

Abstract
In the present study, we investigated whether saliva from Phlebotomus papatasi and Phlebotomus duboscqi inhibited antigen-induced neutrophil migration and the mechanisms involved in these effects. The pretreatment of immunized mice with salivary gland extracts (SGE) of both phlebotomines inhibited OVA challenge-induced neutrophil migration and release of the neutrophil chemotactic mediators, MIP-1α, TNF-α, and leukotriene B4 (LTB4). Furthermore, SGE treatment enhanced the production of anti-inflammatory mediators, IL-10 and PGE2. SGE treatments failed to inhibit neutrophil migration and MIP-1α and LTB4 production in IL-10−/− mice, also failing in mice treated with nonselective (indomethacin) or selective (rofecoxibe) cyclooxygenase (COX) inhibitors. COX inhibition resulted in diminished SGE-induced IL-10 production, and PGE2 release triggered by SGE remained increased in IL-10−/− mice, suggesting that prostanoids are acting through an IL-10-dependent mechanism. SGE treatments in vivo reduced the OVA-induced lymphoproliferation of spleen-derived cells. Further, the in vitro incubation of bone marrow-derived dendritic cells (DC) with SGE inhibited the proliferation of CD4+T cells from OVA-immunized mice, which was reversed by indomethacin and anti-IL-10 antibody treatments. Supporting these results, SGE induced the production of PGE2 and IL-10 by DC, which were blocked by COX inhibition. These effects were associated with the reduction of DC-membrane expression of MHC-II and CD86 by SGE treatment. Altogether, the results showed that Phlebotomine saliva inhibits immune inflammation-induced neutrophil migration by an autocrine DC sequential production of PGE2/IL-10, suggesting that the saliva constituents might be promising therapeutic molecules to target immune inflammatory diseases.
Keywords: antigen presentation, anti-inflammatory mediators, inflammatory diseases, cytokines, insect saliva, sand fly
INTRODUCTION
Although neutrophils have a protective role during many pathogen infections [1], they are also implicated in the pathogenesis of a number of inflammatory disorders, including rheumatoid arthritis (RA), multiple sclerosis, inflammatory bowel diseases, glomerulonephritis, immune vasculite, and psoriasis [2,3,4,5]. The deleterious role of the neutrophils in these disorders is a result of the ability of these cells to produce significant amounts of proinflammatory cytokines as well as substances such as reactive oxygen and nitrogen species, lysosomal enzymes, and metalloproteases [6, 7]. Indeed, pharmacologic strategies to limit neutrophil trafficking and/or activation have received attention as a potential treatment of autoimmune diseases.
During the inflammatory response, the migration and activation of neutrophils depend on several proinflammatory mediators [5]. In fact, TNF-α, IL-1β, platelet-activation factor, C5a, leukotriene B4 (LTB4), and a variety of chemokines, such as IL-8, growth-related oncogene-α, MIP-1α, and stromal cell-derived factor, are chemotactic mediators involved in the recruitment of neutrophils to sites of inflammation and their activation [8,9,10]. Previously, our group demonstrated that antigen challenge in immunized mice induces neutrophil migration that requires CD4+T cell-derived TNF-α. The release of TNF-α in this model depends on antigen presentation and MIP-1α production. Moreover, TNF-α promotes neutrophil recruitment through a LTB4-dependent mechanism [11,12,13].
Saliva from several species of blood-feeding arthropods arranges a formidable set of sophisticated and redundant mechanisms to overcome the hemostatic and inflammatory system of the vertebrate host during blood meal [14]. Vasodilators, anticoagulants, inhibitors of platelet aggregation, and anti-inflammatory and immunomodulatory molecules are part of this salivary mixture [15, 16]. These active molecules also participate in the transmission as well as the establishment of some arthropod-borne diseases, including Leishmaniasis and Lyme disease [17, 18]. For instance, in Leishmaniasis, there is evidence that the saliva of the vectors Lutzomyia and Phlebotomus enhances the disease [19,20,21,22]. These exacerbated effects are associated with the saliva’s capacity to selectively inhibit several macrophage functions, including antigen presentation, NO, and hydrogen peroxide production, thus inhibiting the ability of macrophages to kill intracellular Leishmania major [23,24,25,26,27,28,29]. Furthermore, vector saliva inhibits the production of protective type 1 cytokines such IL-12 and IFN-γ [30,31,32], and it enhances the production of IL-10, IL-4, IL-6, and PGE2, all of which enhance survival of the Leishmania parasite [33,34,35].
Undoubtedly, Phlebotomine saliva contains several potent pharmacologic factors. Among those properties, identification of the anti-inflammatory and immunomodulatory moieties could be useful in the development of drugs to treat inflammatory diseases. Recently, our group demonstrated that the systemic pretreatment of mice with salivary gland extract (SGE) from the New World vector Lutzomyia longipalpis inhibited neutrophil migration during OVA-induced immune peritonitis. This effect was associated with inhibition of the production of the neutrophil chemoattract mediators, MIP-1α and TNF-α [11, 36]. On the other hand, SGE treatment increased the local production of IL-10 and IL-4, which are described as anti-inflammatory cytokines in the context of immune response [36]. However, the specific site of saliva action was not addressed in the previous study. In the present study, we investigated whether salivary gland homogenates from Phlebotomus papatasi and Phlebotomus duboscqi inhibit neutrophil migration in immune inflammation as well as the mechanisms involved.
MATERIALS AND METHODS
Mice
Female BALB/c and C57BL/6 mice and mice with a targeted disruption of IL-10 (C57BL/6 IL-10−/−), weighing 18–22 g, were housed in temperature-controlled rooms (22–25°C) and received water and food ad libitum in the animal facility of the Department of Pharmacology or Immunology, School of Medicine of Ribeirão Preto, University of São Paulo (Brazil).
Breeding pairs of IL-10−/− were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Breeding stocks backcrossed to C57BL/6 were obtained and housed in a sterile laminar flow until experiments were conducted. The genetic status was confirmed by PCR. All experiments were conducted in accordance with National Institutes of Health (NIH) guidelines on the welfare of experimental animals and with the approval of the Ribeirão Preto School of Medicine Ethics Committee.
Sand fly SGE
Salivary glands were prepared from 7- to 10-day-old laboratory-bred females of P. papatasi and P. duboscqi from the Laboratory of Malaria and Vector Research at the NIH (Bethesda, MD, USA) as described previously [20]. Briefly, 50 pairs of salivary glands were dissected under sterile conditions in endotoxin-free PBS, placed in 50 μl sterile PBS buffer, and kept at −70°C until needed. Immediately before use, the glands were disrupted by sonication using a Sonifer 450 homogenizer (Branson, Danbury, CT, USA). Endotoxin levels were evaluated using the QCL-1000® chromogenic Limulus amoebocyte lysate endpoint assay kit (Lonza, Switzerland), resulting in negligible levels of endotoxin in the salivary gland supernatant.
Procedures for active sensitization with OVA
On Day 0, mice received a single s.c. injection of OVA (100 μg) in 0.2 mL of an emulsion containing 0.1 mL PBS and 0.1 mL CFA. The mice were given booster injections of OVA in IFA on Days 7 and 14. Control mice (sham-immunized) were injected s.c. with 0.2 mL of an emulsion containing equal volumes of PBS and CFA, followed by boosters of an emulsion of PBS and IFA without OVA on Days 7 and 14. On Day 21, immunized and control animals were challenged with an i.p. injection of OVA (10 μg) or PBS.
Leukocyte migration induced by OVA and LPS
Immunized or control (sham-immunized) mice were i.p.-challenged with PBS (0.1 mL/cavity) or OVA (10 μg/cavity). Some naïve mice received an i.p. injection of LPS (100 ng/cavity). The total leukocytes that migrated to the peritoneal cavity were harvested by an injection of 3 ml PBS plus EDTA 1 mM at 6 h and/or 48 h post-stimulus. Total counts were performed on a cell counter, and differential cell counts (200 cells total) were carried out on cytocentrifuge slides stained with Rosenfeld. The results are presented as the number of neutrophils per cavity.
Determination of leukocyte migration into the peritoneal cavity by flow cytometry
Samples of 106 cells obtained from peritoneal exudates were suspended and incubated for 30 min at 4°C in PBS containing 2% of BSA and FcγRI block mAb (CD16/CD32) to avoid nonspecific background staining. After the blocking step, cells were identified by characteristic size (forward-scatter) and granulosity (side-scatter) combined with two-color analysis. Briefly, the lymphocytes were identified as CD19+, CD3+CD4+ and CD3+CD8+; dendritic cells (DC) were identified as CD11c+CD11b+; macrophage as CD11b+CD11c−; and neutrophils as Gr1+ (BD Biosciences PharMingen, San Diego, CA, USA). The isotype controls used were rat IgG2b and rat IgG2a (BD Biosciences PharMingen). After staining, cells were fixed with 1% paraformaldehyde and analyzed by flow cytometry (FACScan™ and CELLQuest™ software, BD Biosciences PharMingen).
Effect of SGE on OVA-induced cytokine and LTB4 production
The immunized and sham-immunized (control) animals treated with SGE or PBS were challenged (i.p.) with 10 μg OVA. After 6 h, the animals were killed, and the peritoneal exudates harvested with 1 mL PBS containing 1 mM EDTA. Peritoneal exudates were centrifuged, and their supernatants were collected and stored at −70°C for determination of MIP-1α, TNF-α, IL-10, and LTB4 by ELISA and PGE2 by radioimmunoassay (RIA).
Measurements of MIP-1α, TNF- α, LTB4, and IL-10 in the peritoneal exudates
The concentrations of MIP-1α, TNF-α, LTB4, and IL-10 in the peritoneal exudates were determined by using a double-ligand ELISA. Briefly, a flat-bottomed 96-well microtiter plate was coated with 100 μL antibody directed to one of the above mediators at a dilution of 2 μg/mL (MIP-1α, TNF-α, LTB4) or 1 μg/mL (IL-10) in coating buffer and incubated overnight at 4°C. After washing and nonspecific-binding blockade, samples and standards were loaded into plates and incubated overnight. Subsequently, the plates were washed, and the appropriate biotinylated polyclonal anti-mediator antibody or mAb anti-mediator was added. After 1 h, the plates were washed, avidin-peroxidase (diluted 1:5000) was then added to each well for 15 min, and the plates were washed thoroughly again. Next, substrate (0.4 mg o-phenylenediamine and 0.4 L H2O2 in 1 mL substrate buffer) was added, the reaction was stopped with H2SO4 (1 M), and the OD was measured at 490 nm. The results were expressed as pg cytokines per mL supernatant.
Measurements of PGE2 by RIA
Samples of peritoneal exudates of OVA-immunized and sham-immunized mice were harvested and lyophilized. A RIA kit was used to determine the PGE2 levels, according to the manufacturer’s instructions (DuPont NEN® Research Products, Boston, MA, USA).
Treatments
Mice were treated with SGE 48 h before OVA or LPS i.p. injection. Some groups also received treatments with a nonselective cyclooxygenase (COX) inhibitor (indomethacin; 5 mg/kg, diluted in 0.1 mM Tris/HCl buffer, pH 8.0) or COX-2-selective inhibitor (rofecoxibe; 3 mg/kg, diluted in PBS plus Tween 80 5%) or vehicles. Drug treatments consisted of 1× during 3 days, as the first and last treatments were performed 30 min before SGE treatment or OVA challenge, respectively.
DC generation and T cell purification
DC were generated in vitro from bone marrow cells from 6- to 8-week-old wild-type BALB/c mice as described previously [37, 38]. Briefly, femurs and tibias were flushed with RPMI 1640 (Gibco-BRL Life Technologies, Grand Island, NY, USA) to release the bone marrow cells that were cultured in 24-well culture plates in RPMI-1640 (Gibco) supplemented with 10% heat-inactivated FCS, 100 μg/ml penicillin, 100 μg/ml streptomycin, 5 × 10−5 M 2-ME (all from Sigma Chemical Co., St. Louis, MO, USA), murine GM-CSF (30 ng/ml), and IL-4 (10 ng/ml; Peprotech, Rocky Hill, NJ, USA). On Days 3 and 6, the supernatants were gently removed and replaced with the same volume of supplemented medium. On Day 9, the nonaherent cells were collected and submitted to a positive selection using anti-CD11c magnetic beads, according to the manufacturer’s instructions (Miltenyi Biotec, Auburn, CA, USA), to eliminate the residual macrophage contamination. Flow cytometric evaluation of purified DC shows that 91% of cells express CD11cinterm or high (marker of DC).
DC (1×106/ml) in RPMI 1640 supplemented with 10% FBS were incubated with P. papatasi SGE (0.5 gland), IL-10 (100 μg/ml), or PGE2 (1 μM) alone, or anti-IL-10 (α-IL-10; 10 μg/ml), indomethacin (10 μg/ml), or medium was added to the culture in the presence or absence of SGE. Briefly, these cells were incubated overnight at 37°C in 5% CO2. In some experiments, DC were treated overnight (37°C in 5% CO2) using the treatment described above before LPS (2 μg/ml) stimulation for 24 h. The cells were then harvested, and surface expression was characterized by flow cytometry using antibodies against MHC class-II, CD80, CD86, and CD40 conjugated to PE or FITC, as well as isotype controls.
To purify CD4+T cells, splenocytes from OVA-immunized mice were incubated with beads coated with antibodies against L3T4 and isolated using biomagnetic separation (Dynabeads, Dynal A.S., Oslo, Norway). The procedures were performed in accordance with the manufacturer’s instructions. The isolated CD4+T lymphocytes (1×106/ml) were cultured with or without SGE in RPMI 1640 10% FSB overnight at 37°C in 5% CO2.
Lymphoproliferation assay
To assess the influence of SGE treatment in lymphoproliferation, OVA-immunized mice were treated in vivo with PBS or SGE from P. papatasi or P. duboscqi, and the spleens were harvested for T cell isolation for the proliferation assay. Briefly, each spleen was removed and washed twice with PBS separately. Tissues were minced, and the cells were filtered through a cell strainer and centrifuged at 500 G at 4°C for 10 min. The cell pellet was resuspended in RPMI-1640 medium to a concentration of 2.5 × 106 cells/ml, and 5 × 105 cells/well were added in 96-well microtiter plates and cultured with OVA (10 μg/ml) or medium for 72 h. Twelve hours before the termination of culture, 0.5 μCi [3H]thymidine (NEN, Boston, MA, USA) was added to each well to determine CD4+T proliferation. To perform in vitro culture assay, purified OVA-primed CD4+T cells, pretreated with or without SGE (5×105 cells/well of a 96-well plate), were incubated with purified DC (5×104 cells/well) and treated with SGE or medium. In some experiments, freshly isolated OVA-primed CD4+T cells were added to wells, where the DC were pretreated previously with different stimuli, as described above, to the cell ratio of 10:1. In all of the experiments, OVA (10 μg/ml) or medium were added to the culture and incubated for 72 h in a total volume of 20 μL per condition. Similar to DC, CD4+ T lymphocytes washed twice in PBS followed the treatment before the coculture assay to avoid the interference of drug action. Before the CD4+T addition, the supernatant from DC culture was harvested to measure IL-10 and PGE2 levels by ELISA and RIA, respectively, and proliferation was measured by overnight [3H]thymidine incorporation.
Statistical analysis
Data are reported as mean ± sem and are representative of two to four independent experiments. The results of an individual experiment were not combined, but they were analyzed individually. The means from different groups were compared by ANOVA followed by Bonferroni’s t-test. Statistical significance was set at P < 0.05.
RESULTS
P. papatasi or P. duboscqi SGE inhibit immune peritonitis-induced neutrophil migration
In a previous study, we demonstrated that the OVA i.p. challenge in immunized mice induced a dose (3–30 μg/cavity)- and time-dependent neutrophil migration. This migration peaked 6 h after OVA challenge, decreasing thereafter and returning to the control level by the 24th hour [11]. Confirming these observations, in the present study, the i.p. administration of OVA (10 μg/cavity) in immunized mice induced a significant neutrophil migration at 6 h after challenge (Fig. 1A) compared with the control group (sham-immunized mice). The pretreatment of immunized mice (48 h before OVA challenge) with SGE (one salivary gland/mouse i.v., ∼1 μg total protein) from P. papatasi or P. duboscqi abolished the OVA-induced neutrophil migration (Fig. 1A). SGE pretreatment also reduced the recruitment of other leukocyte subtypes such as B, CD4+T, and CD8+T lymphocytes and macrophages. On the other hand, SGE pretreatments induced statistically significant eosinophil migration to the peritoneal cavity (Table 1).
Figure 1.

Effects of pretreatments with P. papatasi and P. duboscqi salivary extracts on OVA-induced immune inflammation. OVA-immunized and control (sham-immunized) mice were treated with PBS or SGE from P. papatasi (Papa) or P. duboscqi (Dub) vector (one salivary gland/i.v./animal) 48 h before i.p. challenge with PBS or OVA (10 μg). The neutrophil migration (A) and MIP-1α (B), TNF-α (C), and LTB4 (D) production were determined in the peritoneal exudates 6 h after challenge. Data are the mean ± sem and are representative of two different experiments with four (ELISA) to six (neutrophil migration) mice per group per experiment; #, P < 0.05, compared with control group and immunized mice challenged with PBS; *, P < 0.05, compared with the PBS-pretreated, OVA-immunized group and challenge with OVA (PBS+OVA).
TABLE 1.
Effect of Sand Fly SGE on Leukocyte Migration Induced by OVA in Immunized Mice
| Cells | False-immunized |
OVA-immunized |
|||
|---|---|---|---|---|---|
| PBS (i.v.) | PBS (i.v.) | OVA (i.p.) | |||
| OVA (i.p.) | PBS (i.p.) | PBS (i.v.) | P. papatasil (i.v.) | P. duboscqi (i.v.) | |
| Total leukocytes | 4.33 × 106 ± 1.2 | 5.25 × 106 ± 3.6 | 12.33 × 106 ± 3.1a | 9.42 × 106 ± 3.7b | 8.50 × 106 ± 3.8b |
| Neutrophils | 12.0 ± 0.14 (29%) | 0.06 ± 0.0003 (1.1%) | 71 ± 0.31 (58%)a | 0.65 ± 0.015 (6.6%)b | 0.8 ± 0.20 (9.3%)b |
| Eosinophils | 2.0 ± 0.01 (4.6%) | 3.0 ± 0.02 (5.7%) | 20 ± 0.1 (16.2%)a | 57.1 ± 7.0 (60.3%)b | 43.0 ± 6.0 (50.3%)b |
| Macrophages | 1.0 ± 0.1 (2.1%) | 0.4 ± 0.1 (0.7%) | 3.2 ± 0.8 (2.6%)a | 0.32 ± 0.6 (0.33%)b | 0.6 ± 0.04 (0.72%)b |
| B cells | 0.25 ± 0.02 (0.6%) | 0.23 ± 0.04 (0.2%) | 2.1 ± 0.1 (1.7%)a | 0.2 ± 0.06 (0.2%)b | 0.5 ± 0.2 (0.7%)b |
| CD4+T | 0.4 ± 0.2 (0.9%) | 0.2 ± 0.09 (0.36%) | 1.2 ± 0.3 (0.9%)a | 0.2 ± 0.04 (0.2%)b | 0.4 ± 0.08 (0.5%)b |
| CD8+T | 0.002 ± 0.0004 (0.004%) | 0.003 ± 0.001 (0.006%) | 0.014 ± 0.004a (0.01%) | 0.0052 ± 0.002b (0.005%) | 0.0023 ± 0.001b (0.003%) |
The data are mean ± sd of the number of leukocytes recovered from the peritoneal cavity of OVA-immunized and control (sham-immunized) mice treated with PBS or SGE from P. papatasi or P. duboscqi vector (one gland/i.v./animal) 48 h before i.p. challenge with PBS or OVA (10 μg). The values inside parenthesis mean the percentage of each cellular type relative to the total leukocyte number. The values are ×105 per peritoneal cavities. Experimental groups included four to five animals.
P < 0.05 compared with control groups;
P < 0.05 compared with the PBS-pretreated OVA-immunized group and challenge with OVA (PBS + OVA).
SGE inhibit the release of neutrophil chemotactic mediators
Considering that the OVA challenge-induced neutrophil migration in immunized mice is mediated by the sequential release of MIP-1α (CCL3), TNF-α, and LTB4 [12], we analyzed the effects of SGE on the peritoneal release of these mediators. The pretreatment of immunized mice with SGE from P. papatasi and P. duboscqi inhibited OVA-induced MIP-1α (Fig. 1B), TNF-α (Fig. 1C), and LTB4(Fig. 1D) production in the peritoneal cavity.
Inhibition of OVA-induced neutrophil migration by SGE depends on sequential release of PGE2 and IL-10
Next, we addressed whether SGE treatments inhibited neutrophil migration by inducing the production of the anti-inflammatory cytokines IL-10 or IL-4. OVA challenge (10 μg/cavity) by itself did not induce a significant release of IL-10 in PBS-pretreated mice. However, the pretreatment of OVA-immunized mice with SGE from P. papatasi or P. duboscqi significantly increased IL-10 production after OVA challenge (Fig. 2A). The IL-4 levels were similar in all groups analyzed (data not shown). Confirming the involvement of IL-10 in the inhibitory effects of SGE upon neutrophil migration, the SGE pretreatments failed to inhibit OVA-induced neutrophil migration in immunized IL-10−/− mice (Fig. 2B). Furthermore, the inhibitory effects of SGE on the peritoneal releases of MIP-1α (Fig. 2C) and LTB4(Fig. 2D) induced by OVA challenge were not observed in IL-10−/− mice.
Figure 2.

The anti-inflammatory effect of P. papatasi and P. duboscqi salivary extracts depends on IL-10 production. Wild-type (WT) and IL-10−/− mice were immunized against OVA or sham-immunized mice (Control). Mice were treated with PBS or SGE from P. papatasii or P. duboscqi vector (one salivary gland/i.v./animal) 48 h before i.p. challenge with PBS or OVA (10 μg). Six hours after challenge, peritoneal exudates were collected, and we determined the effect of SGE pretreatment on IL-10 levels (A) in wild-type mice and the effect of P. papatasii on wild-type and IL-10−/− mice upon neutrophil migration (B), MIP-1α (C), and LTB4 (D) production. Data are the mean ± sem and are representative of two experiments; n = 4–6 per group; *, P < 0.05, compared with the PBS-pretreated, OVA-immunized group and challenge with OVA.
Taking into account previous results showing that PGE2 is able to induce IL-10 production [39], we investigated the involvement of this eicosanoid on the SGE effects. Pretreatment of the OVA-immunized mice with SGE promoted a significant increase of OVA-induced PGE2 production compared with PBS-pretreated mice (Fig. 3A). Moreover, the s.c. treatment of immunized mice with nonselective COX (indomethacin) or selective (rofecoxibe) COX-2 inhibitors prevented the inhibitory effects of SGE (48 h before OVA challenge) on OVA-induced neutrophil migration (Fig. 3B), in addition to MIP-1α (Fig. 3C) and LTB4(Fig. 3D) production. Further, the treatment of the immunized mice with indomethacin or rofecoxibe also prevented the increase of IL-10 production by SGE administration (Fig. 4A). On the other hand, the increase of OVA-induced PGE2 production by P. papatasi SGE pretreatment was not altered in IL-10−/− mice (Fig. 4B). Taken together, these results clearly demonstrate that the PGE2/IL-10 sequential pathway is involved in the inhibitory effects of the Phlebotomine salivary extracts upon OVA-induced neutrophil migration in immunized mice.
Figure 3.

The anti-inflammatory effect of P. papatasi and P. duboscqi salivary extracts depends on PGE2 production. OVA-immunized or control mice (sham-immunized) were treated with PBS or SGE from P. papatasi or P. duboscqi vector (one salivary gland/i.v./animal) 48 h before i.p. challenge with PBS or OVA (10 μg). Mice were also treated (as described in Materials and Methods) with indomethacin (Indo; 5 mg/Kg/s.c.), rofecoxibe (3 mg/Kg/s.c.), or vehicles before challenge. Six hours after challenge, peritoneal exudates were collected, and we determined the effect of SGE pretreatment on PGE2 (A) levels by RIA and neutrophil migration (B) and the effect of P. papatasi pretreatment on MIP-1α (C) and LTB4 (D) production by ELISA. Data are the mean ± sem and are representative of two experiments (n=5 per group); #, P < 0.05, compared with control group and immunized mice challenged with PBS; *, P < 0.05, compared with the PBS-treated group.
Figure 4.

SGE induce prostanoid-dependent IL-10 production. Wild-type and IL-10−/− mice were immunized against OVA or sham-immunized mice (Control). Mice were treated with PBS or SGE from P. papatasi or P. duboscqi vector (one salivary gland/i.v./animal) 48 h before i.p. challenge with PBS or OVA (10 μg). (A) Wild-type mice were also treated with indomethacin (5 mg/Kg/s.c.), rofecoxibe (3 mg/Kg/s.c.), or vehicles before OVA challenge, and exudate samples were collected 6 h after challenge for IL-10 measurement by ELISA. (B) Peritoneal exudate samples from wild-type and IL-10−/− mice were collected 2 h after OVA challenge for PGE2 measurement by RIA. Data are the mean ± sem and are representative of two independent experiments (n=4–5 per group); #, P < 0.05, compared with control group; *, P < 0.05, compared with the PBS-pretreated, OVA-immunized group and challenge with OVA.
SGE reduce antigen presentation ability of DC: autocrine role of the IL-10/PGE2 pathway
The neutrophil migration observed during OVA-induced immune peritonitis depends on antigen presentation by APC and consequently, CD4+ T cell activation [11]. In an attempt to determine the mechanism by which SGE are inhibiting OVA-induced neutrophil migration, we investigated the effects of SGE on OVA presentation by DC and an OVA-induced lymphoproliferative response. Figure 5A shows that spleen cells from OVA-immunized mice presented an intense lymphoproliferative response induced by OVA. There was a significant reduction in the lymphoproliferation when spleen cells were obtained from mice that had received a systemic injection (one salivary gland/mouse) of SGE 48 h before. Considering that both SGE showed similar results concerning their anti-inflammatory activity, the P. papatasi SGE was used in all further experiments.
Figure 5.

The suppressive effect of SGE on OVA-induced lymphoproliferation depends on PGE2 and IL-10. OVA-immunized and sham-immunized mice (Control) were i.v.-treated with PBS or SGE from P. papatasi or P. duboscqi vector (one salivary gland/i.v./animal) 48 h before splenocyte harvesting. (A) Splenocytes were cultured with or without OVA (10 μg/mL) during 72 h, and the proliferation assay was performed by [3H]thymidine incorporation. (B) CD4+T or DC (1×106 cells/ml) from OVA-immunized and naïve mice, respectively, were incubated overnight with or without P. papatasii SGE (0.5 gland), and coculture of CD4+T and DC (10:1) was performed for 72 h in the presence of OVA (same dose). In another set of experiments, DC (1×106 cells) from naïve mice were incubated overnight with SGE (0.5 gland), IL-10 (100 μg/ml), PGE2 (1 μM), antibody against IL-10 (α-IL-10; 10 μg/ml), or indomethacin (10 μg/ml). α-IL-10 or indomethacin was also added in the presence or absence of SGE. Afterwards, CD4+T cells of OVA-immunized mice were added, followed by stimulation with or without OVA. The lymphoproliferation was assessed by [3H]thymidine incorporation (C), PGE2 by RIA (D), and IL-10 by ELISA (E). The results are expressed as the mean ± sem of at least two separate experiments made in triplicate (n=3 per group); #, P < 0.05, compared with the medium stimulus; *, P < 0.05, compared with the saliva control (PBS) and DC + CD4+T groups.
To assess which immune cell is inhibited by SGE, purified OVA-primed CD4+T cells or DC obtained from bone marrow were incubated with PBS or SGE (0.5 gland/1×106 cells) overnight. Afterwards, the lymphoproliferative response was assessed in the coculture of these cells. It was observed that the lymphoproliferation was reduced when DC were incubated with SGE, whereas the incubation of OVA-CD4+T cells with SGE did not alter this response (Fig. 5B). These results suggest that SGE suppresses the immune response by acting preferentially on APCs.
Further, investigating the mechanism by which SGE inhibited DC function, we observed that the preincubation of DC with indomethacin or α-IL-10 prevented the inhibitory effect of SGE on the OVA-induced lymphoproliferation. These results suggest that PGs and IL-10 mediate the immunosuppressive action of SGE on APC. Corroborating this hypothesis, the treatment of DC with exogenous PGE2 or IL-10 also inhibited OVA-induced lymphoproliferation (Fig. 5C). Furthermore, SGE induced significant production of PGE2(Fig. 5D) and of IL-10 (Fig. 5E) by DC. In addition, the IL-10 production by SGE-treated DC was inhibited by incubation with indomethacin (Fig. 5E). In accordance with the previous results, IL-10 did not induce the release of PGE2, nor did α-IL-10 prevent SGE stimulation of PGE2 production by DC (Fig. 5E).
Inhibitory effect of SGE on DC surface molecule expression
In an attempt to determine the mechanism by which SGE inhibits the ability of DC to present antigen, we evaluated its effect on expression of DC surface molecules involved in antigen presentation. The expression of MHC-II and the costimulatory molecule CD86 in the membranes of LPS-stimulated DC was not homogeneous. However, this finding is relatively common in the literature [40,41,42]. The SGE treatment inhibited the LPS-induced DC expression of these molecules by 59% and 75%, respectively (Fig. , 6Aand 6B). On the other hand, the expression of CD80 or CD40 was not altered by SGE treatment. The low expression of these molecules in the membrane of LPS-induced DC could be a consequence of the period of DC differentiation. In fact, there is evidence in literature, including the articles that describe the methods to generate DC from bone marrow cells, where the expression of CD80 and CD40 is low in DC stimulated by LPS during 24 h [37, 40, 41]. Supporting the idea that the inhibition of neutrophil migration by SGE was associated with their ability to inhibit DC-presenting antigen, SGE from P. papatasi or P. duboscqi did not alter LPS-induced neutrophil migration to the peritoneal cavity (Fig. 7).
Figure 6.

SGE induces down-regulation of DC surface molecule expression. DC cells previously incubated overnight with SGE from P. papatasi or PBS were cultured in the presence of LPS during 24 h. Surface molecules were labeled with FITC or PE conjugated with anti-MHC class-II, anti-CD80, anti-CD86, or anti-CD40 antibodies. (A) The representative geometric mean fluorescence intensity (MFI) of DC staining from LPS or LPS + SGE cultures is shown in each box. The filled histograms represent cells labeled with the specific mAb; the empty histograms represent the same cell suspension labeled with isotypic control mAb. (B) The bars display the relative MFI obtained from one of four independent experiments made in duplicate (n=2); *, P < 0.05, compared with the LPS stimulation.
Figure 7.
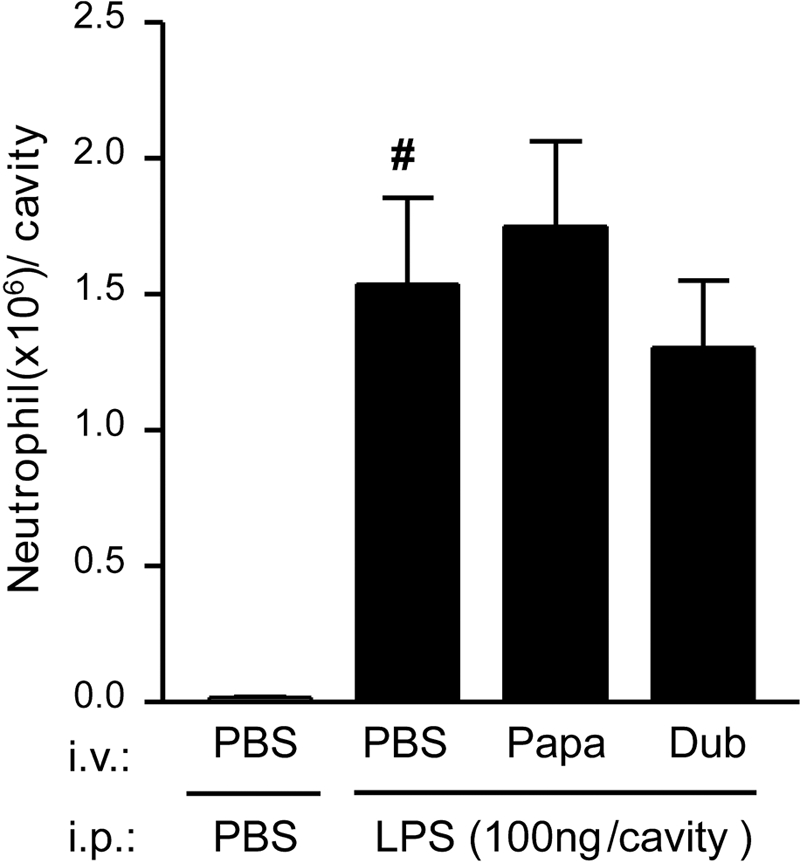
Effect of SGE on LPS-induced neutrophil migration. Mice were treated with PBS or SGE from P. papatasi or P. duboscqi (one salivary gland/i.v./animal) before i.p. injection of LPS (100 ng). The neutrophil migration was determined 6 h after the i.p. administration of LPS as described in Materials and Methods. Data are mean ± sem and are representative of two different experiments (n=5); #, P < 0.05, compared with PBS (vehicle of LPS) i.p. stimulus.
DISCUSSION
Several studies have reported that Phlebotomine saliva contains numerous substances with pharmacological properties that include vasodilatation, anticoagulation, anti-inflammation, and immunomodulation [22, 26]. In this context, our group described previously that salivary extracts of the New World vector L. longipalpis present anti-inflammatory properties by inhibiting the neutrophil migration during the effector phase of a Th1-like immune response [36]. In the present study, we demonstrated that the saliva from Phlebotomines from Old World species P. papatasi and P. duboscqi inhibited the ability of DC to present antigen, and as a consequence, it reduced neutrophil migration during specific antigen-induced inflammation. The inhibitory effects of the SGE depend on sequential production of PGE2 and IL-10 by DC, which in turn act in an autocrine manner, reducing the antigen-presenting ability of DC.
The development of an inflammatory response and consequently, neutrophil migration after OVA challenge in immunized mice result from the activation of a specific OVA-CD4+ T cell by APCs [11]. The activation of both cell types is responsible for the sequential release of neutrophil chemotactic mediators MIP-1α, TNF-α, and LTB4 at the inflammatory foci [11,12,13]. Therefore, the inhibition of the release of these mediators by SGE explains their ability to inhibit OVA-induced neutrophil migration. The inhibitory effect of SGE on the production of these neutrophil chemotactic mediators is dependent on the production of PGE2 and IL-10. In fact, genetic ablation of IL-10 or pharmacologic inhibition of PG production prevent the inhibitory effect of SGE on neutrophil migration and the release of neutrophil chemotactic mediators MIP-1α, TNF-α, and LTB4. Moreover, the PGE2 and IL-10 production by DC occurs in a sequential manner. It has been demonstrated consistently that IL-10 is an anti-inflammatory cytokine that plays an important role in limiting tissue injury during the specific immune inflammatory response by down-regulation of the inflammatory reactions [43]. Its anti-inflammatory activity involves the inhibition of cytokine production by T cells (e.g., IL-2), NK cells (e.g., IFN-γ), and monocyte/macrophages and DC (e.g., IL-1α and IL-1β, IL-6, IL-8, IL-12, TNF-α, and GM-CSF) [43, 44]. IL-10 also inhibits CC (MCP-1, MCP-5, MIP-1α, MIP-1β, MIP-3α, MIP-3β, RANTES, and macrophage-derived chemokine) and CXC chemokine (IL-8, IFN-inducible protein 10, MIP-2, and keratinocyte-derived chemokine) production by different cell types [45,46,47,48]. Although there is evidence that P. papatasi SGE used in the present study contains compounds that increase IL-10 levels such as adenosine/adenosine monophosphate (AMP) [49], we have demonstrated that IL-10 release depends on previous PGE2 production by DC. Furthermore, P. duboscqi salivary extracts do not contain adenosine [50]. Corroborating our results, there is evidence that PGE2 stimulates the production of IL-10 by macrophages, T cells [51,52,53], or bone marrow DC. It has been suggested that the molecular mechanism involved in the PGE2-induced IL-10 production depends on the activation of EP2/EP4 PG receptors, which leads to an increase in the intracellular cAMP levels, ultimately responsible for IL-10 production via STAT3 [54, 55].
Our results also show that SGE treatment enhances OVA-induced eosinophil influx. Similarly, it has already been demonstrated that SGE enhances the eosinophil migration to Leishmania infection sites [56, 57]. It was not the aim of the present study to determine the mechanism involved in SGE-induced eosinophil recruitment. However, in our experimental conditions, it could be a consequence of the increase in the production of Th2 cytokines at the inflammatory focus [58]. There is evidence in literature that SGE enhances the release of cytokines and chemokines of the Th2 pattern, such as IL-4, IL-5, IL-13, and MCP-1, which are chemotactic to eosinophils [31, 57].
Taking into account the immunomodulatory effects of PGE2 and IL-10 released by SGE, we addressed the mechanism by which they could be inhibiting the production of neutrophil chemotactic mediators in our model. As mentioned before, the effector phase of OVA challenge-induced neutrophil migration depends on local antigen presentation by DC to CD4+ T cells, which in turn proliferate and release the chemotactic factor as well as TNF-α [11]. Treatment of immunized mice with SGE reduced the OVA-induced proliferation of spleen-derived T cells. This inhibitory effect of SGE was mediated by the sequential release of PGE2 and IL-10. This conclusion is supported by the blockade of SGE inhibition of spleen T cell proliferation by indomethacin or antibody against IL-10. Moreover, PGE2 and IL-10 mimicked the SGE inhibitory effect, and the eicoisanoid effect was inhibited by antibody against IL-10. The efficient activation of CD4+T cells required their engagement with APC provided by interactions of TCR with MHC molecule/peptide complexes and associated costimulatory molecules, as well as with cytokines released by both cell types [59]. All of these processes can be down-modulated by IL-10 and PGE2 as already described [60, 61]. In our system, it seems that DC are the source of SGE-stimulated PGE2 and IL-10 production, as SGE inhibited OVA-induced CD4+T cell proliferation when incubated with DC but not with CD4+T cells. Additionally, SGE-treated DC released PGE2 and IL-10. Thus, DC sequentially released PGE2 and IL-10, which act in an autocrine manner, inhibiting their antigen-presentation ability.
The reduced ability of SGE-treated DC to present antigen could be associated with reduced expression of MHC-II as well as CD86 costimulatory molecules in the DC membrane. Consistently, agonists of the EP2 and EP4 PGE2 receptors significantly induced the production of IL-10 by DC, which acting in an autocrine manner, decreased MHC class-II molecule expression [61,62,63]. The interaction of CD80 and CD86 molecules with CD28 expressed by CD4+T cells augments and sustains CD4+T cell activation [64, 65]. Likewise, the CD40–CD40 ligand interaction is also important for the maintenance of T cell immunity [66, 67]. The SGE treatment, although it did inhibit the expression of MHC-II and CD86 costimulatory molecules, affected neither CD80 nor CD40 expression.
In addition to the autocrine role of DC-derived IL-10/PGE2, which inhibits the ability of DC to present antigen, these substances could be acting directly on CD4+T cells in a paracrine manner to inhibit their activation. In fact, as described in literature, PGE2 and IL-10 induce suppression of T cell proliferation, which is related to the down-regulation of the expression of IL-2R and IL-2 production [68]. In our experimental conditions, the proliferation of CD4+T cells to OVA was inhibited by exogenous administration of PGE2 in the culture but not of IL-10, suggesting that only the prostanoid is acting in an autocrine and paracrine manner (data not shown). There is also an in vitro demonstration that the saliva of other blood-feeding arthropods, such as the tick Ixodes scapularis, inhibits DC maturation and function in a PGE2-dependent mechanism. The saliva of I. scapularis as well as PGE2 induces a concomitant inhibition of TNF-α and induction of IL-10 production by DC. Furthermore, although PGE2 is a common immunomodulatory mediator among the saliva of I. scapularis, P. papatasi, and P. duboscqi, the mechanism seems to be different, as PGE2 is a constituent of I. scapularis saliva, whereas P. papatasi and P. duboscqi saliva might contain substances that induce PGE2 production. Another important difference is that the saliva of I. scapularis did not alter the expression of presentation molecules in contrast to the present data [69].
Further supporting that Phlebotomine SGE affect antigen presentation, SGE did not inhibit LPS-induced neutrophil migration, which does not depend on presentation but on direct activation of TLR with consequent production of neutrophil-chemotactic mediators. Thus, considering the novel mechanism demonstrated herein, which does not affect host defense (the response to LPS remains), we extrapolate that the anti-inflammatory effect of saliva has clinical potential, as it would inhibit only autoimmune inflammation.
Others have also demonstrated the immunosuppressive effect of Phlebotomine saliva. L. longipalpis SGE suppresses T cell proliferation in response to SRBCs [25], and Maxadilan, the vasodilatory peptide of L. longipalpis, modulates cytokine production [70,71,72] and T cell proliferation [73]. In the present study, Maxadilan is not involved in the SGE effects, as it is not a constituent of Phlebotomine saliva from Old World flies, P. papatasi or P. duboscqi [14, 19, 74]. In addition, in a previous experiment, we demonstrated that Maxadilan did not inhibit the OVA-induced neutrophil migration in immunized mice [36].
Recently, it was described in the literature that neutrophils recruited to the site of Leishmania infection internalize the parasite [75, 76] and that SGE enhances the neutrophil migration process [32, 56, 57]. It was also observed that the parasite internalization delays the neutrophil apoptotic death program and induces MIP-1β release, which recruits macrophages to the infection sites. The migrated macrophages ingest the infected apoptocic neutrophils, a process that stimulates the TGF-β and PGE2 release, which down-modulates the macrophage activation and as a consequence, contributes to Leishmania infection establishment [75, 76]. Together, these findings suggest that the parasites use the granulocytes as “Trojan horses” to invade the macrophages [75]. In this context, the inhibition of DC function by SGE described in the present investigation may also represent an additional mechanism to explain the ability of Phlebotomine saliva to exacerbate the Leishmania infection. It is important to point out that our results seem to be apparently controversial with the above-described ability of the SGE to enhance the neutrophil migration to the infection site. However, it is important to mention that the routes of SGE administration are different; whereas in the present investigation, SGE was administered systemically, in the mentioned studies, it was injected locally [32, 56, 57, 77]. It has been well demonstrated that systemic administration of inflammatory stimuli (i.e., LPS) or of neutrophil chemotactic mediators (i.e, IL-8 and TNF-α) inhibits the neutrophil migration induced by different stimuli, whereas the local administration of the substances induces the neutrophil migration [78, 79].
To summarize, in the present study, we demonstrated a novel, anti-inflammatory mechanism in which saliva inhibits neutrophil migration during the effector phase of specific antigen-induced inflammation. These anti-inflammatory effects seem to be dependent on a sequential production of PGE2 and IL-10, which are released by DC and act in an autocrine manner, inhibiting antigen presentation by these cells and preventing the release of neutrophil chemotactic factors with no effect on the host defense. Taking into account that the tissue damage observed in several autoimmune diseases (i.e., RA) is a consequence of the neutrophil emigration, we believe that the active constituents of the saliva could be a prototype to the development of new drugs to prevent the tissue lesion observed in such diseases. Our group is currently working on the isolation of phlebotomine salivary compounds to look for the active principles that are responsible for the anti-inflammatory activity. Thus, this will probably open perspectives for the development of novel drugs for the treatment of immune inflammatory diseases. However, these proceedings require laborious approaches concerning the reduced volume of saliva obtained.
Acknowledgments
J.G.V., J.M.C.R., and D-E.E. were supported in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH (Bethesda, MD, USA). We are thankful to Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Fundação de Apoio ao Ensino, Pesquisa e Assistência do Hospital des Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (FAEPA) for financial support and to Giuliana Bertozi for help with ELISAs. We are grateful to Nancy Shulman for editorial assistance.
References
- Malech H. L., Gallin J. I. Current concepts: immunology Neutrophils in human diseases. N Engl J Med. 1987;317:687–694. doi: 10.1056/NEJM198709103171107. [DOI] [PubMed] [Google Scholar]
- Haynes B. Vasculitis: phatogenic mechanisms of vessel eamage. Gallin J., Goldstain I. M., Snyderman R., editors. Raven Press; New York, NY, USA: Inflammation: Basic Principles and Clinical Correlates. 1992:921–941. [Google Scholar]
- Holdsworth S. R., Bellomo R. Differential effects of steroids on leukocyte-mediated glomerulonephritis in the rabbit. Kidney Int. 1984;26:162–169. doi: 10.1038/ki.1984.150. [DOI] [PubMed] [Google Scholar]
- Wandall J. H. Function of exudative neutrophilic granulocytes in patients with Crohn’s disease or ulcerative colitis. Scand J Gastroenterol. 1985;20:1151–1156. doi: 10.3109/00365528509088887. [DOI] [PubMed] [Google Scholar]
- Weissmann G., Korchak H. Rheumatoid arthritis The role of neutrophil activation. Inflammation. 1984;8(Suppl.):S3–S14. doi: 10.1007/BF00915708. [DOI] [PubMed] [Google Scholar]
- Rutgers A., Heeringa P., Tervaert J. W. The role of myeloperoxidase in the pathogenesis of systemic vasculitis. Clin Exp Rheumatol. 2003;21:S55–S63. [PubMed] [Google Scholar]
- Hampton M. B., Kettle A. J., Winterbourn C. C. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- Binder R., Kress A., Kirschfink M. Modulation of C5a-mediated effector functions of human polymorphonuclear leukocytes by tumor necrosis factor α and granulocyte macrophage colony-stimulating factor. Exp Clin Immunogenet. 1999;16:212–225. doi: 10.1159/000019113. [DOI] [PubMed] [Google Scholar]
- Drost E. M., MacNee W. Potential role of IL-8, platelet-activating factor and TNF-α in the sequestration of neutrophils in the lung: effects on neutrophil deformability, adhesion receptor expression, and chemotaxis. Eur J Immunol. 2002;32:393–403. doi: 10.1002/1521-4141(200202)32:2<393::AID-IMMU393>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z., Kim J., Koch A. E. Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol. 2003;15:15–21. doi: 10.1016/s1044-5323(02)00124-0. [DOI] [PubMed] [Google Scholar]
- Canetti C., Silva J. S., Ferreira S. H., Cunha F. Q. Tumor necrosis factor-α and leukotriene B(4) mediate the neutrophil migration in immune inflammation. Br J Pharmacol. 2001;134:1619–1628. doi: 10.1038/sj.bjp.0704403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos C. D., Canetti C., Souto J. T., Silva J. S., Hogaboam C. M., Ferreira S. H., Cunha F. Q. MIP-1α[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-α and LTB4. J Leukoc Biol. 2005;78:167–177. doi: 10.1189/jlb.0404237. [DOI] [PubMed] [Google Scholar]
- Verri W. A., Jr, Cunha T. M., Ferreira S. H., Wei X., Leung B. P., Fraser A., McInnes I. B., Liew F. Y., Cunha F. Q. IL-15 mediates antigen-induced neutrophil migration by triggering IL-18 production. Eur J Immunol. 2007;37:3373–3380. doi: 10.1002/eji.200737488. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M. Role of saliva in blood-feeding by arthropods. Annu Rev Entomol. 1987;32:463–478. doi: 10.1146/annurev.en.32.010187.002335. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M., Weis J. J., Telford S. R., III Saliva of the tick Ixodes dammini inhibits neutrophil function. Exp Parasitol. 1990;70:382–388. doi: 10.1016/0014-4894(90)90121-r. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M., Francischetti I. M. Role of arthropod saliva in blood feeding: sialome and post-sialome perspectives. Annu Rev Entomol. 2003;48:73–88. doi: 10.1146/annurev.ento.48.060402.102812. [DOI] [PubMed] [Google Scholar]
- Warburg A., Schlein Y. The effect of post-bloodmeal nutrition of Phlebotomus papatasi on the transmission of Leishmania major. Am J Trop Med Hyg. 1986;35:926–930. doi: 10.4269/ajtmh.1986.35.926. [DOI] [PubMed] [Google Scholar]
- Donnelly K. B., Lima H. C., Titus R. G. Histologic characterization of experimental cutaneous Leishmaniasis in mice infected with Leishmania braziliensis in the presence or absence of sand fly vector salivary gland lysate. J Parasitol. 1998;84:97–103. [PubMed] [Google Scholar]
- Champagne D. E. The role of salivary vasodilators in bloodfeeding and parasite transmission. Parasitol Today. 1994;10:430–433. doi: 10.1016/0169-4758(94)90173-2. [DOI] [PubMed] [Google Scholar]
- Titus R. G., Ribeiro J. M. Salivary gland lysates from the sand fly Lutzomyia longipalpis enhance Leishmania infectivity. Science. 1988;239:1306–1308. doi: 10.1126/science.3344436. [DOI] [PubMed] [Google Scholar]
- Gillespie R. D., Mbow M. L., Titus R. G. The immunomodulatory factors of bloodfeeding arthropod saliva. Parasite Immunol. 2000;22:319–331. doi: 10.1046/j.1365-3024.2000.00309.x. [DOI] [PubMed] [Google Scholar]
- Targett G. A. Parasites, arthropod vectors, and immune responses. Parasite Immunol. 2006;28:117–119. doi: 10.1111/j.1365-3024.2006.00825.x. [DOI] [PubMed] [Google Scholar]
- Titus R. G., Kelso A., Louis J. A. Intracellular destruction of Leishmania tropica by macrophages activated with macrophage activating factor/interferon. Clin Exp Immunol. 1984;55:157–165. [PMC free article] [PubMed] [Google Scholar]
- Titus R. G., Theodos C. M., Shankar A. H., Hall L. R. Interactions between Leishmania major and macrophages. Immunol Ser. 1994;60:437–459. [PubMed] [Google Scholar]
- Titus R. G. Salivary gland lysate from the sand fly Lutzomyia longipalpis suppresses the immune response of mice to sheep red blood cells in vivo and concanavalin A in vitro. Exp Parasitol. 1998;89:133–136. doi: 10.1006/expr.1998.4272. [DOI] [PubMed] [Google Scholar]
- Titus R. G., Bishop J. V., Mejia J. S. The immunomodulatory factors of arthropod saliva and the potential for these factors to serve as vaccine targets to prevent pathogen transmission. Parasite Immunol. 2006;28:131–141. doi: 10.1111/j.1365-3024.2006.00807.x. [DOI] [PubMed] [Google Scholar]
- Urioste S., Hall L. R., Telford S. R., III, Titus R. G. Saliva of the Lyme disease vector, Ixodes dammini, blocks cell activation by a nonprostaglandin E2-dependent mechanism. J Exp Med. 1994;180:1077–1085. doi: 10.1084/jem.180.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela J. G., Belkaid Y., Garfield M. K., Mendez S., Kamhawi S., Rowton E. D., Sacks D. L., Ribeiro J. M. Toward a defined anti-Leishmania vaccine targeting vector antigens: characterization of a protective salivary protein. J Exp Med. 2001;194:331–342. doi: 10.1084/jem.194.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitumbi J., Warburg A. Phlebotomus papatasi saliva inhibits protein phosphatase activity and nitric oxide production by murine macrophages. Infect Immun. 1998;66:1534–1537. doi: 10.1128/iai.66.4.1534-1537.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima H. C., Titus R. G. Effects of sand fly vector saliva on development of cutaneous lesions and the immune response to Leishmania braziliensis in BALB/c mice. Infect Immun. 1996;64:5442–5445. doi: 10.1128/iai.64.12.5442-5445.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbow M. L., Bleyenberg J. A., Hall L. R., Titus R. G. Phlebotomus papatasi sand fly salivary gland lysate down-regulates a Th1, but up-regulates a Th2, response in mice infected with Leishmania major. J Immunol. 1998;161:5571–5577. [PubMed] [Google Scholar]
- Belkaid Y., Kamhawi S., Modi G., Valenzuela J., Noben-Trauth N., Rowton E., Ribeiro J., Sacks D. L. Development of a natural model of cutaneous Leishmaniasis: powerful effects of vector saliva and saliva preexposure on the long-term outcome of Leishmania major infection in the mouse ear dermis. J Exp Med. 1998;188:1941–1953. doi: 10.1084/jem.188.10.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P., Artis D., Uzonna J., Zaph C. The development of effector and memory T cells in cutaneous Leishmaniasis: the implications for vaccine development. Immunol Rev. 2004;201:318–338. doi: 10.1111/j.0105-2896.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- Sacks D., Anderson C. Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunol Rev. 2004;201:225–238. doi: 10.1111/j.0105-2896.2004.00185.x. [DOI] [PubMed] [Google Scholar]
- De Freitas L. A., Mbow L. M., Estay M., Bleyenberg J. A., Titus R. G. Indomethacin treatment slows disease progression and enhances a Th1 response in susceptible BALB/c mice infected with Leishmania major. Parasite Immunol. 1999;21:273–277. doi: 10.1046/j.1365-3024.1999.00211.x. [DOI] [PubMed] [Google Scholar]
- Monteiro M. C., Nogueira L. G., Almeida Souza A. A., Ribeiro J. M., Silva J. S., Cunha F. Q. Effect of salivary gland extract of Leishmania vector, Lutzomyia longipalpis, on leukocyte migration in OVA-induced immune peritonitis. Eur J Immunol. 2005;35:2424–2433. doi: 10.1002/eji.200526160. [DOI] [PubMed] [Google Scholar]
- Lutz M. B., Kukutsch N., Ogilvie A. L., Rossner S., Koch F., Romani N., Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- Oliveira C. J., Cavassani K. A., Moré D. D., Garlet G. P., Aliberti J. C., Silva J. S., Ferreira B. R. Tick saliva inhibits the chemotactic function of MIP-1α and selectively impairs chemotaxis of immature dendritic cells by down-regulating cell-surface CCR5. Int J Parasitol. 2007;38:705–716. doi: 10.1016/j.ijpara.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Harizi H., Gualde N. Eicosanoids: an emerging role in dendritic cell biology. Arch Immunol Ther Exp (Warsz) 2004;52:1–5. [PubMed] [Google Scholar]
- Xu Y., Zhan Y., Lew A. M., Naik S. H., Kershaw M. H. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol. 2007;179:7577–7584. doi: 10.4049/jimmunol.179.11.7577. [DOI] [PubMed] [Google Scholar]
- Johnson L. M., Scott P. STAT1 expression in dendritic cells, but not T cells, is required for immunity to Leishmania major. J Immunol. 2007;178:7259–7266. doi: 10.4049/jimmunol.178.11.7259. [DOI] [PubMed] [Google Scholar]
- Kang K., Kim H., Kim K. I., Yang Y., Yoon D. Y., Kim J. H., Ryu J. H., Noh E. J., Jeon S. D., Lim J. S. SK-126, a synthetic compound, regulates the production of inflammatory cytokines induced by LPS in antigen-presenting cells. Biochem Pharmacol. 2008;75:1054–1064. doi: 10.1016/j.bcp.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Moore K. W., de Waal Malefyt R., Coffman R. L., O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Ding Y., Chen D., Tarcsafalvi A., Su R., Qin L., Bromberg J. S. Suppressor of cytokine signaling 1 inhibits IL-10-mediated immune responses. J Immunol. 2003;170:1383–1391. doi: 10.4049/jimmunol.170.3.1383. [DOI] [PubMed] [Google Scholar]
- Berkman N., John M., Roesems G., Jose P. J., Barnes P. J., Chung K. F. Inhibition of macrophage inflammatory protein-1 α expression by IL-10 Differential sensitivities in human blood monocytes and alveolar macrophages. J Immunol. 1995;155:4412–4418. [PubMed] [Google Scholar]
- Nicod L. P., el Habre F., Dayer J. M., Boehringer N. Interleukin-10 decreases tumor necrosis factor α and β in alloreactions induced by human lung dendritic cells and macrophages. Am J Respir Cell Mol Biol. 1995;13:83–90. doi: 10.1165/ajrcmb.13.1.7598941. [DOI] [PubMed] [Google Scholar]
- Gruber M. F., Williams C. C., Gerrard T. L. Macrophage-colony-stimulating factor expression by anti-CD45 stimulated human monocytes is transcriptionally up-regulated by IL-1 β and inhibited by IL-4 and IL-10. J Immunol. 1994;152:1354–1361. [PubMed] [Google Scholar]
- Rossi D. L., Vicari A. P., Franz-Bacon K., McClanahan T. K., Zlotnik A. Identification through bioinformatics of two new macrophage proinflammatory human chemokines: MIP-3α and MIP-3β. J Immunol. 1997;158:1033–1036. [PubMed] [Google Scholar]
- Ribeiro J. M., Modi G. The salivary adenosine/AMP content of Phlebotomus argentipes Annandale and Brunetti, the main vector of human kala-azar. J Parasitol. 2001;87:915–917. doi: 10.1645/0022-3395(2001)087[0915:TSAACO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Kato H., Jochim R. C., Lawyer P. G., Valenzuela J. G. Identification and characterization of a salivary adenosine deaminase from the sand fly Phlebotomus duboscqi, the vector of Leishmania major in sub-Saharan Africa. J Exp Biol. 2007;210:733–740. doi: 10.1242/jeb.001289. [DOI] [PubMed] [Google Scholar]
- Strassmann G., Patil-Koota V., Finkelman F., Fong M., Kambayashi T. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J Exp Med. 1994;180:2365–2370. doi: 10.1084/jem.180.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-ishi S., Utsunomiya I., Yamamoto T., Komuro Y., Hara Y. Effects of prostaglandins and cyclic AMP on cytokine production in rat leukocytes. Eur J Pharmacol. 1996;300:255–259. doi: 10.1016/0014-2999(96)00005-2. [DOI] [PubMed] [Google Scholar]
- Demeure C. E., Yang L. P., Desjardins C., Raynauld P., Delespesse G. Prostaglandin E2 primes naive T cells for the production of anti-inflammatory cytokines. Eur J Immunol. 1997;27:3526–3531. doi: 10.1002/eji.1830271254. [DOI] [PubMed] [Google Scholar]
- Harizi H., Gualde N. Pivotal role of PGE2 and IL-10 in the cross-regulation of dendritic cell-derived inflammatory mediators. Cell Mol Immunol. 2006;3:271–277. [PubMed] [Google Scholar]
- Cheon H., Rho Y. H., Choi S. J., Lee Y. H., Song G. G., Sohn J., Won N. H., Ji J. D. Prostaglandin E2 augments IL-10 signaling and function. J Immunol. 2006;177:1092–1100. doi: 10.4049/jimmunol.177.2.1092. [DOI] [PubMed] [Google Scholar]
- Monteiro M. C., Lima H. C., Souza A. A., Titus R. G., Romao P. R., Cunha F. Q. Effect of Lutzomyia longipalpis salivary gland extracts on leukocyte migration induced by Leishmania major. Am J Trop Med Hyg. 2007;76:88–94. [PubMed] [Google Scholar]
- Teixeira C. R., Teixeira M. J., Gomes R. B., Santos C. S., Andrade B. B., Raffaele-Netto I., Silva J. S., Guglielmotti A., Miranda J. C., Barral A., Brodskyn C., Barral-Netto M. Saliva from Lutzomyia longipalpis induces CC chemokine ligand 2/monocyte chemoattractant protein-1 expression and macrophage recruitment. J Immunol. 2005;175:8346–8353. doi: 10.4049/jimmunol.175.12.8346. [DOI] [PubMed] [Google Scholar]
- Giembycz M. A., Lindsay M. A. Pharmacology of the eosinophil. Pharmacol Rev. 1999;51:213–340. [PubMed] [Google Scholar]
- Mueller D. L., Jenkins M. K., Schwartz R. H. Clonal expansion versus functional clonal inactivation: a costimulatory signaling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- Medzhitov R., Janeway C., Jr Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A., Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- Kambayashi T., Wallin R. P., Ljunggren H. G. cAMP-elevating agents suppress dendritic cell function. J Leukoc Biol. 2001;70:903–910. [PubMed] [Google Scholar]
- Harizi H., Grosset C., Gualde N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J Leukoc Biol. 2003;73:756–763. doi: 10.1189/jlb.1002483. [DOI] [PubMed] [Google Scholar]
- de Boer M., Kasran A., Kwekkeboom J., Walter H., Vandenberghe P., Ceuppens J. L. Ligation of B7 with CD28/CTLA-4 on T cells results in CD40 ligand expression, interleukin-4 secretion and efficient help for antibody production by B cells. Eur J Immunol. 1993;23:3120–3125. doi: 10.1002/eji.1830231212. [DOI] [PubMed] [Google Scholar]
- Bachmann M. F., McKall-Faienza K., Schmits R., Bouchard D., Beach J., Speiser D. E., Mak T. W., Ohashi P. S. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity. 1997;7:549–557. doi: 10.1016/s1074-7613(00)80376-3. [DOI] [PubMed] [Google Scholar]
- Diehl L., Den Boer A. T., van der Voort E. I., Melief C. J., Offringa R., Toes R. E. The role of CD40 in peripheral T cell tolerance and immunity. J Mol Med. 2000;78:363–371. doi: 10.1007/s001090000126. [DOI] [PubMed] [Google Scholar]
- O'Sullivan B., Thomas R. CD40 and dendritic cell function. Crit Rev Immunol. 2003;23:83–107. doi: 10.1615/critrevimmunol.v23.i12.50. [DOI] [PubMed] [Google Scholar]
- Ding L., Shevach E. M. IL-10 inhibits mitogen-induced T cell proliferation by selectively inhibiting macrophage costimulatory function. J Immunol. 1992;148:3133–3139. [PubMed] [Google Scholar]
- Sa-Nunes A., Bafica A., Lucas D. A., Conrads T. P., Veenstra T. D., Andersen J. F., Mather T. N., Ribeiro J. M., Francischetti I. M. Prostaglandin E2 is a major inhibitor of dendritic cell maturation and function in Ixodes scapularis saliva. J Immunol. 2007;179:1497–1505. doi: 10.4049/jimmunol.179.3.1497. [DOI] [PubMed] [Google Scholar]
- Bozza M., Soares M. B., Bozza P. T., Satoskar A. R., Diacovo T. G., Brombacher F., Titus R. G., Shoemaker C. B., David J. R. The PACAP-type I receptor agonist Maxadilan from sand fly saliva protects mice against lethal endotoxemia by a mechanism partially dependent on IL-10. Eur J Immunol. 1998;28:3120–3127. doi: 10.1002/(SICI)1521-4141(199810)28:10<3120::AID-IMMU3120>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Soares M. B., Titus R. G., Shoemaker C. B., David J. R., Bozza M. The vasoactive peptide Maxadilan from sand fly saliva inhibits TNF-α and induces IL-6 by mouse macrophages through interaction with the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor. J Immunol. 1998;160:1811–1816. [PubMed] [Google Scholar]
- Rogers K. A., Titus R. G. Immunomodulatory effects of Maxadilan and Phlebotomus papatasi sand fly salivary gland lysates on human primary in vitro immune responses. Parasite Immunol. 2003;25:127–134. doi: 10.1046/j.1365-3024.2003.00623.x. [DOI] [PubMed] [Google Scholar]
- Qureshi A. A., Asahina A., Ohnuma M., Tajima M., Granstein R. D., Lerner E. A. Immunomodulatory properties of Maxadilan, the vasodilator peptide from sand fly salivary gland extracts. Am J Trop Med Hyg. 1996;54:665–671. doi: 10.4269/ajtmh.1996.54.665. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M., Vachereau A., Modi G. B., Tesh R. B. A novel vasodilatory peptide from the salivary glands of the sand fly Lutzomyia longipalpis. Science. 1989;243:212–214. doi: 10.1126/science.2783496. [DOI] [PubMed] [Google Scholar]
- van Zandbergen G., Klinger M., Mueller A., Dannenberg S., Gebert A., Solbach W., Laskay T. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol. 2004;173:6521–6525. doi: 10.4049/jimmunol.173.11.6521. [DOI] [PubMed] [Google Scholar]
- Ribeiro-Gomes F. L., Otero A. C., Gomes N. A., Moniz-De-Souza M. C., Cysne-Finkelstein L., Arnholdt A. C., Calich V. L., Coutinho S. G., Lopes M. F., DosReis G. A. Macrophage interactions with neutrophils regulate Leishmania major infection. J Immunol. 2004;172:4454–4462. doi: 10.4049/jimmunol.172.7.4454. [DOI] [PubMed] [Google Scholar]
- Silva F., Gomes R., Prates D., Miranda J. C., Andrade B., Barral-Netto M., Barral A. Inflammatory cell infiltration and high antibody production in BALB/c mice caused by natural exposure to Lutzomyia longipalpis bites. Am J Trop Med Hyg. 2005;72:94–98. [PubMed] [Google Scholar]
- Tavares-Murta B. M., Cunha F. Q., Ferreira S. H. The intravenous administration of tumor necrosis factor α, interleukin 8 and macrophage-derived neutrophil chemotactic factor inhibits neutrophil migration by stimulating nitric oxide production. Br J Pharmacol. 1998;124:1369–1374. doi: 10.1038/sj.bjp.0701965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. J., Ford-Hutchinson A. W., Walker J. R. Anti-inflammatory activity of bacterial endotoxin. J Pharm Pharmacol. 1977;29:702–704. doi: 10.1111/j.2042-7158.1977.tb11441.x. [DOI] [PubMed] [Google Scholar]