

Abstract
Carbohydrate binding modules (CBMs) are found in polysaccharide-targeting enzymes and increase catalytic efficiency. Because only a relatively small number of CBM structures have been solved, computational modeling represents an alternative approach in conjunction with experimental assessment of CBM functionality and ligand-binding properties. An accurate target-template sequence alignment is the crucial step during homology modeling. However, low sequence identities between target/template sequences can be a major bottleneck. We therefore incorporated the predicted hydrophilic aromatic residues (HARs) and secondary structure elements into our feature-incorporated alignment (FIA) algorithm to increase CBM alignment accuracy. An alignment performance comparison for FIA and six others was made, and the greatest average sequence identities and similarities were achieved by FIA. In addition, structure models were built for 817 representative CBMs. Our models possessed the smallest average surface-potential z scores. Besides, a large true positive value for liagnd-binding aromatic residue prediction was obtained by HAR identification. Finally, the pre-simulated CBM structures have been deposited in the Database of Simulated CBM structures (DS-CBMs). The web service is publicly available at http://dscbm.life.nthu.edu.tw/ and http://dscbm.cs.ntou.edu.tw/.
Introduction
Carbohydrate-binding modules (CBMs) are structural domains found within polysaccharide-targeting enzymes but do not contain the active sites. CBMs increase the catalytic efficiencies of their enzymes by bringing the catalytic sites into prolonged and intimate contact with substrates [1], [2]. Currently, CBMs are found among 64 protein families which are defined in CAZy, a regularly updated database (http://www.cazy.org/) [3], according to their homologies and functionalities. In addition to conventional carbohydrate-binding functions, CBMs have been reported to participate in an immune system-related allergic reaction [4]. Historically, several binding modules that were found to bind cellulose were named cellulose-binding domains (CBDs) [5], [6]. With the subsequent identification of CBMs that bind a wide range of polysaccharides, including crystalline cellulose, hemicelluloses, such as glucan, xylan, mannan, and glucomannan as well as insoluble and soluble starches [7], the generalized term CBM has evolved. CBMs represent all of the non-catalytic sugar-binding modules derived from glycoside hydrolases. Furthermore, ligand-binding site properties and structural topologies for CBMs have been summarized and reviewed [8]. CBMs are classified into three types in terms of ligand-binding function: surface-binding (type A), glycan-chain binding (type B) and small-sugar binding (type C) [9]. Type A CBMs possess a platform-like or horizontal hydrophobic surface consisting of aromatic residues. The planar conformation of the type A binding site interacts with the flat surfaces of crystalline polysaccharides. The binding site architecture of type B CBMs form a cleft or groove shape in which aromatic residues interact with free single polysaccharide chains. Type C CBMs are characterized by steric restrictions in the binding site and only binds mono-, di-, or tri-saccharides. Regardless of the three types, aromatic residues contribute to stacking interactions with the sugar rings leading to van der Waals interactions and the side chain of polar residues may provide hydrogen bonds with the sugar ligand [10]. Despite of their low sequence identity, CBMs are structurally characterized by a β-sandwich fold and seven fold types have been observed: β-sandwich, β-trefoil, cysteine knot, unique, OB fold, hevein fold and hevein-like fold [7]. Among these seven, β-sandwich and β-trefoil foldings are found in most CBM families. The best studied system to date are the starch-binding CBMs whose structural, functional and evolutionary relationships has been analyzed based on the comprehensive understanding of CBM20 [11]. Starch-binding CBMs have been identified in ten CBM families (20, 21, 25, 26, 34, 41, 45, 48, 53 and 58).
X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy (EM) have been used to determine the three-dimensional (3D) structures of CBMs [12], [13], [14]. In general, a 3D structure reveals certain of the chemical and physical characteristics of a protein, which can increase our knowledge of substrate-protein, ligand-protein, and/or protein-protein interactions and structural folding motifs. Usually, hydrophilic residues are found on protein surfaces and are the residues that interact with substrates, ligands, or other proteins. Conversely, hydrophobic residues are usually located in the core of a protein and stabilize the structure. Given the spatial coordinates of a protein, in silico investigations, e.g., molecular docking, can be performed before attempting more timely, costly, and labor intensive “wet” experiments [15]. However, as of August, 2011, more than 16 million sequences were available in the UniProtKB/TrEMBL database (http://www.ebi.ac.uk/uniprot/TrEMBLstats/), whereas fewer than 76 thousand 3D structures had been deposited in the Protein Data Bank (http://www.rcsb.org/). Additionally, the number of sequenced proteins is increasing more rapidly than is the number of solved structures. Although the technical aspects of the methods used to determine 3D protein structures have substantially improved over the years and grown more sophisticated, their execution remains expensive and time-consuming. For protein structures that have not been solved experimentally, in silico modeling, e.g., homology modeling [16], fold recognition [17], and ab initio prediction [18] can be used instead. Of these three approaches, homology modeling is the most accurate [19] and it involves three major steps: template selection, target-template sequence alignment, and model building. In practice, the target-template sequence alignment and template selection are the most critical steps for accurate homology modeling. 30% sequence identity is the minimum percentage that is necessary for accurate homology modeling [16], because accurate target-template sequence alignment is sensitive to high sequence identities. Less than 5% of CBMs has experimentally solved structures, and the sequence identities among families are usually less than 30%. Fortunately however, CBM family members have similar secondary structures and conserved potential solvent-accessible aromatic residues, which we refer to as hydrophilic aromatic residues (HARs), which are often responsible for ligand-binding function. The prototype of this idea was successfully applied to predict an in silico structure for Rhizopus oryzae glucoamylase (CBM21) in low sequence identity condition and the predicted ligand-binding residues were experimentally verified [20]. When the positions of the conserved HARs and the secondary structure elements of CBMs were integrated into a feature-incorporated alignment (FIA) algorithm [21], we found an ∼5% improvement in the average sequence similarity and identity compared with target-template alignments obtained using six leading alignment algorithms. The improved alignments were used to identify conserved ligand-binding aromatic residues in CBM domains for which 3D structures were unavailable. For the study reported herein, we were dedicated to construct in silico structures for CBMs referred as targets. Therefore, 93 non-redundant experimentally determined CBM structures and 817 representative CBM sequences for which the corresponding structures have yet to be solved were used as templates and targets, respectively. A template filter algorithm was developed to rank the likelihood that a template structure would be a good match for a given target by assessing the proposed identity level and similarity level of the template-target sequence alignments produced by the FIA algorithm in the preceding step (the proposed identity level and similarity level are defined in Materials and Methods). Then, in silico structures were built using single-template and combinations of double templates. Finally, for each target the best in silico structure was identified according to its surface-potential z score, which was the lowest among the structures
Results
Functional correlation
The goal of this work reported herein was to establish reliable in silico structures for CBMs by improving the target-template sequence alignment procedure and template filter steps. In addition, our long-range goal is to apply the results of our in silico models to biological applications that involve CBMs. Table 1 contained the profile information for four CBMs from different families and their identified HARs. Figure 1 showed that known ligand-binding aromatic residues Tyr524 and Trp540 in CBM20 Aspergillus oryzae glucoamylase [22]; Trp545, Trp561 and Typ588 in CBM20 Bacillus sp. TS-23 α-amylase [23]; Trp15 and Trp22 in CBM22 Nicotiana tabacum Nictaba [24]; and Trp543, Trp580 and Trp594 in CBM49 Solanum lycopersicum endo-β-1,4-D-glucanase [25] have been experimentally validated, even though the corresponding in vitro structures are not available. Among ten known ligand-binding aromatic residues highlighted in ball and stick, five colored in green in Figure 1 were predicted as HARs with a 50% true positive rate. The lower surface-potential z score indicated that the structures are more stabilized and reliable. These four in silico structures each possessed low surface-potential z score for the CBM and the known ligand-binding HARs were located on the surfaces of the in silico CBM structures.
Table 1. Profile summaries for selected CBMs containing known ligand-binding aromatic residues.
| Family | UniProt | Protein | Position | Organism | HARs | Template(s) | Identity | Z score |
| CBM20 | AMYG | Glucoamylase | 511–606 | Aspergillus oryzae | Y524, W540, Y553, W560, W586 | 1pam–1d3c | 39.1 | –0.545 |
| CBM20 | Q59222 | α-amylase | 515–608 | Bacillus sp.TS-23 | W545, Y558, Y606 | 1cyg–1ac0 | 50.5 | –0.838 |
| CBM22 | Q94EW1 | Nictaba | 1–165 | Nicotiana tabacum | W5, W15, Y45, Y59, F99, W121, F129, F130, W151, F160 | 1dyo | 30.1 | 0.445 |
| CBM49 | Q9ZSP9 | Endo-β-1,4-D-glucanase | 529–625 | Solanum lycopersicum | W543, Y585, F589, Y622 | 2j1v–2orz | 31.4 | –0.186 |
Figure 1. CBMs containing known ligand-binding aromatic residues and their in silico structures.

The four selected CBMs with experimentally determined ligand-binding aromatic residues are Aspergillus oryzae glucoamylase (AMYG, CBM20, [22]), Bacillus sp. TS-23 α-amylase (Q59222, CBM20, [23]), Nicotiana tabacum Nictaba (Q94EW1, CBM22, [24]), and Solanum lycopersicum endo-β-1,4-D-glucanase (Q9ZSP9, CBM49, [25]). Known ligand-binding aromatic residues are highlighted in ball and stick model. HARs and non-HARs are texted in green and blue, respectively. Five out of ten known ligand-binding residues are predicted as HARs. The 3D structures were rendered by Jmol (http://www.jmol.org/).
Prediction analysis
We could use the locations of the known ligand-binding residues in the template structures to further characterize the ligand-binding functions with respect to conserved HARs in in silico structures. Table 2 listed four selected CBMs from different CBM families for which neither ligand-binding abilities nor in vitro structures have been experimentally determined. The aromatic residues conserved to reported ligand-binding aromatic residues were texted in green in Figure 2. (An additional sixteen examples are given in Supporting Information Table S1 and Figure S1). Tyr79 in Bacteroides ovatus arabinosidase (CBM4), Tyr472, Phe481, Trp520 and Phe538 in Caldocellum saccharolyticum β-1,4-mannanase (CBM6), Trp330 in Plasmodium falciparum LCCL domain-containing protein (CBM32), and Trp309, Phe314 and Tyr352 in Phaseolus vulgaris starch synthase III (CBM53) were identified as HARs positioned on in silico structural surface, indicating that they could be potential ligand-binding residues. Nine of eleven conserved aromatic residues were identified as HARs, hence a true positive rate of 81.8% was achieved. Notably, the average sequence identity and similarity obtained using FIA were greater than those obtained using the other alignment algorithms (see Figure 3). Moreover, none of their target-template alignments had sequence identities >28%, a value which is found for the most difficult examples of homology modeling [16]. Even though the sequence identities were always <30%, the predicted HARs were conserved in the alignments and located on the surfaces of the in silico structures. Our in silico structures support the idea that these predicted HARs can be potential ligand-binding residues.
Table 2. Profile summaries for selected CBMs containing aromatic residues conserved to known ligand-binding residues.
| Family | UniProt | Protein | Position | Organism | HARs | Template(s) | Identity | Z score |
| CBM4 | Q59218 | Arabinosidase | 59–197 | Bacteroides ovatus | W76, Y79, F116, F117 | 2zex | 26.9 | –0.463 |
| CBM6 | P77847 | β-1,4-mannanase | 453–575 | Caldocellum saccharolyticum | W461, Y472, Y477, F481, Y499, Y514, W520, F538, Y564 | 1w9s–1uxx | 27.9 | –0.454 |
| CBM32 | Q8IK83 | LCCL domain-containing protein | 305–420 | Plasmodium falciparum | Y307, Y329, W330, F335, Y339, F380 | 2j1v–2j7m | 26.7 | –0.510 |
| CBM53 | A4F2M4 | Starch synthase III | 280–365 | Phaseolus vulgaris | F306, W309, F314, W327, F346, Y352 | 2v8l–2c3w | 24.3 | –0.367 |
The italics denote aromatic residues conserved to reported ligand-binding residues in corresponding template(s). No experimental data concerning their ligand-binding abilities is available for the unannotated HARs. The used template structures are 2zex [44], 1w9s [45], 1uxx [46], 2j1v [24], 2j7m [47] and 2v8l [48], 2c3w [49].
Figure 2. CBMs containing aromatic residues conserved to known ligand-binding residues and their in silico structures.

The four selected CBMs with aromatic residues conserved to known ligand-binding residues are Bacteroides ovatus arabinosidase (Q59218, CBM4), Caldocellum saccharolyticum β-1,4-annanase (P77847, CBM6), Plasmodium falciparum LCCL domain-containing protein (Q8IK83, CBM32), and Phaseolus vulgaris starch synthase III (A4F2M4, CBM53). Known ligand-binding aromatic residues are highlighted in ball and stick model. HARs and non-HARs are texted in green and blue, respectively. Nine out of eleven aromatic residues conserved to known ligand-binding residues are predicted as HARs. The 3D structures were rendered by Jmol (http://www.jmol.org/).
Figure 3. Average sequence identities and similarities for CBMs from seven sequence alignment methods.

The sequence identities (similarities) are averaged from 75,981 (817 * 93) target-template pairwise sequence alignments. The used alignment programs are FIA [21], MUSCLE [32], ClustalW2 [33], ProbCons [36], T-COFFEE [35], DIALIGN-TX [34], and MAFFT [37].
Performance comparison
The alignment performances for FIA and the six leading alignment programs were compared. Figure 3 showed the average number of sequence similarities and identities for the FIA, MUSCLE, ClustalW2, ProbCons, T-COFFEE, DIALIGN-TX, and MAFFT alignments. Each of the 817 target sequences was individually aligned with each of the 93 template sequences for a total of 75,981 (817 * 93) target-template sequence alignments. For the FIA alignments, the greatest average sequence similarity (45.9%) and average sequence identity (28.1%) were found, whereas the smallest average sequence similarity (28.5%) was found for the DIALIGN-TX alignments, and the smallest average sequence identity (16.4%) was found for the ClustalW2 alignments. Figure 4 plotted the average z scores for the in silico structures derived from alignments of various alignment tools. The modeling procedures used to produce the in silico structures were identical except for the target-template sequence alignments, which used different alignment tools. The z scores reported in Figure 4 were the average of the five smallest z scores for each target CBM. For FIA, the smallest average z score is –0.280, whereas that for DIALIGN-TX is –0.206. The relative numbers (as percentages) of structures build using single templates and double templates were given in Figure 5. For the in silico structures, between 60.8% and 63.0% of the top five candidates were derived from double-template modeling indicating that double-template modeling produced smaller z scores than the single-template approach. In summary, the performance of FIA was superior for both sequence alignment and structure building. The inclusions of conserved secondary structure elements and HAR positions in FIA improved model building and identification of ligand-binding residues.
Figure 4. Average z scores for in silico structures built based on seven sequence alignment methods.

The z scores are averaged from the lowest z scores of top five in silico structures for each target CBM. The used alignment programs are FIA [21], MUSCLE [32], ClustalW2 [33], ProbCons [36], T-COFFEE [35], DIALIGN-TX [34], and MAFFT [37]. The surface potential z scores are evaluated by PROSA2003 [38].
Figure 5. Percentages of structures built using single- and double-templates based on seven sequence alignment methods.

The percentages indicate the top five structures with lowest z scores for each target CBM are derived from either single-template or double-template homology modeling. The used alignment programs are FIA [21], MUSCLE [32], ClustalW2 [33], ProbCons [36], T-COFFEE [35], DIALIGN-TX [34], and MAFFT [37].
Discussion
Template choice
We expected that multiple-template-based structure modeling would increase the quality of the models. The average z-scores in Figure 4 showed that simulated structures based on double-template ranked in the top five were at least 60%, indicating that double-template modeling produces more accurate structural predictions. An assessment of the raw data, which are not shown, indicates that double-template-based modeling is slightly better than is the single-template-based approach. Specifically, the best combinations of double templates usually consisted of one major template sequence that matched the target sequence as main skeleton and a second compatible or complementary sequence. “Compatible” indicates that the two templates are homologues with few insertions and deletions in their aligned sequences, whereas “complementary” denotes that one template includes an aligned sequence that is mismatched in the other template-target sequence alignment. Therefore, a key factor for successful homology modeling is the ability to choose compatible or complementary combinations of templates rather than considering as many templates as possible, i.e., the use of two or more dissimilar templates may decrease the modeling quality. Another disadvantage associated with multiple-template-based homology modeling is the greater computational time as more combinations of template structures must be matched to the target and evaluated.
Correlation of hydrophilic aromatic residue
Given that aromatic residues are known to participate in ligand-binding function in CBMs, the identification of HARs was introduced into our homology-modeling scheme as they could serve ligand-binding aromatic residues. The occurrence times for the two upstream and downstream polar residues flanking 97 known ligand-binding aromatic residues were determined (see Table 3). In addition to the polar residues, the occurrence times for glycine and alanine were also determined. The large occurrence times for glycine and alanine can be rationalized on the basis of their relatively small sizes, which would minimize steric conflicts with their neighboring aromatic residues. Moreover, the HAR identification procedure can be used as a simple but effective sequence-based ligand-binding residue predictor. 97 known ligand-binding aromatic residues and 558 aromatic residues without experimental ligand-binding abilities in 49 CBM structure templates were used as positive and negative sets, respectively (see Supporting Information Table S2). In comparison with a sequence-based ligand-binding residue predictor, FRcons-lig [26], which combined information of amino acid conservation, secondary structure and relative solvent accessibility, the number of true positives for any given number of false positives was larger for HAR-based predictor than FRcons-lig as shown in Figure 6. With the cut-off threshold for the sum of weighted scores set to 97, the true positive and false positive percentages were 74.2% and 46.2%, respectively. Because some of the “real” ligand-binding aromatic residues have yet to be determined, the false and true positive percentage is an overestimation and underestimation, respectively. When more experimentally determined ligand-binding aromatic residues become available, the false and true positive percentage are expected to be decreased and increased, respectively. Furthermore, our intention was not just to build reliable in silico structures but also to be able to correlate structural aspects of CBMs with the corresponding experimental functional assays. Here, we mainly focused on the prediction of the HARs of CBMs that possibly bind substrate polysaccharides. In fact, a typical CBM ligand-binding site contains multiple aromatic and polar residues. To fully characterize the ligand-binding sites of CBMs, polar residues in the vicinity of the predicted HARs in the in silico structures may also be involved in ligand-binding function.
Table 3. Occurrence times for residues that flank known ligand-binding aromatic residues.
| G | N | S | T | D | A | Q | I | E | K | V | L | Y | P | C | M | F | H | R | W |
| 49 | 43 | 34 | 34 | 33 | 30 | 27 | 22 | 22 | 20 | 16 | 13 | 9 | 8 | 6 | 6 | 6 | 4 | 3 | 3 |
The occurrence times were derived from the two upstream and downstream residues flanking 97 known aromatic ligand-binding residues and were transformed into weighted scores for HAR prediction.
Figure 6. Receiver operating characteristic (ROC) curves of ligand-binding residue prediction.

97 known ligand-binding aromatic residues and 558 aromatic residues without experimental ligand-binding abilities in 49 template structures (see Supporting Information Table S2) are as positive and negative sets for prediction of ligand-binding aromatic residues, respectively. The ROC curves are generated by HAR-based and FRcons-lig [26] predictions.
How to use DS-CBMs
All in silico structures simulated in this study have been deposited into DS-CBMs at http://dscbm.life.nthu.edu.tw and http://dscbm.cs.ntou.edu.tw. The 817 target CBMs can be searched for by their CBM family, protein, or organism names, and by keywords. In answer to a query, matched CBMs with brief profiles as shown in Figure 7(A) are returned. Additionally, the CBM entry, organism, and structure template(s) are accessible via cross-database hyperlinks. Finally, the in silico structure with the best surface-potential z score is identified. Directly clicking on the structural preview image allows users to switch into the interactive 3D visualization interface where the structure can be manipulated as shown in Figure 7(B). Rotation and shift functions are provided in the control panel and all operating functions can be accessed by clicking the right mouse button. At the bottom, HARs and known ligand-binding residues are annotated in the sequence view. Finally, clicking on a specific amino acid within the target sequence highlights its position in the corresponding in silico structure. Researchers may also submit a CBM sequence to the on-line structure modeling system. When the modeling procedure is completed, a page similar to that shown in Figure 7(B) will be generated.
Figure 7. Snapshots of DS-CBMs system.

(A) The DS-CBMs search results for the CBM20 family. The CBM entry, organism, and structure template(s) are accessible via cross-database hyperlinks. (B) Simulated 3D structure visualization for Aspergillus oryzae glucoamylase (CBM20). The CBM profiles, 3D structure manipulation, HAR identification and known ligand-binding residue annotation are provided.
Conclusion
We developed an automated and generalized homology modeling procedure for CBMs. A total of 817 in silico structures with the minimum z scores were generated and deposited in the DS-CBMs. The major challenge of CBM homology model building is that the target and template sequences contain only a small number of homologues, i.e., low sequence identity, so that conventional homology modeling procedures may fail to build reliable in silico structures. Our main contribution has been to improve target-template sequence alignment by incorporating the conserved positions of secondary structure elements and HARs into the FIA algorithm, which is used to provide accurate target-template alignment for homology model building. Additionally, using a single template to build the model may not be the best strategy. The template filter step was incorporated to discover multiple homologous templates for model building and double-template-based structure model building conducted lower surface-potential z scores for in silico structures. Finally, low surface-potential z scores and assessment of the in silico structures suggest that the structures likely have been “correctly” built and are functionally relevant. HAR identification demonstrated its higher true positive rate and lower false positive rate for liagnd-binding aromatic residue prediction for CBMs. In conclusion, more than 95% of the CBMs do not have solved structures, and the ligand specificity of a particular CBM is mainly determined by the positions and orientations of the aromatic ligand-binding residues. The in silico CBM structures can be integrated with current databases like CAZy and Pfam to discover potential ligand-binding residues. The integrated in silico and in vitro resources would facilitate the comprehension of functional similarities and diversities among all CBMs.
Materials and Methods
System definition and overview
The objective of this study was to build accurate in silico structures for CBMs without manual intervention. Figure 8 illustrated the flowchart for the modeling procedure and the mathematical definitions are defined as follows. Given a target CBM sequence of t and a set of m non-redundant CBM templates denoted P, the goal was to generate the most reliable in silico structure s* for t by identifying the minimum surface-potential z score after the sequence of t was considered each of the templates in P. Secondary structure prediction and identification of hydrophilic aromatic residues (HARs) were performed in the first two steps. For the third step, a set of pairwise alignments denoted A between a sequence of t and each of the template sequences in P was made using feature-incorporated alignment (FIA). For the fourth step, the set of candidate target-template alignments denoted CA was ranked from the top k-matched template among A according to the proposed identity level and similarity level. Subsequently, a CSS set was build that contained the candidate in silico structures of t derived from single- and double-template alignments in the CA. s* was identified as the in silico structure of t in the CSS that had the minimum surface-potential z score. The implementation of each step is detailed in the following sections.
Figure 8. Flowchart for FIA-based homology modeling.

The proposed structure modeling procedure for CBMs comprises of five modules: secondary structure prediction, hydrophilic aromatic residue (HAR) identification, target-template alignment, template filter, and structure building and evaluation. Details are described in Materials and Methods.
Secondary structure prediction
CBM domains have been classified into seven fold families, i.e., β-sandwiches, β-trefoils, cysteine knots, unique, OB folds, hevein folds, and hevein-like folds [8]. The β-sandwich and β-trefoil folds have been found in CBMs, and these β-strand folds are conserved among CBM families. We first predicted the secondary structure elements of the CBMs using the Discrimination of protein Secondary structure Class (DSC) algorithm [27]. When a residue was predicted to be α-helical or β-stranded with a probability of <50%, it was annotated as a loop residue (Only high confident predictions for helix and strand were labeled for latter sequence alignment.). On average, a three-state (helix, strand and loop) accuracy of 70.1% was obtained by DSC.
Hydrophilic aromatic residue identification
CBM ligand-binding sites are of three types: surface binding (type A), glycan-chain binding (type B), and small-sugar binding (type C) [7]. Type A CBMs possess a platform-like hydrophobic surface consisted of aromatic residues. In contrast, the binding site architecture of type B CBMs shapes a cleft or groove arrangement in which aromatic residues interact with free single polysaccharide chains. Due to stereo restriction in the binding site, Type C CBMs lacks of the cleft form as in type B CBMs only bind mono-, di-, or trisaccharides. In general, CBM ligand-binding sites contain aromatic and polar residues. In terms of their ligand-binding interactions, aromatic residues are involved in aromatic stacking, whereas polar residues form hydrogen bonds with ligands [8]. Additionally, polar residues adjacent to aromatic residues can enlarge the surface area of neighboring regions thereby increasing the contact area(s) between the binding residues and ligands. In our previous study, the number of preferred occurrence time that polar residues flanked an aromatic residue was determined for starch-binding CBMs [21], and the aromatic residues that were flanked on both sides by polar residues were defined as HARs. For this study, the occurrence time for two upstream and downstream amino acids that flank 97 known ligand-binding aromatic residues was summarized in Table 3 (the structure template profiles including known ligand-binding aromatic residues are shown in Supporting Information Table S2). Interestingly, the preferred occurrence times for Asn (N), Ser (S), Asp (D), Thr (T), Gln (Q), Glu (E), and Lys (K) are observed and consistent with their ligand-binding functionality for polar residues in CBMs. To identify potential HARs, the weighted scores (directly derived from the occurrence times) of the sets of two upstream and downstream flanking residues for each aromatic residue were summed. When the sum was greater than or equal to the cut-off threshold of 97 (see Discussion), these aromatic residues were defined as HARs. For example, the motif GSWNP had a score of 134 (49 + 34 + 43 + 8), and therefore, the central tryptophan was identified as an HAR. Conversely, the motif PTYKA had a score of 92 (8 + 34 + 20 + 30), and the central tyrosine was therefore not identified as an HAR. Step 1 and step 2 identified the core-conserved sequence signatures associated with secondary structure prediction and HARs that were then used for sequence alignment.
Target-template alignment by FIA
To evaluate the target-template sequence matching, a target sequence t was aligned one by one with the templates in P as described by Equation 1, where FIA represents the feature-incorporated alignment [21], and A is the set of preliminary alignments used in the template filter step.
| (1) |
The FIA algorithm adopted the affine gap-penalty model and the Blosum62 matrix [28], [29]. The individual residues of a full-length CBM were not uniformly weighted by the FIA, which emphasized alignment of the secondary structure elements and conserved HAR positions.
Template filter
Usually, conventional template selection that is used during homology modeling attempts to identify the best template for subsequent in silico modeling. However, the criteria used for sequence matching may conflict with that used for structure building and assessment, e.g., the surface-potential z score for the modeled target structure. Therefore, we used a template filter that had been designed to select templates according to the proposed identity level and similarity level for t' and p' as defined in Equation 2 and Equation 3, respectively. Here, ident(t', p'), sim(t', p'), and gap(t', p') represent the number of aligned identical residues, similar residues, and opening gaps, respectively, in t' and p', where t' and p' denote the sequences of t and p aligned using FIA. The proposed identity level (similarity level) charged extra opening penalties. The larger the identity level (similarity level) is, the larger the sequence homology is and the fewer gaps present. Subsequently, Equation 4 was used to filter homologue target-template sequence alignments for CA, where top_il(A) and top_sl(A) represent the top k target-template alignments in A for the identity level and similarity level, respectively. In practice, between k and 2k target-template alignments were contained in CA for use in model building.
| (2) |
| (3) |
| (4) |
Structure model building and evaluation
With the use of the template filter, CA was obtained to build the CSS for the sequence of t. Intuitively, multiple-template-based homology modeling would be expected to improve the accuracy of a modeled structure, but that is not always the case. Larsson reported that double-template and triple-template modeling was more accurate than was considering four or more templates [30]. Multiple-template-based manner here refers to model construction based on the integration of multiple templates instead of averaging individual models from individual templates. To ensure the quality of the model and the efficiency of the process, we incorporated only single and combinations of double templates into the modeling process. We did not include triple templates because their computational costs were extremely high. The CSS were generated using single templates, combinations of double templates and Equation 5 where s_modeling(a) and d_modeling(b, c) are for modeling using a single template, a, and double templates, b and c, respectively. For quality assessment, the quality of each in silico structure was evaluated according to its surface-potential z score. The most reliable structure s* in CSS was chosen as the one that had the lowest average surface-potential z score as determined by Equation 6, where z_score(s) is the z score for candidate simulated structure s.
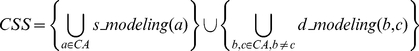 |
(5) |
| (6) |
Materials
CBM with domain sizes exceeding 85 residues in length were identified in the Pfam database and grouped according to their CBM classification [31]. A total of 93 non-redundant templates were then selected from 18 CBM families. Of these, 3, 2, 4, 11, 22, 2, 9, 3, 1, 25, 1, 5, 3, and 2 were from the CBM families CBM2 (PF00553), CBM3 (PF00942), CBM4&9&16&22 (PF02018), CBM6 (PF03422), CBM13 (PF00652), CBM17&28 (PF03424), CBM20 (PF00686), CBM21 (PF03370), CBM25 (PF03423), CBM32 (PF00754), CBM33 (PF03067), CBM34 (PF02903), CBM40 (PF02973), and CBM51 (PF08305), respectively. The criteria for their selection were as follows. If multiple crystal structures were available for a CBM, the one with the best resolution was used. If more than one structure of the same CBM had the same resolution, which was also the best, the one with the smaller R-factor was used. For the ensemble of NMR solution structures for a CBM, which were used when a corresponding crystal structure was unavailable, the structure used was randomly selected from the ensemble. A total of 817 representative CBM sequences were used as targets to predict their in silico structures. The targets all had pairwise sequence identities below 80% and were from 19 CBM families among which 15, 25, 45, 36, 92, 1, 4, 97, 78, 33, 174, 131, 39, 4, and 43 were selected, respectively, from CBM2, CBM3, CBM4&9&16&22, CBM6, CBM13, CBM15 (PF03426), CBM17&28, CBM20, CBM21, CBM25, CBM32, CBM33, CBM34, CBM40, and CBM51.
The alignment performance for the 817 target sequences against the 93 template sequences was compared among FIA and six alignment tools, MUSCLE v3.8.31 [32], ClustalW2 v2.1 [33], DIALIGN-TX v1.0.2 [34], T-COFFEE v5.31 [35], ProbCons v1.12 [36], and MAFFT v6.850 [37] with default parameter settings used. The structure model building engine was Modeller v9.8. The 3D visualization and the preview images were rendered by Jmol (http://www.jmol.org/) and PyMol (http://www.pymol.org/), respectively. Surface-potential z scores were calculated by PROSA2003 [38].
Supporting Information
CBMs containing aromatic residues conserved to known ligand-binding residues and their in silico structures. Sixteen CBMs without in vitro structures are predicted. O30421, Caldocellum saccharolyticum xylanase; O30426, Caldocellum saccharolyticum xylanase; O88043, Streptomyces coelicolor putative secreted arabinosidase; Q60043, Thermoanaero bacterium endoxylanase; O69822, Streptomyces coelicolor putative secreted protein; Q9KBL8, Bacillus halodurans glucan 1,4-β-glucosidase; Q1ENB1, Guillardia theta putative starch binding domain protein; Q6R608, Solanum tuberosum 4-α-glucanotransferase; PPR3C, Danio rerio protein phosphatase 1 regulatory subunit 3C; Q89ZX7, Bacteroides thetaiotaomicron putative uncharacterized protein; Q8LEV3, Arabidopsis thaliana putative uncharacterized protein; Q9U5D0, Drosophila melanogaster hemolectin; Q82M60, Streptomyces avermitilis putative secreted protein; A8GAL3, Serratia proteamaculans α-amylase catalytic region; Q8A1R7, Bacteroides thetaiotaomicron α-galactosidase; and Q9XGC0, Vigna unguiculata starch synthase isoform SS III. Known ligand-binding aromatic residues are highlighted in ball and stick model. HARs and non-HARs are texted in green and blue, respectively. The 3D structures were rendered by Jmol (http://www.jmol.org/).
(TIF)
Profile summaries for CBMs containing aromatic residues conserved to reported ligand-binding residues. Sixteen CBMs with target-template sequence identity less than 30% are selected for prediction analysis. The HARs highlighted italics denote aromatic residues conserved to known ligand-binding residues in corresponding template(s). No experimental data concerning their ligand-binding abilities is available for the unannotated HARs.
(DOC)
Profiles for non-redundant CBM structure templates. 93 non-redundant structures are selected as templates. The HARs with bold fonts indicate experimentally determined ligand-binding residues. No experimental data concerning their ligand-binding abilities is available for the unannotated HARs.
(DOC)
Acknowledgments
The authors thank Jeffrey Spencer for proofreading, Tsan-Huang Shih for computational supports and Chih-Ying Ho for website art design. We also credit Ee-Ling Low, Sim-Kun Ng, Yuan-Pei Ci, and Chien-Jung Chen for CBM data collection and discussions.
Footnotes
Competing Interests: Dr. Wei-I Chou contributed to this study as a PhD student of the corresponding author in Institute of Molecular and Cellular Biology and Department of Medical Science, National Tsing Hua University. Dr. Chou served in Simpson Biotech Co., Ltd from 2007 to 2010, and is currently employed by Reber Genetics Co., Ltd. The DS-CBMs database is open to the public for internet access. The authors declare that this study is free from any commercial or financial interest. The employment of Dr. Wei-I Chou does not alter the authors' adherence to all the PLoS ONE policies on sharing data and materials..
Funding: This work was funded by National Science Council, R.O.C. grant numbers NSC100-2628-B-007-003-MY3 to Margaret Dah-Tsyr Chang; NSC100-2627-B-019-006 and NSC98-2221-E-019-031-MY2 to Tun-Wen Pai; NSC99-2627-B-126-001 to Chuan-Yi Tang; and NSC98-2917-I-007-105 to Wei-Yao Chou. This work was also supported by Toward world-class university project to NTHU, Simpson Biotech Corporation grant number 100A0157V7 to Margaret Dah-Tsyr Chang, and Ju-Chen Shen Tsing Hua Fellowship to Ting-Ying Jiang. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bolam DN, Ciruela A, McQueen-Mason S, Simpson P, Williamson MP, et al. Pseudomonas cellulose-binding domains mediate their effects by increasing enzyme substrate proximity. Biochem J. 1998;331(Pt 3):775–781. doi: 10.1042/bj3310775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomme P, Warren RA, Gilkes NR. Cellulose hydrolysis by bacteria and fungi. Adv Microb Physiol. 1995;37:1–81. doi: 10.1016/s0065-2911(08)60143-5. [DOI] [PubMed] [Google Scholar]
- 3.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shani N, Shani Z, Shoseyov O, Mruwat R, Shoseyov D. Oxidized cellulose binding to allergens with a carbohydrate-binding module attenuates allergic reactions. J Immunol. 2011;186:1240–1247. doi: 10.4049/jimmunol.1000640. [DOI] [PubMed] [Google Scholar]
- 5.Tomme P, Van Tilbeurgh H, Pettersson G, Van Damme J, Vandekerckhove J, et al. Studies of the cellulolytic system of Trichoderma reesei QM 9414. Analysis of domain function in two cellobiohydrolases by limited proteolysis. Eur J Biochem. 1988;170:575–581. doi: 10.1111/j.1432-1033.1988.tb13736.x. [DOI] [PubMed] [Google Scholar]
- 6.Gilkes NR, Warren RA, Miller RC, Jr, Kilburn DG. Precise excision of the cellulose binding domains from two Cellulomonas fimi cellulases by a homologous protease and the effect on catalysis. J Biol Chem. 1988;263:10401–10407. [PubMed] [Google Scholar]
- 7.Hashimoto H. Recent structural studies of carbohydrate-binding modules. Cell Mol Life Sci. 2006;63:2954–2967. doi: 10.1007/s00018-006-6195-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillen D, Sanchez S, Rodriguez-Sanoja R. Carbohydrate-binding domains: multiplicity of biological roles. Appl Microbiol Biotechnol. 2010;85:1241–1249. doi: 10.1007/s00253-009-2331-y. [DOI] [PubMed] [Google Scholar]
- 9.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pell G, Williamson MP, Walters C, Du H, Gilbert HJ, et al. Importance of hydrophobic and polar residues in ligand binding in the family 15 carbohydrate-binding module from Cellvibrio japonicus Xyn10C. Biochemistry. 2003;42:9316–9323. doi: 10.1021/bi0347510. [DOI] [PubMed] [Google Scholar]
- 11.Christiansen C, Abou Hachem M, Janecek S, Vikso-Nielsen A, Blennow A, et al. The carbohydrate-binding module family 20—diversity, structure, and function. FEBS J. 2009;276:5006–5029. doi: 10.1111/j.1742-4658.2009.07221.x. [DOI] [PubMed] [Google Scholar]
- 12.Mitsuoka K. Obtaining high-resolution images of biological macromolecules by using a cryo-electron microscope with a liquid-helium cooled stage. Micron. 2011;42:100–106. doi: 10.1016/j.micron.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Shin J, Lee W. Structural proteomics by NMR spectroscopy. Expert Rev Proteomics. 2008;5:589–601. doi: 10.1586/14789450.5.4.589. [DOI] [PubMed] [Google Scholar]
- 14.Parker MW. Protein structure from X-ray diffraction. Journal of Biological Physics. 2003;29:341–362. doi: 10.1023/A:1027310719146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliveira OV, Freitas LC, Straatsma TP, Lins RD. Interaction between the CBM of Cel9A from Thermobifida fusca and cellulose fibers. J Mol Recognit. 2009;22:38–45. doi: 10.1002/jmr.925. [DOI] [PubMed] [Google Scholar]
- 16.Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16:172–177. doi: 10.1016/j.sbi.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Jones D, Thornton J. Protein fold recognition. J Comput Aided Mol Des. 1993;7:439–456. doi: 10.1007/BF02337560. [DOI] [PubMed] [Google Scholar]
- 18.Bonneau R, Baker D. Ab initio protein structure prediction: progress and prospects. Annu Rev Biophys Biomol Struct. 2001;30:173–189. doi: 10.1146/annurev.biophys.30.1.173. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Tang GW, Capriotti E. Comb Chem High Throughput Screen; 2011. Comparative Modeling: The State of the Art and Protein Drug Target Structure Prediction. [DOI] [PubMed] [Google Scholar]
- 20.Chou WI, Pai TW, Liu SH, Hsiung BK, Chang MD. The family 21 carbohydrate-binding module of glucoamylase from Rhizopus oryzae consists of two sites playing distinct roles in ligand binding. Biochem J. 2006;396:469–477. doi: 10.1042/BJ20051982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou WY, Chou WI, Pai TW, Lin SC, Jiang TY, et al. Feature-incorporated alignment based ligand-binding residue prediction for carbohydrate-binding modules. Bioinformatics. 2010;26:1022–1028. doi: 10.1093/bioinformatics/btq084. [DOI] [PubMed] [Google Scholar]
- 22.Paldi T, Levy I, Shoseyov O. Glucoamylase starch-binding domain of Aspergillus niger B1: molecular cloning and functional characterization. Biochem J. 2003;372:905–910. doi: 10.1042/BJ20021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo HF, Chiang WY, Chi MC, Hu HY, Lin LL. Site-directed mutagenesis of the conserved threonine, tryptophan, and lysine residues in the starch-binding domain of Bacillus sp. strain TS-23 alpha-amylase. Curr Microbiol. 2004;48:280–284. doi: 10.1007/s00284-003-4198-y. [DOI] [PubMed] [Google Scholar]
- 24.Boraston AB, Wang D, Burke RD. Blood group antigen recognition by a Streptococcus pneumoniae virulence factor. J Biol Chem. 2006;281:35263–35271. doi: 10.1074/jbc.M607620200. [DOI] [PubMed] [Google Scholar]
- 25.Watson JN, Newstead S, Dookhun V, Taylor G, Bennet AJ. Contribution of the active site aspartic acid to catalysis in the bacterial neuraminidase from Micromonospora viridifaciens. FEBS Lett. 2004;577:265–269. doi: 10.1016/j.febslet.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 26.Fischer JD, Mayer CE, Soding J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 2008;24:613–620. doi: 10.1093/bioinformatics/btm626. [DOI] [PubMed] [Google Scholar]
- 27.King RD, Sternberg MJ. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 1996;5:2298–2310. doi: 10.1002/pro.5560051116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gotoh O. An improved algorithm for matching biological sequences. J Mol Biol. 1982;162:705–708. doi: 10.1016/0022-2836(82)90398-9. [DOI] [PubMed] [Google Scholar]
- 30.Larsson P, Wallner B, Lindahl E, Elofsson A. Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 2008;17:990–1002. doi: 10.1110/ps.073344908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn RD, Mistry J, Tate J, Coggill P, Heger A, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 34.Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol Biol. 2008;3:6. doi: 10.1186/1748-7188-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 36.Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Briefings in Bioinformatics. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 38.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harata K, Haga K, Nakamura A, Aoyagi M, Yamane K. X-ray structure of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011. Comparison of two independent molecules at 1.8 A resolution. Acta Crystallogr D Biol Crystallogr. 1996;52:1136–1145. doi: 10.1107/S0907444996008438. [DOI] [PubMed] [Google Scholar]
- 40.Uitdehaag JC, Kalk KH, van Der Veen BA, Dijkhuizen L, Dijkstra BW. The cyclization mechanism of cyclodextrin glycosyltransferase (CGTase) as revealed by a gamma-cyclodextrin-CGTase complex at 1.8-A resolution. J Biol Chem. 1999;274:34868–34876. doi: 10.1074/jbc.274.49.34868. [DOI] [PubMed] [Google Scholar]
- 41.Sorimachi K, Le Gal-Coeffet MF, Williamson G, Archer DB, Williamson MP. Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to beta-cyclodextrin. Structure. 1997;5:647–661. doi: 10.1016/s0969-2126(97)00220-7. [DOI] [PubMed] [Google Scholar]
- 42.Charnock SJ, Bolam DN, Turkenburg JP, Gilbert HJ, Ferreira LM, et al. The X6 “thermostabilizing” domains of xylanases are carbohydrate-binding modules: structure and biochemistry of the Clostridium thermocellum X6b domain. Biochemistry. 2000;39:5013–5021. doi: 10.1021/bi992821q. [DOI] [PubMed] [Google Scholar]
- 43.Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, et al. Structural basis for ligand and heparin binding to neuropilin B domains. Proc Natl Acad Sci U S A. 2007;104:6152–6157. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bae B, Ohene-Adjei S, Kocherginskaya S, Mackie RI, Spies MA, et al. Molecular basis for the selectivity and specificity of ligand recognition by the family 16 carbohydrate-binding modules from Thermoanaerobacterium polysaccharolyticum ManA. J Biol Chem. 2008;283:12415–12425. doi: 10.1074/jbc.M706513200. [DOI] [PubMed] [Google Scholar]
- 45.van Bueren AL, Morland C, Gilbert HJ, Boraston AB. Family 6 carbohydrate binding modules recognize the non-reducing end of beta-1,3-linked glucans by presenting a unique ligand binding surface. J Biol Chem. 2005;280:530–537. doi: 10.1074/jbc.M410113200. [DOI] [PubMed] [Google Scholar]
- 46.Pires VM, Henshaw JL, Prates JA, Bolam DN, Ferreira LM, et al. The crystal structure of the family 6 carbohydrate binding module from Cellvibrio mixtus endoglucanase 5a in complex with oligosaccharides reveals two distinct binding sites with different ligand specificities. J Biol Chem. 2004;279:21560–21568. doi: 10.1074/jbc.M401599200. [DOI] [PubMed] [Google Scholar]
- 47.Ficko-Blean E, Boraston AB. The interaction of a carbohydrate-binding module from a Clostridium perfringens N-acetyl-beta-hexosaminidase with its carbohydrate receptor. J Biol Chem. 2006;281:37748–37757. doi: 10.1074/jbc.M606126200. [DOI] [PubMed] [Google Scholar]
- 48.Tung JY, Chang MD, Chou WI, Liu YY, Yeh YH, et al. Crystal structures of the starch-binding domain from Rhizopus oryzae glucoamylase reveal a polysaccharide-binding path. Biochem J. 2008;416:27–36. doi: 10.1042/BJ20080580. [DOI] [PubMed] [Google Scholar]
- 49.Boraston AB, Healey M, Klassen J, Ficko-Blean E, Lammerts van Bueren A, et al. A structural and functional analysis of alpha-glucan recognition by family 25 and 26 carbohydrate-binding modules reveals a conserved mode of starch recognition. J Biol Chem. 2006;281:587–598. doi: 10.1074/jbc.M509958200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CBMs containing aromatic residues conserved to known ligand-binding residues and their in silico structures. Sixteen CBMs without in vitro structures are predicted. O30421, Caldocellum saccharolyticum xylanase; O30426, Caldocellum saccharolyticum xylanase; O88043, Streptomyces coelicolor putative secreted arabinosidase; Q60043, Thermoanaero bacterium endoxylanase; O69822, Streptomyces coelicolor putative secreted protein; Q9KBL8, Bacillus halodurans glucan 1,4-β-glucosidase; Q1ENB1, Guillardia theta putative starch binding domain protein; Q6R608, Solanum tuberosum 4-α-glucanotransferase; PPR3C, Danio rerio protein phosphatase 1 regulatory subunit 3C; Q89ZX7, Bacteroides thetaiotaomicron putative uncharacterized protein; Q8LEV3, Arabidopsis thaliana putative uncharacterized protein; Q9U5D0, Drosophila melanogaster hemolectin; Q82M60, Streptomyces avermitilis putative secreted protein; A8GAL3, Serratia proteamaculans α-amylase catalytic region; Q8A1R7, Bacteroides thetaiotaomicron α-galactosidase; and Q9XGC0, Vigna unguiculata starch synthase isoform SS III. Known ligand-binding aromatic residues are highlighted in ball and stick model. HARs and non-HARs are texted in green and blue, respectively. The 3D structures were rendered by Jmol (http://www.jmol.org/).
(TIF)
Profile summaries for CBMs containing aromatic residues conserved to reported ligand-binding residues. Sixteen CBMs with target-template sequence identity less than 30% are selected for prediction analysis. The HARs highlighted italics denote aromatic residues conserved to known ligand-binding residues in corresponding template(s). No experimental data concerning their ligand-binding abilities is available for the unannotated HARs.
(DOC)
Profiles for non-redundant CBM structure templates. 93 non-redundant structures are selected as templates. The HARs with bold fonts indicate experimentally determined ligand-binding residues. No experimental data concerning their ligand-binding abilities is available for the unannotated HARs.
(DOC)