playback analysis. Rolling leukocytes were defined as those cells moving at a velocity less than that of erythrocytes and visibly interacting with the endothelial surface within a given vessel. Leukocyte rolling flux was defined as the number of rolling cells/min and was measured in each venule, typically in 30- to 60-s segments to avoid undue exposure of the tissue to fluorescence illumination. When multiple venules were examined, data were averaged to generate a single data point per animal per time-point. Leukocyte rolling velocity was determined by measuring the time required for a leukocyte to roll along a 100-μm length of venule. Rolling velocity was determined for 20 leukocytes at each time interval. In some animals, however, less than 20 leukocytes were observed rolling in a vessel during the period of recording. In these animals, the velocity of each of the leukocytes observed to be rolling was measured. Leukocytes were considered adherent to the venular endothelium if they remained stationary for 30 s or longer and are shown as the number of cells/mm2 pial postcapillary venule surface area (calculated assuming cylindrical vessel geometry).
Dermal microvasculature
The microcirculation of the ventral abdominal skin was prepared for microscopy as described previously [16, 26]. Briefly, a midline abdominal incision was made, extending from the level of the diaphragm to the pelvic region. The skin was carefully separated from the underlying tissue, remaining attached laterally to ensure the blood supply remained intact. The area of skin was then extended over a viewing pedestal and secured along the edges using a 3-0 suture. The loose connective tissue on the dermal undersurface was removed carefully by dissection under an operating microscope. The exposed dermal microvasculature was immersed in normal saline and covered with a coverslip, held in place with vacuum grease. To visualize leukocytes, animals were injected (i.v.) with 50 μL 0.05% rhodamine 6G immediately prior to microscopy. Leukocyte rolling flux, rolling velocity, and adhesion were quantitated as for the cerebral microcirculation. In regards to leukocyte rolling, to correct for significant differences in circulating leukocyte counts between mouse strains, leukocyte rolling flux was also examined after normalization to leukocyte count in each animal (expressed as cells/min/106 cells).
Assessment of VCAM-1 expression
VCAM-1 expression in the dermal and pial microvasculatures was quantitated using a modified version of a previously published technique [20]. Briefly, anti-VCAM-1 antibody (6C7.1) was conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA), and anti-KLH was conjugated to Alexa Fluor 594 as a control mAb. Tissue distribution of antibodies conjugated to Alexa Fluor 488 was determined by epi-illumination at 450–490 nm with a 515-nm emission filter. Alexa Fluor 594-associated fluorescence was detected by epi-illumination at 530–585 nm excitation filter with a 615-nm emission filter (Carl Zeiss Filter Set 00). Images were visualized using a SIT video camera (Dage-MTI VE-1000) on predefined gain and black-level settings and recorded for subsequent playback analysis using a videocassette recorder. Mice were anesthetized, and the right carotid artery and the left jugular vein were cannulated. To detect expression of VCAM-1, mice received 90 μg 6C7.1ALEXA 488 and 20 μg anti-KLHALEXA 594 i.v. Antibodies were allowed to circulate for 5 min, and then the mouse was exsanguinated via the carotid artery with simultaneous infusion of bicarbonate-buffered saline via the jugular vein. An additional 15-mL buffer was subsequently backflushed through the carotid artery after severing the abdominal vena cava. The pial and dermal microvasculatures were surgically exposed in the same manner as for intravital microscopy, and each microcirculation was visualized in turn to assess VCAM-1 expression.
The dermal and cerebral microvasculatures were first assessed for nonspecific (anti-KLHALEXA 594) labeling. Vessels containing detectable Alexa Fluor 594-associated fluorescence were deemed to have been inadequately exsanguinated and were excluded from analysis. In most experiments, this applied to a maximum of one to two vessels. Analyses were performed on captured video frames using Scion Image analysis software. The intensity of 6C7.1ALEXA 488-specific staining in individual vessels was determined for four to 10 vessels per preparation, imaged via 20× objective (LD Achroplan 20X/0.40 NA, Carl Zeiss) fields. The intensity of Alexa Fluor 488-derived fluorescence associated with the vascular wall, and that in the avascular surrounding tissue, were measured. The intensity of extravascular fluorescence was subtracted from that present in the vessel wall, and the resultant intensity readings were averaged over the length of the vascular wall assessed. Final data are shown as average intensity in the vascular segments examined.
As an alternative approach to assessment of VCAM-1 expression, simultaneous quantitation of intravascular VCAM-1 and PECAM-1 staining was performed. 6C7.1ALEXA 488 (90 μg) was used to label VCAM-1 and Alexa Fluor 594-conjugated MEC 13.3 (50 μg) used to label PECAM-1. Mice were injected with antibodies and exsanguinated after 5 min, and the tissues were exteriorized as already described. Dermal and cerebral microvessels were visualized using a Leica SP5 multiphoton confocal microscope equipped with a SpectraPhysics MaiTai laser tuned to 790 nm. Epifluorescence images were collected using nondescanned detectors and a FITC/tetramethylrhodamine-isothiocyanate dichroic beam-splitter. Predefined settings for laser power and detector gain were used for all experiments. Images of vessels with positive staining for PECAM-1 and VCAM-1 were captured, and mean pixel intensity within defined vascular segments was determined. The average background intensity was determined in unstained extravascular regions, and this value was subtracted from the intravascular intensities of adhesion molecule staining. Data are expressed as a ratio of the staining intensity of VCAM-1:PECAM-1.
Histology
Morphology of lesion-prone areas of dorsal skin was determined histologically using H&E staining of 6 μm sections from formalin-fixed, paraffin-embedded tissue. Sections were examined in a blinded manner by two operators, and the following parameters were assessed: epidermal changes including proliferation, rarefaction, and ulceration and leukocyte infiltration in dermis and deeper s.c. tissue, which was rated as absent, diffuse, moderate, or intense.
Statistics
Data are presented as mean ± sem. For comparisons involving two groups, Student’s unpaired t-tests or if appropriate, nonparametric Mann Whitney U tests were used. For comparisons involving three groups, ANOVA, followed by Scheffe post-hoc tests or Student’s t-tests, followed by Bonferroni’s multiple comparisons test were used. Paired t-tests were used for comparison of adhesion molecule expression in dermal and cerebral vessels and for analysis of the effects of pre- and post-function-blocking antibody treatment. For analysis of dermal pathology, the rates of incidence of macroscopic lesions and histological end-points were compared using χ2 analysis. Statistical significance was set at P< 0.05.
RESULTS
ICAM-1 is critical to leukocyte adhesion in the dermal vasculature of lupus-prone mice
We first examined a role for ICAM-1 in mediating leukocyte adhesion in the dermal vasculature of MRL/faslpr mice. In previous studies, we have reported that leukocyte adhesion is increased in dermal postcapillary venules of 16-week MRL/faslpr mice, relative to MRL+/+ mice of the same age [16]. Similar findings were made in the present experiments (Fig. 1). However, in ICAM-1−/− MRL/faslpr mice, leukocyte adhesion was almost completely absent at the same time-point (Fig. 1). Rolling was also investigated in these mice, as there is evidence that ICAM-1 plays a role in mediating rolling under some inflammatory conditions [27]. Leukocyte rolling was altered in the dermal vasculature of 16-week ICAM-1+/+ MRL/faslpr mice, via a significant reduction in leukocyte rolling velocity without any marked changes in rolling flux (Fig. ,2A and B). In contrast, in MRL/faslpr mice lacking ICAM-1, leukocyte rolling velocity was similar to that in the nondiseased MRL+/+ mice, suggesting a role for ICAM-1 in contributing to the reduction in leukocyte rolling velocity in MRL/faslpr mice. However, leukocyte rolling flux in ICAM-1−/− MRL/faslpr mice was elevated significantly relative to that of MRL+/+ and ICAM-1+/+ MRL/faslpr mice (Fig. 2B). In addition, the number of circulating leukocytes in the ICAM-1−/− mice was elevated greater than twofold (P<0.05) relative to ICAM-1+/+ MRL/faslpr mice (Table 1). Following correction of leukocyte rolling flux for the number of circulating leukocytes, rolling was no longer significantly different between ICAM-1+/+ and ICAM-1−/− MRL/faslpr mice (ICAM-1+/+, 8.85±2.5 cells/min/106 cells, vs. ICAM-1−/−, 4.66±0.69 cells/min/106 cells; P=0.159). This indicated that the increase in flux in ICAM-1−/− mice was a result of the increase in the number of circulating leukocytes in these mice.
Figure 1.
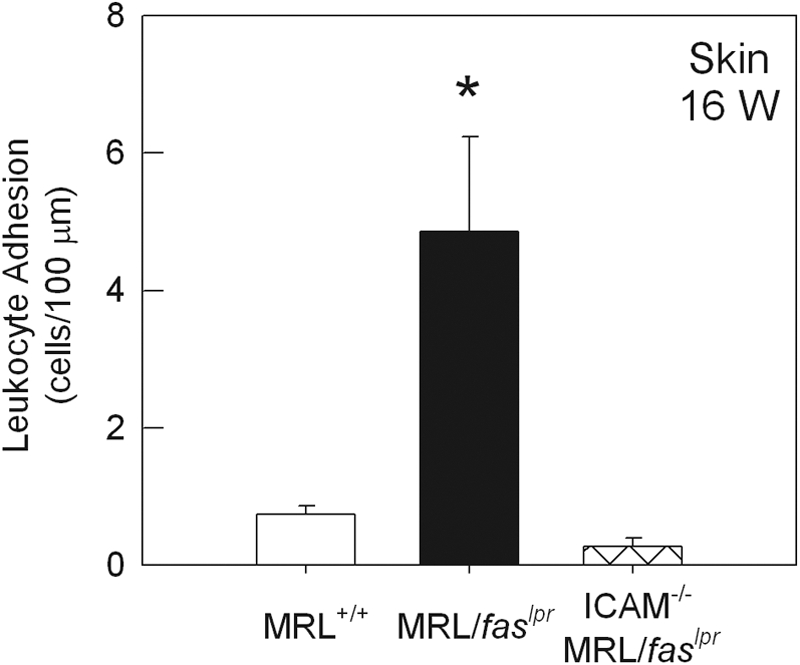
Leukocyte adhesion in dermal postcapillary venules of 16-week (16 W) MRL+/+ (n=6), MRL/faslpr (n=6), and ICAM-1−/− MRL/faslpr (n=10) mice. Data were collected from an average of four to five venules per animal. *, P < 0.05, versus MRL+/+.
Figure 2.
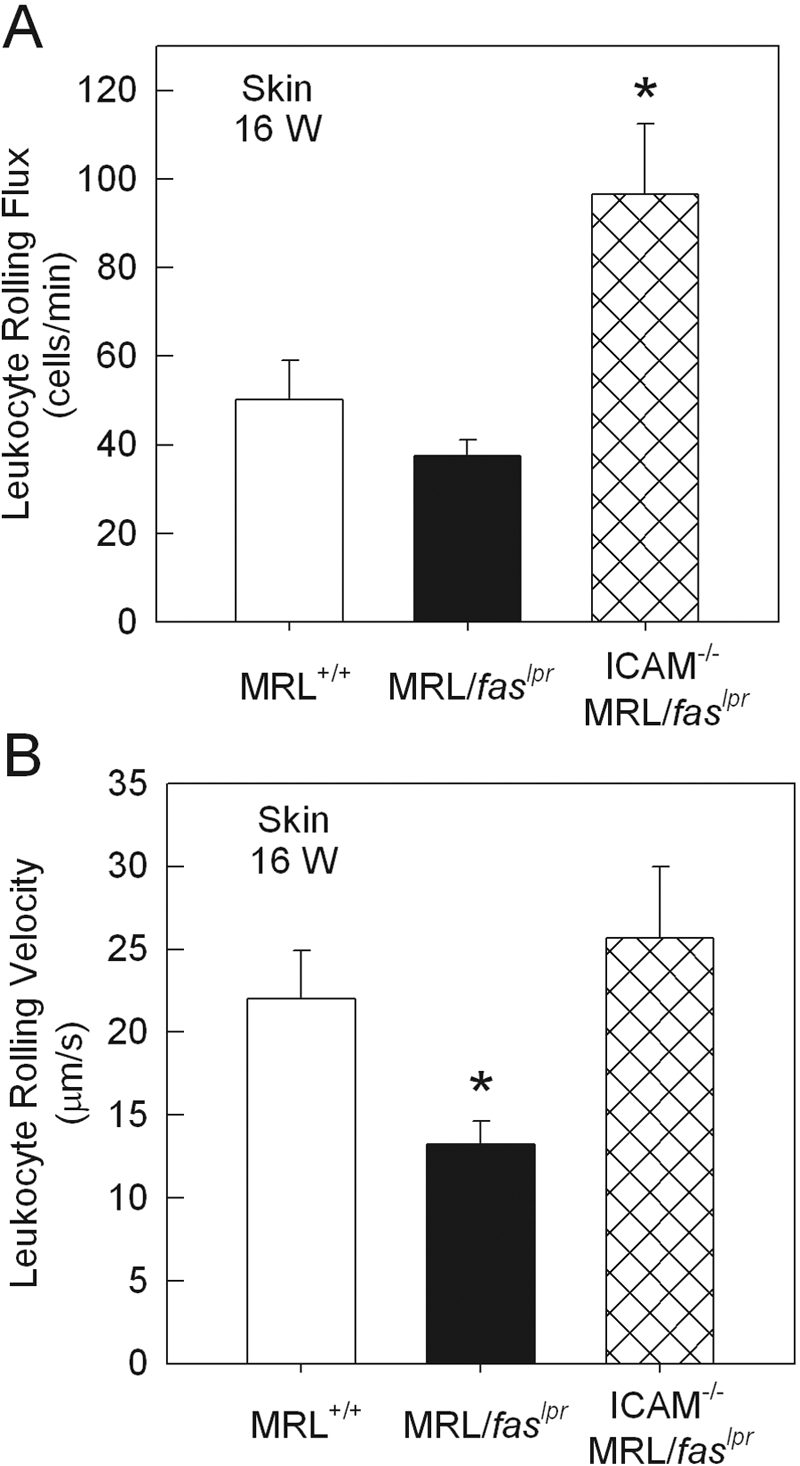
Leukocyte rolling flux (A) and rolling velocity (B) in dermal postcapillary venules of 16-week MRL+/+ (n=6), MRL/faslpr (n=6), and ICAM-1−/− MRL/faslpr (n=10) mice. Data were collected from an average of four to five venules per animal. *, P < 0.05, versus MRL+/+.
TABLE 1.
Circulating Leukocyte Counts in 16-Week MRL+/+, MRL/faslpr, and ICAM-1–/– MRL/faslpr Mice
| Strain | Circulating leukocytes (cells/mL×106) |
|---|---|
| MRL+/+ (n = 6) | 3.6 ± 0.6 |
| MRL/faslpr (n = 6) | 5.2 ± 0.8 |
| ICAM−/− MRL/faslpr (n = 14) | 11.6 ± 1.2a |
P < 0.01 versus MRL+/+ and MRL/faslpr.
ICAM-1 contributes to dermal pathology in lupus-prone mice
The absence of ICAM-1 was associated with a significant reduction in the incidence of macroscopic skin lesions in MRL/faslpr mice. In ICAM-1+/+ MRL/faslpr mice at 16 weeks, ∼30% of mice displayed macroscopic dermal lesions, as opposed to ∼10% of ICAM-1−/− MRL/faslpr mice (P<0.05; Table 2). Similarly at 20 weeks, lesion incidence was 61% in ICAM-1+/+ MRL/faslpr mice versus only 29% in ICAM-1−/− MRL/faslpr mice (P<0.05; Table 2). These data support earlier findings in these mice [10].
TABLE 2.
Macroscopic and Morphological Assessment of Dermal Lesions in 16- and 20-Week ICAM-1+/+ and ICAM-1–/– MRL/faslpr Micea
| Macroscopic lesions |
Epidermal hyperplasia (20 week) | Dermal leukocyte infiltration (20 week) | S.C. leukocyte infiltration (20 week) | ||
|---|---|---|---|---|---|
| (16 week) | (20 week) | ||||
| ICAM-1+/+ | 29/101 | 37/61 | 7/12 | 4/12 | 7/12 |
| ICAM-1–/– | 7/75 | 12/42 | 0/7 | 0/7 | 1/7 |
| Pb | <0.001 | <0.001 | <0.016 | <0.05 | <0.05 |
“Macroscopic lesions” denotes areas of the skin in which loss of hair, redness, and bleeding were apparent macroscopically. The histological endpoints assessed were “Epidermal hyperplasia”, denoting lesions in which the epidermis was intact but had undergone substantial proliferation; “Dermal leukocyte infiltration” denotes lesions in which intense leukocyte infiltration was present within the dermis; “S.C. leukocyte infiltration” denotes lesions in which intense leukocyte infiltration was present within the deeper s.c. tissue.
Comparison of incidence of various endpoints in affected skin region, as assessed by χ2 analysis. Histological sections were examined in a blinded manner.
To further assess the role of ICAM-1 in dermal leukocyte recruitment, skin from ICAM-1+/+ and ICAM-1−/− MRL/faslpr mice was examined histologically. At 16 weeks of age, the epidermis of lesion-prone dorsal skin from ICAM-1+/+ MRL/faslpr mice was predominantly intact, but occasional mice showed regions of rarefaction (thinning) and ulceration. Intense leukocyte infiltration was rare (one of nine) in the dermis but more commonly observed (four of nine) in the deeper connective tissue (data not shown). However, at 20 weeks of age, more than 50% of the lesions displayed a hyperproliferative epidermis (Fig. 3 A), and the remaining animals displayed rarefaction and ulceration. The rate of intense leukocyte infiltration had increased in the dermis (four of 12) and the s.c. connective tissue (7/12; Fig. ,3Aand 3C, and Table 2). Below areas of raised/thickened epithelium, intense cellular infiltration was observed in five of seven wild-type mice.
Figure 3.

Histological comparison of lesion-prone areas of skin in 20-week ICAM-1+/+ and ICAM-1−/− MRL/faslpr mice. Shown are low (A and B)- and high (C and D)-power fields of skin from ICAM-1+/+ (A and C) and ICAM-1−/− (B and D) MRL/faslpr mice. In ICAM-1+/+ mice, the epidermis was often observed to be highly proliferative and the dermis expanded (A), and intense leukocyte infiltration was present within the deeper s.c. tissue (C). In contrast, in MRL/faslpr mice lacking ICAM-1, the epidermis retained a regular, nonproliferative morphology (B), and leukocyte infiltration was reduced greatly (D). Micrographs are representative of samples from n = 12 (ICAM-1+/+) and n = 7 (ICAM-1−/−) mice.
In 16-week ICAM-1−/− MRL/faslpr mice, the epidermis was mostly intact and nonproliferative, but many animals (six of nine) displayed rarefaction and ulceration. In most cases, leukocyte infiltration was minimal (data not shown). At 20 weeks, epidermal proliferation was not observed (zero of seven mice), but rarefaction or ulceration was seen in three of seven mice (Fig. 3B). In addition, leukocyte infiltration in the dermis and the deeper connective tissues was moderate or minimal, and only one animal displayed intense infiltration in the connective tissue (Fig. ,3Band 3D, and Table 2).
ICAM-1 is not required for leukocyte adhesion in the cerebral vasculature of lupus-prone mice
Previous experiments have indicated that the α4-integrin/VCAM-1 pathway is critical to the elevated leukocyte adhesion in the cerebral microvasculature of MRL/faslpr mice [17]. However, the role of ICAM-1 has not been assessed. As seen previously, we observed an increase in adhesion in cerebral postcapillary venules of 16-week MRL/faslpr mice, relative to similarly aged MRL+/+ mice (Fig. 4). However, in contrast to the dermal vasculature, adhesion in the pial microvasculature was significantly elevated in ICAM-1−/− MRL/faslpr mice (Fig. 4). Comparison of leukocyte rolling in MRL+/+ and MRL/faslpr mice revealed a significant increase in leukocyte rolling flux in MRL/faslpr mice (Fig. 5 A), as described previously [17]. Leukocyte rolling flux in pial postcapillary venules of ICAM-1−/− MRL/faslpr mice was not significantly different from that in ICAM-1+/+ MRL/faslpr mice (Fig. 5A). In addition, leukocyte rolling velocity in the pial microvasculature did not differ in the three strains of mice (Fig. 5B).
Figure 4.
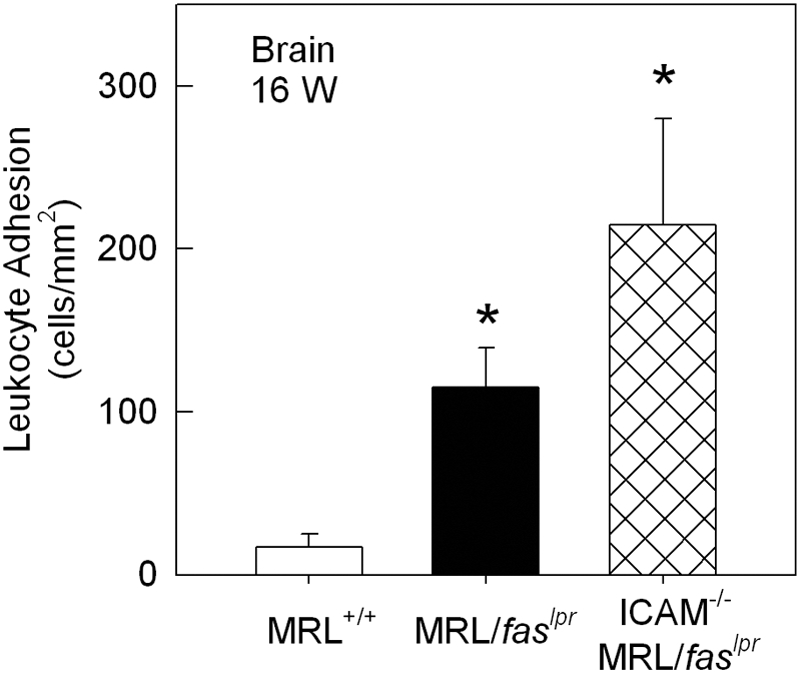
Leukocyte adhesion in pial postcapillary venules of 16-week MRL+/+ (n=6), MRL/faslpr (n=9), and ICAM-1−/− MRL/faslpr (n=6) mice. Data were collected from an average of three to four venules per animal. *, P < 0.05, versus MRL+/+.
Figure 5.
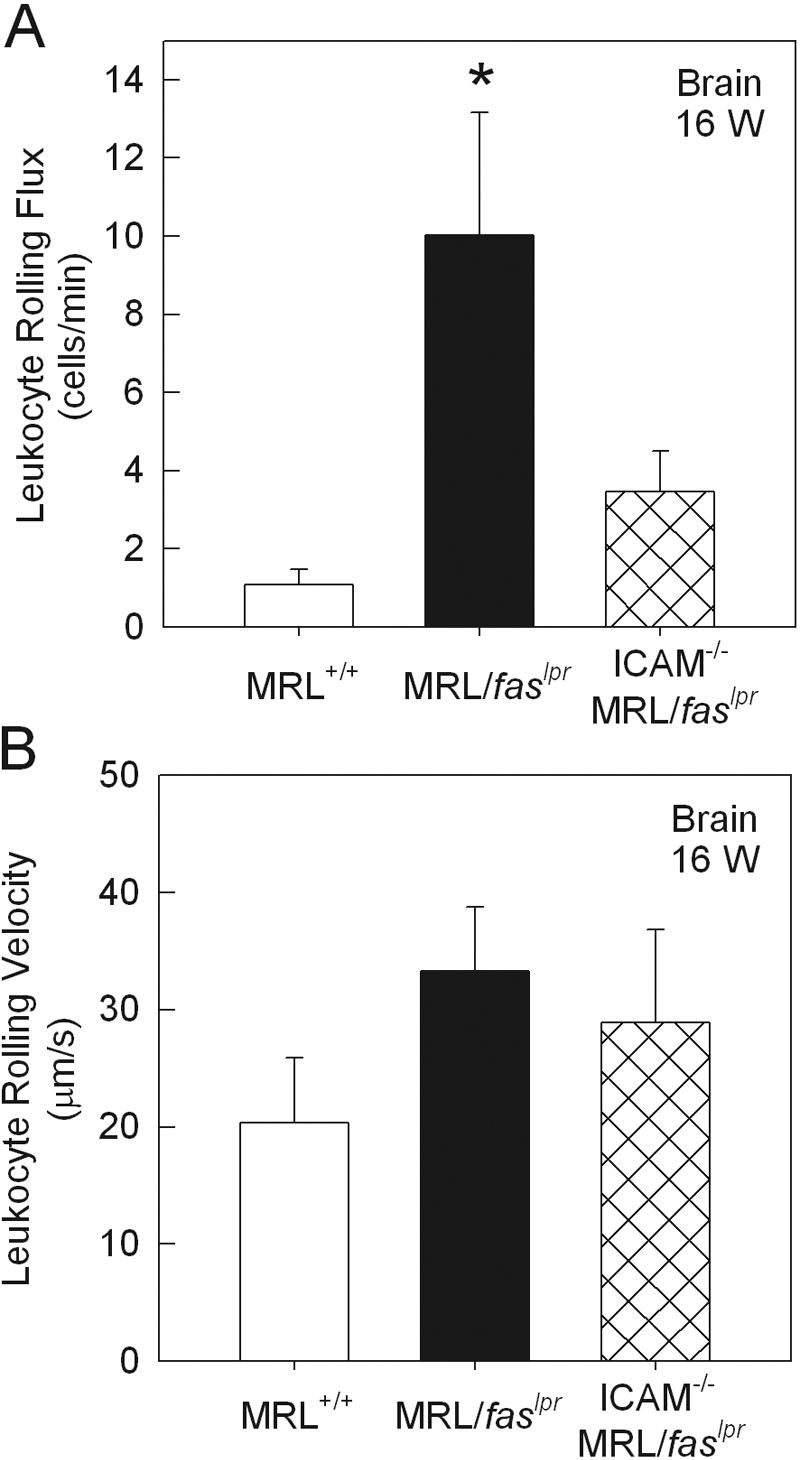
Leukocyte rolling flux (A) and rolling velocity (B) in pial postcapillary venules of 16-week MRL+/+ (n=6), MRL/faslpr (n=9), and ICAM-1−/− MRL/faslpr (n=6) mice. Data were collected from an average of three to four venules per animal. *, P < 0.05, versus MRL+/+.
VCAM-1 expression in skin and brain of MRL/faslpr mice
The ability of the cerebral microcirculation to readily support leukocyte interactions in the absence of ICAM-1 suggested the involvement of an alternative adhesion pathway. We therefore examined expression of VCAM-1 in pial microvessels and compared it with that in the dermal microvasculature. In the skin, a low but consistent level of VCAM-1 expression was detectable in postcapillary venules of MRL+/+, ICAM-1+/+ MRL/faslpr, and ICAM-1−/− MRL/faslpr mice (Fig. 6). However, in all three strains, the intensity of VCAM-1 staining in pial vessels was twice as high as that in dermal vessels, although only in MRL+/+ and ICAM-1−/− MRL/faslpr mice was this difference statistically significant (Fig. 6A). It was notable that in the pial vasculature, the level of VCAM-1 staining did not differ between MRL+/+ and ICAM-1+/+ MRL/faslpr mice, despite the fact that VCAM-1 has been observed to support a greater number of interactions in the lupus-prone strain [17]. In addition, in ICAM-1−/− MRL/faslpr mice, pial expression of VCAM-1 was almost twice that in ICAM+/+ MRL/faslpr mice.
Figure 6.
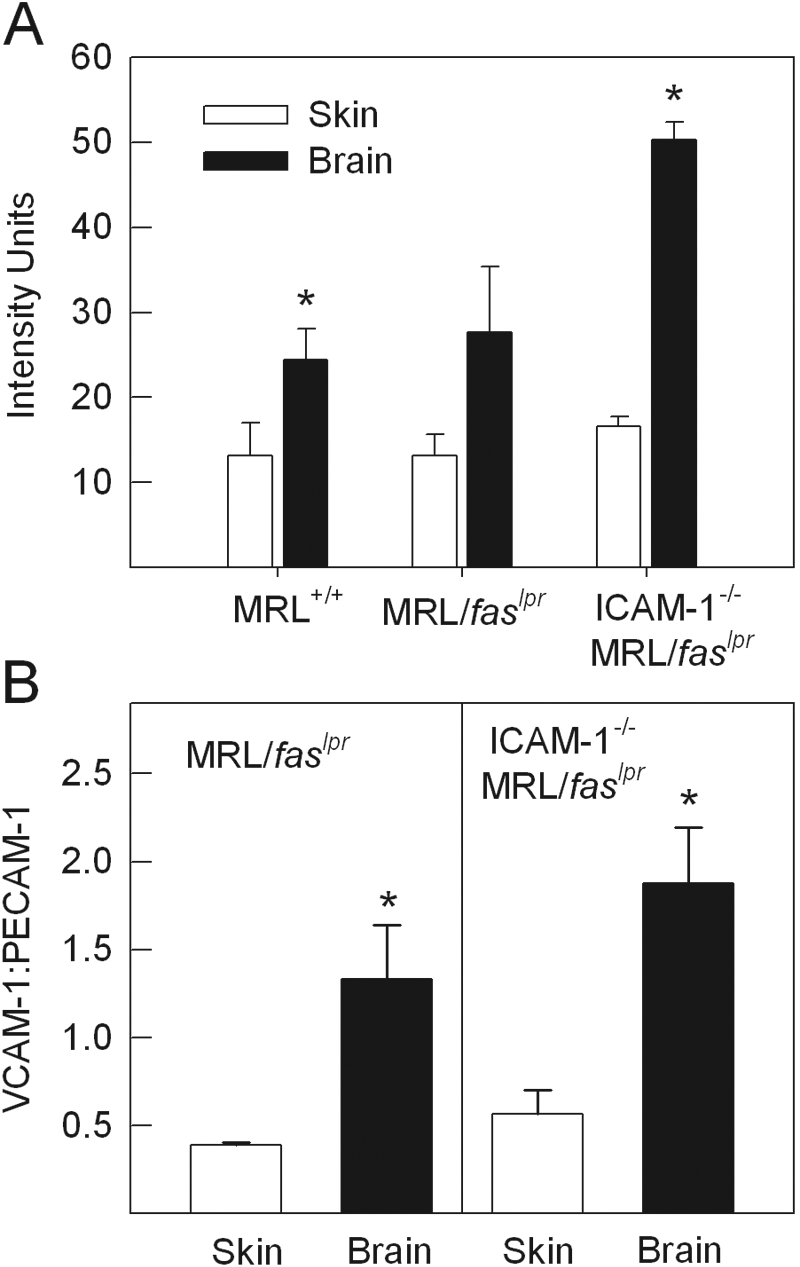
Analysis of VCAM-1 expression in the dermal and pial microvasculatures of MRL+/+, MRL/faslpr, and ICAM-1−/− MRL/faslpr mice. (A) Expression of VCAM-1, as assessed using i.v. administration of Alexa Fluor 488-conjugated anti-VCAM-1 and a nonbinding control antibody. Mice were exsanguinated 5 min after antibody administration, the microvasculature imaged using conventional epifluorescence microscopy, and vascular fluorescence assessed via image analysis (n=4/group). Data are shown as mean intensity units (above background) of the examined vascular segments. (B) Ratio of staining intensities of VCAM-1 and PECAM-1 in dermal and cerebral vessels of MRL/faslpr and ICAM-1−/− MRL/faslpr mice, as assessed following visualization via multi-photon confocal microscopy and image analysis (n=4/group). *, P < 0.05, versus skin via paired t-test.
Given that dermal and cerebral tissue generated substantial autofluorescence, which hampered the sensitivity of the assay, an alternative approach was used to examine this issue. Published work indicates that endothelial expression of PECAM-1 does not differ significantly across tissues, when assessed using an in vivo-radiolabeled immunoassay [28]. Therefore, it was reasoned that PECAM-1 staining could serve as a reference intravascular molecule to allow comparison of expression of simultaneously labeled VCAM-1. This was achieved using Alexa Fluor 488-conjugated anti-VCAM-1 and Alexa Fluor 594-conjugated anti-PECAM-1 and visualization of exsanguinated tissue via in vivo multi-photon confocal microscopy. Comparison of intensity of PECAM-1 staining in dermal and cerebral vessels confirmed that PECAM-1 expression within individual microvessels did not differ significantly between these tissues (skin, 11.3±4.2, vs. brain, 13.2±2.0, intensity units, n=4). When VCAM-1 expression was expressed as a ratio of intensity of VCAM-1:PECAM-1 staining, the level of VCAM-1 in dermal vessels in ICAM+/+ and ICAM-1−/− MRL/faslpr mice was less than 50% that of PECAM-1 (Fig. 6B). In contrast, in cerebral vessels, VCAM-1 expression in ICAM-1+/+ and ICAM-1−/− MRL/faslpr mice was approximately threefold that in dermal vessels (P<0.05 vs. dermal vessels; Fig. 6B). Representative confocal microscopy images of dermal and cerebral vessels simultaneously stained for VCAM-1 and PECAM-1 in ICAM-1−/− MRL/faslpr mice are shown in Figure 7.
Figure 7.

Representative confocal microscopy images of vascular labeling of VCAM-1 and PECAM-1 in cerebral and dermal vessels of an ICAM-1−/− MRL/faslpr mouse. (A and B) Cerebral vessel stained for VCAM-1 (green, A) and PECAM-1 (red, B). (C and D) Dermal vessels stained for VCAM-1 (green, C) and PECAM-1 (red, D). VCAM-1 expression is higher in cerebral vessels, whereas PECAM-1 staining is similar in the two tissues. Original scale bar = 50 μm.
Role of VCAM-1 in cerebral leukocyte adhesion in ICAM-1−/− MRL/faslpr mice
We next assessed the effect of VCAM-1 blockade in ICAM-1−/− MRL/faslpr mice. Anti-VCAM-1 treatment resulted in a significant reduction in leukocyte adhesion in pial postcapillary venules, an effect not seen in mice treated with isotype control antibody (Fig. 8). This treatment also caused a significant reduction in the number of rolling leukocytes (pre-antibody, 6.8±1.9, vs. post-antibody, 1.6±1.5, cells/min; P<0.05 via paired t-test), an effect not observed in mice treated with isotype control antibody (data not shown). These findings are similar to previous observations in ICAM-1+/+ MRL/faslpr mice, indicating an important role for the α4-integrin/VCAM-1 pathway in mediating leukocyte endothelial-cell interactions in the cerebral vasculature of lupus-prone mice, but also suggesting the existence of an additional pathway mediating adhesion in this system [17].
Figure 8.
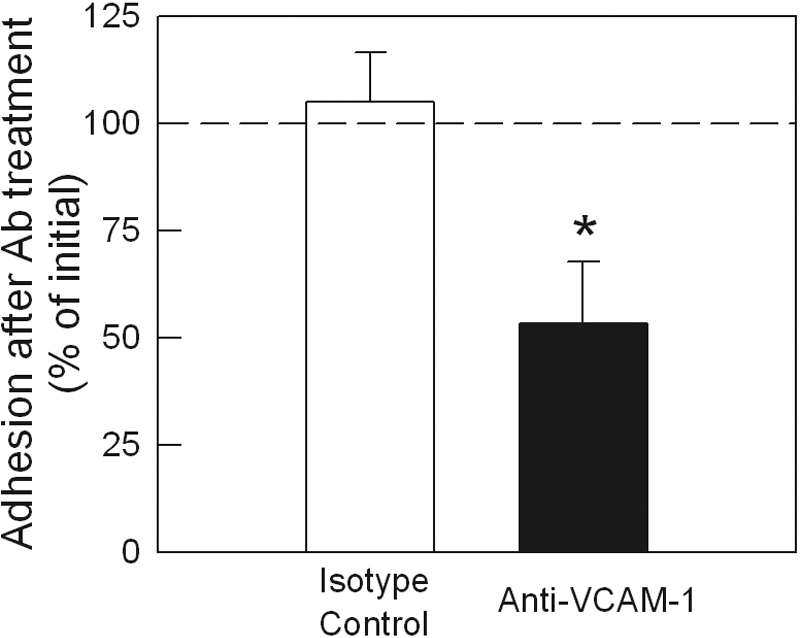
Effect of VCAM-1 blockade on leukocyte adhesion in ICAM-1−/− MRL/faslpr mice. An initial reading of leukocyte adhesion was made, and then mice were treated with anti-VCAM-1 (6C7.1, 90 μg i.v., n=8) or isotype control antibody (n=6), and interactions were assessed 15 min later. Data are shown as mean ± sem of the percentage of the adhesion observed immediately prior to antibody administration. A dotted line at 100% is used to indicate the initial level of adhesion. *, P < 0.05, versus pre-antibody data via paired t-test.
DISCUSSION
Previous studies have shown that ICAM-1 deficiency affords substantial protection against tissue inflammation and lethality in MRL/faslpr mice. However, the relative importance of the trafficking function of ICAM-1 in mediating this protective effect was not clear. Moreover, whether ICAM-1 contributed to leukocyte trafficking in all tissues affected by the systemic inflammatory response in these mice was unknown. In the present studies, we sought to address the question of ICAM-1 involvement in recruitment directly by visualizing leukocyte-endothelial interactions in the microvasculature of two sites affected by leukocyte recruitment in MRL/faslpr mice: the skin and brain. In these animals, the brain, in particular, has been shown to increase expression of ICAM-1 and VCAM-1 progressively over the course of the response, but the relative roles of these adhesion molecules remain unclear [8, 29]. The present experiments revealed that different adhesion molecules were involved in leukocyte trafficking into the skin versus the brain. Leukocyte adhesion and recruitment into the skin of ICAM−/− MRL/faslpr mice were markedly reduced compared with ICAM+/+ MRL/faslpr mice, indicating an important role for ICAM-1 in mediating leukocyte trafficking into the skin during this systemic autoimmune response. In contrast, in the brains of MRL/faslpr mice, leukocyte adhesion was not affected by the absence of ICAM-1 but dependent on VCAM-1.
ICAM-1 is a pleiotropic molecule, with well-established roles in leukocyte-endothelial cell interactions and T cell activation [12, 13, 30, 31]. Either or both of these mechanisms could contribute to the ability of ICAM-1 deficiency to markedly improve survival in MRL/faslpr mice [9, 10]. However, relative to ICAM-1+/+ MRL/faslpr mice, immune responses are not markedly altered in ICAM-1−/− counterparts [9, 10]. In contrast, pulmonary inflammation and vasculitic skin lesions are reduced in these mice, raising the possibility that the major role of ICAM-1 in this response is promotion of leukocyte recruitment. Lloyd et al. [9] explored this hypothesis using adoptive transfer of leukocytes into ICAM-1+/+ and ICAM-1−/− MRL/faslpr mice and found that the absence of ICAM-1 significantly reduced the ability of leukocytes to exit the vasculature and accumulate in the lung. The results of the present study extend this finding in two ways. First, we used direct visualization of affected microvascular beds to identify any alterations in the ability of leukocytes to interact with the endothelial surface in the absence of ICAM-1. Second, we examined two microvascular beds which we have shown previously to respond to this disease model by increasing leukocyte-endothelial cell interactions. This allowed us to unequivocally demonstrate a role for ICAM-1 in mediating leukocyte adhesion in dermal but not pial microvessels in these lupus-prone mice. This is consistent with our previous analysis of leukocyte populations present within the brain of ICAM-1+/+ and ICAM-1−/− MRL/faslpr mice [32]. This study showed that despite clear evidence of robust cerebral ICAM-1 expression in ICAM-1+/+ MRL/faslpr mice, deletion of ICAM-1 did not reduce leukocyte recruitment into the brain [8, 29, 32].
The differential use of ICAM-1 and VCAM-1 by leukocytes in the skin and brain suggests that there may be tissue differences in the expression of these adhesion molecules. We have demonstrated previously that VCAM-1 expression in the brain is higher than that in the skin in noninflamed and inflamed conditions [33]. These findings were made using a dual radiolabeled immunoassay, which measures adhesion molecule expression throughout the brain and skin but were not corrected for regional differences in vascular surface areas between tissues. When VCAM-1 expression was expressed relative to PECAM-1 expression (shown to correlate strongly with vascular surface area), the brain exhibited the highest level of VCAM-1 expression of the tissues examined. This indicated that even in noninflamed tissues, the level of VCAM-1 on a given endothelial cell in brain microvessels is higher than in other tissues [28]. In the present study, we directly visualized binding of anti-VCAM-1 within individual microvessels in the brain and skin and demonstrated that the level of VCAM-1 expression in the pial microvasculature of nondiseased and diseased MRL/faslpr mice was increased relative to that in dermal microvessels in the same animals. The reasons for these regional differences in endothelial adhesion molecule expression are unknown but presumably reflect functional specializations of microvascular endothelial cells specific to each organ. Nevertheless, these findings are consistent with earlier work and provide a potential explanation for the importance of VCAM-1 in leukocyte adhesion in pial but not dermal postcapillary venules.
It has been shown that ICAM-1 is constitutively expressed in the cerebral microvasculature [34]. Furthermore, increased expression of ICAM-1 has been observed in brains of MRL/faslpr mice from as early as 14 weeks of age [8, 29]. Despite this, ICAM-1 was found not to be required for leukocyte trafficking in the brain. The reasons underlying the absence of a role for ICAM-1 in the brain are unclear. This may relate to differences in the relative levels of ICAM-1 and VCAM-1 in the microvasculature of the two tissues. Indeed, in contrast to the observed, specific elevation of VCAM-1 in the brain, expression of ICAM-1 in the cerebral microvasculature has been reported to be similar to that in other tissues when corrected for PECAM-1 expression [28]. Despite this, it is clear from these findings that expression of these adhesion molecules at detectable levels does not necessarily correlate with them having a functional role. Furthermore, coexpression of ICAM-1 and VCAM-1 at these levels does not necessarily lead to them serving redundant, overlapping functions in the microvasculature.
It was noteworthy that in cerebral venules of ICAM-1−/− MRL/faslpr mice, which expressed high levels of VCAM-1, rolling was quite low, but adhesion was at least as high as in ICAM-1+/+ MRL/faslpr mice. These data indicate that the efficiency of converting rolling to adhesion was greater in the ICAM-1−/− mice. Given that we have previously observed a role for VCAM-1 in rolling in these mice [17], it could be seen as unexpected that rolling was also not elevated. However, in inflamed cerebral venules of ICAM-1+/+ MRL/faslpr mice, P-selectin is an additional, important contributor to rolling [17]. Given the observation of reduced rolling in cerebral vessels of ICAM-1−/− mice, it is probable that the contribution of P-selectin is reduced in the ICAM-1−/− mice and that this effect on rolling is greater than the putative “pro-rolling” effect of elevated VCAM-1. However, the high levels of VCAM-1 expression in the ICAM-1−/− mice may have contributed to their greater efficiency of converting rolling to adhesion.
In the present study, acute blockade of VCAM-1 in ICAM-1−/− MRL/faslpr mice was only able to reduce adhesion in cerebral venules by ∼50%. This is similar to our previous findings in ICAM-1+/+ MRL/faslpr mice, in which inhibition of VCAM-1 and its ligand, the α4 integrin, significantly reduced but did not eliminate leukocyte adhesion [17]. One potential explanation for this is that the antibody used (6C7.1) may have been unable to completely inhibit VCAM-1 function in the cerebral microvasculature. However, in our previous work, we have observed that when combined with inhibition of P-selectin, 6C7.1 completely eliminated leukocyte rolling, indicating that it can inhibit VCAM-1 function completely [17]. Alternatively, given that adhesion was only examined 15–30 min after antibody administration, it is possible that the antibody may not have been capable of reversing existing VCAM-1-dependent adhesion contacts of leukocytes which had undergone adhesion prior to antibody administration. Finally, it is also possible that a VCAM-1-independent adhesion pathway is active in this system. Results from the ICAM-1−/− MRL/faslpr mouse indicate that ICAM-1 does not mediate this function. Potential alternative ligands for future investigation would include ICAM-2 and fibrinogen.
SLE remains a disease, where in the past 30 years, few new therapies have been found to be effective [35]. Given the protection in the MRL/faslpr model provided by deletion of some but not all adhesion molecules, inhibition of leukocyte-endothelial cell interactions may provide an effective, novel form of therapy [10, 11, 36]. However, SLE patients display a wide spectrum of disease phenotypes, such that the range of organs affected in individual patients is highly variable. The present observations reveal the importance of elucidating tissue-specific adhesion molecule requirements. Indeed, if anti-adhesion molecule therapy is to be successful in this disease, the targets of the adhesion molecule therapies would need to be selected to reflect the organs most severely affected in individual patients. These data indicate that inhibition of ICAM-1 may be beneficial against dermal complications but not those in the brain. Alternatively, VCAM-1-targeted therapies may be useful in combating cerebral disease but not the highly prevalent skin complications. Improved understanding of the tissue specificity of the mechanisms of leukocyte recruitment will be essential for the implementation of these types of therapies.
Acknowledgments
These studies were supported by the National Health and Medical Research Council (NHMRC; Australia; Program Grant #334067) and the National Institutes of Health (NIH; Bethesda, MD, USA), Grant #RO1AR51807, from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIAMS or NIH. M. U. N. is a NHMRC C. J. Martin Fellow. M. J. H. is a NHMRC Senior Research Fellow. The authors acknowledge Dr. Dan Bullard (University of Alabama at Birmingham, AL, USA) for generous provision of the ICAM-1−/− MRL/faslpr mice and Dr. Dietmar Vestweber (Max Planck Institut, Meunster, Germany) and Dr. Britta Engelhardt (Theodor-Kocher Institut, Bern, Switzerland) for their generous assistance in provision of the 6C7.1 hybridoma.
References
- Lahita R. G. Academic; San Diego, CA, USA: Systemic Lupus Erythematosus. 1999 [Google Scholar]
- Norman M. U., Hickey M. J. Mechanisms of lymphocyte migration in autoimmune disease. Tissue Antigens. 2005;66:163–172. doi: 10.1111/j.1399-0039.2005.00434.x. [DOI] [PubMed] [Google Scholar]
- Pallis M., Robson D. K., Haskard D. O., Powell R. J. Distribution of cell adhesion molecules in skeletal muscle from patients with systemic lupus erythematosus. Ann Rheum Dis. 1993;52:667–671. doi: 10.1136/ard.52.9.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont H. M., Buyon J., Giorno R., Abramson S. Up-regulation of endothelial cell adhesion molecules characterizes disease activity in systemic lupus erythematosus The Shwartzman phenomenon revisited. Arthritis Rheum. 1994;37:376–383. doi: 10.1002/art.1780370311. [DOI] [PubMed] [Google Scholar]
- Takeuchi T., Amano K., Sekine H., Koide J., Abe T. Upregulated expression and function of integrin adhesive receptors in systemic lupus erythematosus patients with vasculitis. J Clin Invest. 1993;92:3008–3016. doi: 10.1172/JCI116924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuthrich R. P., Jevkinar A. M., Takei F., Glimcher L. H., Kelley V. E. Intercellular adhesion molecule-1 (ICAM-1) expression is upregulated in autoimmune murine lupus nephritis. Am J Pathol. 1990;136:441–450. [PMC free article] [PubMed] [Google Scholar]
- Wuthrich R. P. Vascular cell adhesion molecule-1 (VCAM-1) expression in murine lupus nephritis. Kidney Int. 1992;42:903–914. doi: 10.1038/ki.1992.367. [DOI] [PubMed] [Google Scholar]
- McHale J. F., Harari O. A., Marshall D., Haskard D. O. TNF-α and IL-1 sequentially induce ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J Immunol. 1999;163:3993–4000. [PubMed] [Google Scholar]
- Lloyd C. M., Gonzalo J-A., Salant D. A., Just J., Gutierrez-Ramos J-C. Intercellular adhesion molecule-1 deficiency prolongs survival and protects against the development of pulmonary inflammation during murine lupus. J Clin Invest. 1997;100:963–971. doi: 10.1172/JCI119647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard D. C., King P. D., Hicks M. J., Dupont B., Beaudet A. L., Elkon K. B. Intercellular adhesion molecule-1 deficiency protects MRL/MpJ-Faslpr mice from early lethality. J Immunol. 1997;159:2058–2067. [PubMed] [Google Scholar]
- Kevil C. G., Hicks M. J., He X., Zhang J., Ballantyne C. M., Raman C., Schoeb T. R., Bullard D. C. Loss of LFA-1, but not Mac-1, protects MRL/MpJ-Fas(lpr) mice from autoimmune disease. Am J Pathol. 2004;165:609–616. doi: 10.1016/S0002-9440(10)63325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Seventer G. A., Shimizu Y., Horgan K. J., Shaw S. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J Immunol. 1990;144:4579–4586. [PubMed] [Google Scholar]
- Kuhlman P., Moy V. T., Lollo B. A., Brian A. A. The accessory function of murine intercellular adhesion molecule-1 in T lymphocyte activation Contributions of adhesion and co-activation. J Immunol. 1991;146:1773–1782. [PubMed] [Google Scholar]
- Koh D. R., Ho A., Rahemtulla A., Fung-Leung W. P., Griesser H., Mak T. W. Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur J Immunol. 1995;25:2558–2562. doi: 10.1002/eji.1830250923. [DOI] [PubMed] [Google Scholar]
- Chan O. T., Madaio M. P., Shlomchik M. J. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J Immunol. 1999;163:3592–3596. [PubMed] [Google Scholar]
- Hickey M. J., Bullard D. C., Issekutz A., James W. G. Leukocyte-endothelial cell interactions are enhanced in dermal postcapillary venules of MRL/faslpr (lupus-prone) mice: roles of P- and E-selectin. J Immunol. 2002;168:4728–4736. doi: 10.4049/jimmunol.168.9.4728. [DOI] [PubMed] [Google Scholar]
- James W. G., Bullard D. C., Hickey M. J. Critical role of the α 4 integrin/VCAM-1 pathway in cerebral leukocyte trafficking in lupus-prone MRL/fas(lpr) mice. J Immunol. 2003;170:520–527. doi: 10.4049/jimmunol.170.1.520. [DOI] [PubMed] [Google Scholar]
- Engelhardt B., Laschinger M., Schulz M., Samulowitz U., Vestweber D., Hoch G. The development of experimental autoimmune encephalomyelitis in the mouse requires α4-integrin but not α4β7-integrin. J Clin Invest. 1998;102:2096–2105. doi: 10.1172/JCI4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschinger M., Engelhardt B. Interaction of α4-integrin with VCAM-1 is involved in adhesion of encephalitogenic T cell blasts to brain endothelium but not in their transendothelial migration in vitro. J Neuroimmunol. 2000;102:32–43. doi: 10.1016/s0165-5728(99)00156-3. [DOI] [PubMed] [Google Scholar]
- Norman M. U., Lister K. J., Yang Y. H., Issekutz A., Hickey M. J. TNF regulates leukocyte-endothelial cell interactions and microvascular dysfunction during immune complex-mediated inflammation. Br J Pharmacol. 2005;144:265–274. doi: 10.1038/sj.bjp.0706081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory J. L., Morand E. F., McKeown S. J., Ralph J. A., Hall P., Yang Y. H., McColl S. R., Hickey M. J. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol. 2006;177:8072–8079. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- Phillipson M., Heit B., Colarusso P., Liu L., Ballantyne C. M., Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Tavares J., Hickey M. J., Hutchison J., Michaud J., Sutcliffe I. T., Kubes P. A role for platelets and endothelial selectins in tumor necrosis factor-α-induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87:1141–1148. doi: 10.1161/01.res.87.12.1141. [DOI] [PubMed] [Google Scholar]
- Baatz H., Steinbauer M., Harris A. G., Krombach F. Kinetics of white blood cell staining by intravascular administration of rhodamine 6G. Int J Microcirc Clin Exp. 1995;15:85–91. doi: 10.1159/000178955. [DOI] [PubMed] [Google Scholar]
- Hickey M. J., Sharkey K. A., Sihota E. G., Reinhardt P. H., MacMicking J. D., Nathan C., Kubes P. Inducible nitric oxide synthase (iNOS)-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J. 1997;11:955–964. doi: 10.1096/fasebj.11.12.9337148. [DOI] [PubMed] [Google Scholar]
- Hickey M. J., Kanwar S., McCafferty D-M., Granger D. N., Eppihimer M. J., Kubes P. Varying roles of E-selectin and P-selectin in different microvascular beds in response to antigen. J Immunol. 1999;162:1137–1143. [PubMed] [Google Scholar]
- Steeber D. A., Campbell M. A., Basit A., Ley K., Tedder T. F. Optimal selectin-mediated rolling of leukocytes during inflammation in vivo requires intercellular adhesion molecule-1 expression. Proc Natl Acad Sci USA. 1998;95:7562–7567. doi: 10.1073/pnas.95.13.7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppihimer M. J., Russell J., Langley R., Vallien G., Anderson D. C., Granger D. N. Differential expression of platelet-endothelial cell adhesion molecule-1 (PECAM-1) in murine tissues. Microcirculation. 1998;5:179–188. [PubMed] [Google Scholar]
- Zameer A., Hoffman S. A. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol. 2003;142:67–74. doi: 10.1016/s0165-5728(03)00262-5. [DOI] [PubMed] [Google Scholar]
- Diamond M. S., Staunton D. E., Marlin S. D., Springer T. A. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- Broide D. H., Humber D., Sullivan S., Sriramarao P. Inhibition of eosinophil rolling and recruitment in P-selectin- and intracellular adhesion molecule-1-deficient mice. Blood. 1998;91:2847–2856. [PubMed] [Google Scholar]
- James W. G., Hutchinson P., Bullard D. C., Hickey M. J. Cerebral leukocyte infiltration in lupus-prone MRL/MpJ-faslpr mice—roles of intercellular adhesion molecule-1 and P-selectin. Clin Exp Immunol. 2006;144:299–308. doi: 10.1111/j.1365-2249.2006.03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey M. J., Granger D. N., Kubes P. Inducible nitric oxide synthase (iNOS) and regulation of leukocyte/endothelial cell interactions: studies in iNOS-deficient mice. Acta Physiol Scand. 2001;173:119–126. doi: 10.1046/j.1365-201X.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- Henninger D. D., Panes J., Eppihimer M. J., Russell J., Gerritsen M. E., Anderson D. C., Granger D. N. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997;158:1825–1832. [PubMed] [Google Scholar]
- Couzin J. Drug development Lupus drug company asks FDA for second chance. Science. 2005;307:835. doi: 10.1126/science.307.5711.835. [DOI] [PubMed] [Google Scholar]
- He X., Schoeb T. R., Panoskaltsis-Mortari A., Zinn K. R., Kesterson R. A., Zhang J., Samuel S., Hicks M. J., Hickey M. J., Bullard D. C. Deficiency of P-selectin or P-selectin glycoprotein ligand-1 leads to accelerated development of glomerulonephritis and increased expression of CC chemokine ligand 2 in lupus-prone mice. J Immunol. 2006;177:8748–8756. doi: 10.4049/jimmunol.177.12.8748. [DOI] [PubMed] [Google Scholar]