

Abstract
p53 is well known as a “guardian of the genome” for differentiated cells, in which it induces cell cycle arrest and cell death after DNA damage and thus contributes to the maintenance of genomic stability. In addition to this tumor suppressor function for differentiated cells, p53 also plays an important role in stem cells. In this cell type, p53 not only ensures genomic integrity after genotoxic insults but also controls their proliferation and differentiation. Additionally, p53 provides an effective barrier for the generation of pluripotent stem cell-like cells from terminally differentiated cells. In this review, we summarize our current knowledge about p53 activities in embryonic, adult and induced pluripotent stem cells.
Keywords: p53, Embryonic stem cells, Adult stem cells, Induced pluripotent stem cells, Cell differentiation
INTRODUCTION
p53 is one of the most well-known and most intensively investigated tumor suppressor proteins. p53 is not a critical protein for survival, as mice and men can develop in the absence of p53 or when the tumor suppressor protein is mutated. The benefit merely comes into play when cells are exposed to conditions that bear an elevated risk of acquiring mutations, such as irradiation or nucleotide deprivation. Then, p53 halts the cell cycle to allow time for repair of damaged DNA, or it initiates a cell death program to eliminate cells with damaged or mutated DNA from the cell population. Functionally, p53 is a transcription factor. After activation, it binds to the promoters of target genes and stimulates transcription of certain genes, while repressing others[1-3]. In addition, p53 can induce apoptosis in a non-transcriptional manner via direct interaction with pro- and antiapoptotic proteins[4].
Due to its antiproliferative activity, p53 is under tight control. The rapid degradation of p53 in 26S proteasomes ensures that its abundance in non-stressed cells is low. However, when its activity is required, p53 is protected from degradation and accumulates to high levels, while an array of post-translational modifications fine-tunes its activity[5,6]. The major regulator of p53 abundance is the oncoprotein Mdm2. Mdm2 mediates the polyubiquitination of p53 and its association with 26S proteasomes, resulting in p53 degradation[5,7]. Nonetheless, p53 can also be degraded by other ubiquitin ligases and by ubiquitin-independent pathways[5].
The p53 tumor suppressor protein is part of a multigene family that also includes p63 (TAp63) and p73 (Tap73), as well as several splice variants of these proteins[8]. Although p53, p63 and p73 share a significant degree of homology and regulate a common set of target genes, they target different cellular activities. The p53 protein has a fundamental role in growth control and maintenance of genomic integrity, whereas mice that are deficient in p73 have abnormalities of the nervous system and suffer from chronic infections, and p63-null mice lack limbs and a wide range of epithelial structures[8].
Stem cells are present throughout embryonic development and in adult organs. Basically, there are two types of stem cells: embryonic stem cells (ESCs) that can be isolated from the inner cell mass of blastocysts, and adult stem cells (ASCs) that are found in various tissues and organs. ESCs are pluripotent and can differentiate into all tissues of an embryo, whereas ASCs are more restricted in their differentiation potential. ASCs are, however, vital for the normal turnover of regenerative organs, such as blood, skin or intestine, and they are necessary for replenishing specialized cells when they are lost, for example, after tissue damage[9]. Since extensive proliferation and differentiation of stem cells can contribute to hyperproliferative disorders, a coordinated control of stem cell self-renewal and differentiation is fundamental for maintaining tissue and organ homeostasis. p53 appears to contribute to this restraint by controlling the proliferation, self renewal and differentiation of embryonic and ASCs. With the increasing interest in stem cell biology in the past few years, these activities of p53 have gained significantly more attention. In this review, we provide an overview of the current knowledge on p53 regulation and activity in embryonic, adult and induced pluripotent stem cells.
In the following sections, we distinguish the observations made with murine ESCs (mESCs) or human ESCs (hESCs).
p53 IN ESCs
The first observation about a potential role of p53 in ESCs dates back to 1980 when Mora et al[10] observed that p53 was highly expressed in primary cell cultures obtained from 12-14-d old mouse embryos but not in cells from 16-d old embryos. One year later, they observed that the amount of p53 protein decreased significantly during embryogenesis[11]. This observation was further supported in 1985 when Rogel et al[12] noticed a considerable reduction in p53 mRNA during embryogenesis from day 11 onwards. Six years later, Schmid et al[13] reported the tissue-specific expression of the p53 gene during development and confirmed the strong decline of p53 mRNA in cells undergoing terminal differentiation. Subsequent publications further substantiated the finding that p53 is highly abundant in mESCs[14,15]. Despite its high abundance and the fact that the tumor suppressor protein was more strongly acetylated at lysine 383 in hESCs compared to differentiated cells, p53 was found to be inactive in stem cells[16,17].
During differentiation, p53 protein and RNA decrease significantly[2,11,12,14,15]. Whether the reduction in p53 protein levels also corresponds to reduced activity is an open question. Although Lin et al[2] observed an increase in p53 activity during differentiation and transcription of its target genes p21, mdm2 and killer/DR5, Sabapathy et al[14] observed a conformational change in the tumor suppressor protein, and concomitantly, a decrease in its ability to bind DNA, which would imply a decrease in the transcriptional activity of p53 instead.
In ESCs, p53 is predominantly found in the cytoplasm[18-20], This mainly cytoplasmic localization may also account for the weak activity of this tumor suppressor protein in differentiated cells.
p53 and the proliferation of ESCs
In differentiated cells, p53 is an important regulator of cell proliferation. By controlling expression of the p21 gene, which encodes a prominent inhibitor of cyclin-dependent kinases, p53 influences transition from G1 into S-phase of the cell cycle[21]. In addition, p53 is able to initiate apoptosis by both the extrinsic and intrinsic pathways[4,22]. In concordance with these activities in differentiated cells, p53 also controls proliferation and cell death in ESCs. Its absence leads to increased proliferation and reduced levels of spontaneous apoptosis of mESCs[14]. Treatment of hESCs with nutlin, an inhibitor of p53 degradation, leads to the rapid accumulation of p21 and to cell cycle arrest at the G1/S boundary[23,24] (Figure 1A). Although removal of the drug after short-term exposure re-establishes normal hESC morphology, extended treatment results in extensive cell death[24]. Conversely, the inhibition of the transcriptional activity of p53 by pifithrin-α, a small molecule inhibitor of p53, or shifting a temperature-sensitive mutant of p53 to the non-permissive temperature reduces apoptosis in mESCs[14,25,26]. Surprisingly, although pifithrin-α reduces both DNA-damage-induced as well as spontaneous apoptosis in mESCs, treatment of hESCs fails to inhibit apoptosis induction by p53[17]. The reason for this discrepancy is unclear. However, since pifithrin-α only inhibits the transcriptional activities of p53 and not its non-transcriptional proapoptotic activities in the cytoplasm[26], this result may indicate that the mitochondrial pathway of apoptosis induction is more important for p53-dependent apoptosis in hESCs than is the “classical” transcription-dependent pathway.
Figure 1.

Differentiation of embryonic stem cells by p53. A: Treatment of embryonic stem cells (ESCs) with nutlin leads to p53 accumulation and transcriptional activation. Activated p53 stimulates transcription of p21, whose gene product initiates cell cycle arrest at the G1/S border and differentiation; B: Treatment of ESCs with retinoic acid (RA) leads to phosphorylation of p53 at serine 315 and repression of nanog followed by differentiation; C: Irradiation of ESCs with UV light or treatment with doxorubicin leads to activation of p53. Activated p53 represses transcription of nanog and oct4, resulting in the differentiation of ESCs.
Role of p53 in the differentiation of ESCs
p53 is a major driving force for the differentiation of ESCs. Spontaneous differentiation of hESCs is significantly reduced when p53 abundance is decreased[17]. The connection between p53 and differentiation became particularly evident when Lin et al[2] found that p53 binds to the promoter of nanog and suppresses its transcription in mESCs (Figure 1B, Table 1). The homeodomain protein Nanog is highly abundant in ESCs and is required for self-renewal and maintenance of an undifferentiated state[27-29]. Suppression of nanog transcription decreases the amount of Nanog protein, and thus, supports ESC differentiation[2]. In addition to the nanog promoter, p53 binds to the oct4 promoter where it also reduces gene transcription[17]. Like Nanog, Oct4 belongs to the group of pluripotency factors that are necessary for maintaining ESCs in an undifferentiated state[27,30]. Treatment of hESCs with nutlin, which is a drug that leads to the strong accumulation of p53, results in decreased nanog and oct4 expression and induction of the differentiation markers gatA4 and gatA6[24].
Table 1.
p53 target genes regulating stem cell behavior
| Gene | Regulation | Cell type | Effect | Ref. |
| nanog | Repression | mESCs | Pro-differentiation | [2] |
| 4-Oct | Repression | mESC | Pro-differentiation | [17] |
| wnt3, wnt3A, wnt 8a, wnt8b, wnt9A, fzd1, fzd 2, fzd6, fzd8, fzd10, lef1 | Activation | mESCs | Anti-differentiation | [31] |
| miRNA-200c | Activation | Mammary epithelial cells | Inhibition of epithelial-mesenchymal transition | [66] |
| osterix | Repression | Mesenchymal stem cells | Inhibition of osteogenic differentiation | [80] |
| runx2 | Repression | Mesenchymal stem cells | Inhibition of osteogenic differentiation | [81] |
| pparγ | Repression | Mesenchymal stem cells | Inhibition of adipogenic differentiation | [82] |
| duoxa1; duox1 | Activation | Neural stem cells | Activation of neurogenesis | [100] |
| gfi1 | Activation | Hematopoietic stem cells | Maintenance of quiescence | [117] |
| necdin | Activation | Hematopoietic stem cells | Maintenance of quiescence | [117] |
| p21 | Activation | Hematopoietic stem cells | Regulation of HSC amount | [117] |
Further evidence for the importance of p53 for ESC differentiation has come from the analysis of retinoic acid (RA)-mediated differentiation. Treatment with RA is a widely used method for differentiating ESCs in culture. This treatment of ESCs with RA also leads to suppression of nanog transcription. Downregulation of nanog after treatment with RA is, however, greatly attenuated in ESCs when p53 is genetically deleted, indicating that p53 plays an important role in RA-mediated suppression of nanog[2]. It is yet unclear how RA is linked to p53, although the phosphorylation of p53 at serine 315 appears to be particularly important for the suppression of nanog transcription (Figure 1B). This phosphorylation enables the recruitment of the corepressor mSin3a to the nanog promoter, which is essential for the full suppression of nanog transcription[2].
In addition to favoring the differentiation of ESCs, p53 also has antidifferentiation activity. The Wnt signaling pathway is extremely important for the maintenance of self-renewal and pluripotency of murine and human ESCs[31,32]. Wnt signaling is activated by binding of Wnt-ligands to their cognate Frizzled receptor, which culminates in the activation of the Lef1/Tcf transcription complex[33]. Activation of p53 also counteracts differentiation by leading to the induction of Wnt ligands and receptors and Lef1[34] (Table 1).
p53 activities in ESCs in response to DNA damage
In differentiated cells, DNA damage leads to the accumulation of p53 in the nucleus and mitochondria and to the transcription of its target genes, including mdm2, p21, bax, puma and noxa, followed by cell cycle arrest at the G1/S boundary and initiation of cell death[35]. Although the elevated abundance of p53 in ESCs is generally accepted, there are contradictory reports about its activity in ESCs in response to DNA damage. Some studies have reported that p53 is only weakly or not activated in response to γ-irradiation of mESCs, or after treatment with n-phosphonacetyl-L-aspartate, and that both its protein level and expression of its target genes p21 and mdm2 should remain unchanged. Other studies, including our own, have reported p53 accumulation in the nucleus of mESCs in response to UV light or γ irradiation, or after treatment with doxorubicin, as well as transcriptional activation of its target genes p21, mdm2, puma and noxa[2,16,18,36].
Despite transcription of the p21 gene after p53 activation, no p21 protein is produced in mESCs[18,37,38]. For hESCs, there are conflicting data regarding p53 activity in response to DNA damage. Qin et al[17] failed to observe an increase in p21 mRNA in response to UV irradiation despite accumulation and phosphorylation of p53, whereas Filion et al[39] reported a significant induction of p21 mRNA in response to ionizing radiation. However, as reported in mESCs, the p21 protein was hardly detectable in hESCs.
Similar to differentiated cells in which p53 halts the cell cycle and drives cells with damaged DNA into apoptosis[40], p53 is regarded as being responsible for the high sensitivity of mESCs to DNA damage. Following its accumulation in response to UV irradiation, p53 rapidly induces apoptosis in mESCs, leading to the death of a majority of the cells[18,41]. Treatment of hESCs with etoposide leads to association of p53 with mitochondria and to increased expression of puma. Subsequently, Bax and Mcl1 are co-localized in perinuclear structures that resemble mitochondrial aggregates, followed by rapid and extensive induction of apoptosis[20]. hESCs stably transduced with an shRNA that is targeted against p53 show significant reduction of bax and puma expression and apoptosis after treatment with etoposide, indicating a requirement of p53 for the induction of cell death in hESCs in response to DNA damage[20]. In mESCs, the colony forming ability after UV irradiation is more strongly reduced in p53-positive mESCs than in those that lack the tumor suppressor protein[41]. Most interestingly, although ESCs rapidly undergo apoptosis in response to UV-irradiation or treatment with etoposide, ionizing radiation is less efficient in inducing cell death. Conflicting observations have, however, been made regarding the regulation of the clonogenic potential by p53 after ionizing irradiation. Corbet et al[41] have observed a stronger reduction in the colony forming ability of p53-positive mESCs after γ irradiation in comparison to p53-deficient mESCs, although we failed to observe this difference after ionizing radiation[18].
Apart from the increase in abundance, the p53 protein is also post-translationally modified in response to DNA damage[5]. Of note, these post-translational modifications differ between ESCs and differentiated cells and between hESCs and mESCs, both in quality and in intensity. In response to UV- or ionizing radiation, p53 from hESCs is barely phosphorylated at serine 9, which is an amino acid that is phosphorylated in response to cellular stress in differentiated cells[42]. Also, phosphorylation of serine 20 (serine 23 of murine p53) is rather weak in mESCs. One explanation for this weak phosphorylation of p53 in mESCs is that Chk2, the kinase that usually phosphorylates this site, is hyperphosphorylated and tethered in aggregates in mESCs, thus limiting its availability for phosphorylating its cellular targets[37,43]. Conversely, phosphorylation on serine 15 (serine 18 of murine p53) of p53 is stronger in hESCs than in differentiated human cells, while its intensity is similar in mESCs and mouse embryonic fibroblasts (MEFs)[17,37].
In contrast to differentiated cells in which damage-induced p53 activities are mostly restricted to the induction of cell cycle arrest and apoptosis, p53 activation also affects differentiation in ESCs as a response to DNA damage. By binding to the promoters of oct4 and nanog, p53 represses the transcription of these pluripotency factors and facilitates the differentiation of damaged ESCs[2,17,19] (Figure 1C). However, p53 can also induce transcription of Wnt ligands and receptors in response to DNA damage, which has antidifferentiation properties[34,44].
Overall, by enhancing the sensitivity to DNA damaging agents, induction of cell death and by encouraging differentiation, p53 acts as a guardian of the genome for ESCs despite its failure to induce G1 arrest in this cell type.
p53 IN ASCs
In addition to the developing embryo, stem cells have also been identified in somatic tissues of adults, including the nervous system, bone marrow, epidermis, skeletal muscle, mammary gland and liver[45-51]. Stem cells in somatic tissue are usually called tissue or ASCs in order to distinguish them from ESCs that are derived from the inner cell mass of blastocysts[52]. Even in adult organs, tissue stem cells retain the potential for self-renewal and differentiation into different cell types, but they have lost pluripotency as well as the capacity to form a complete new organism[53]. In organs, ASCs reside in specific niches where they remain in a quiescent state during most of the host’s lifetime. However, when new cells are required, these tissue stem cells divide postnatally, frequently in an asymmetric way, by which they generate another stem cell as well as a committed progenitor daughter cell. The progenitor cell then proliferates further and produces a pool of differentiated cells. These differentiated cells replenish cells as they die, due to natural wearing away or after injury[54,55]. Therefore, stem cells are intimately involved in maintaining tissue homeostasis.
Similar to ESCs, proliferation, self-renewal and genomic stability are tightly controlled in ASCs. Defects in these parameters contribute to premature aging, to failure to repair tissue injury and to the development of cancer[56-59]. Detailed knowledge about the processes that regulate proliferation, self-renewal and transformation of tissue stem cells is therefore crucial to enable safe usage of these cells for stem-cell-based therapies. The tumor suppressor protein p53 is a key regulator of these processes. In the following sections, we highlight the most important results regarding p53 regulation and function in various types of tissue stem cells.
Mammary gland stem cells
Mammary gland stem cells (MGSCs) direct mammary gland development and functionality, and alterations in the proliferation of these cells result in defects of the mammary gland[51,60-62]. p53 appears to be a critical control component for the development of mammary glands. This appearance became particularly evident when mammary glands of mice that expressed one wild-type and one C-terminally deleted allele of p53 were investigated[63]. Expression of a C-terminally deleted allele of p53 in combination with a wild-type allele confers increased resistance to tumorigenesis and accelerated aging in mice[64]. Mice that express this combination of wild-type and C-terminally deleted p53 show significant defects in the morphogenesis of mammary gland ducts. Moreover, when mammary epithelium from these mice is serially transplanted, the epithelium shows severely reduced transplant capabilities, indicative of early stem cell exhaustion[63]. Conversely, mammary epithelium of mice, where p53 is genetically deleted, contains an increased number of stem cells and is highly susceptible to tumor formation[65,66]. Moreover, MGSCs from p53-/- mice produce a higher number of mammospheres that are larger in size when cultured[67]. Mammary stem cells from p53+/- mice also show a higher number of mammospheres than those from p53+/+ mice, indicating that gene dosage of p53 is important for proliferation[67]. These results strongly suggest that p53 controls the maintenance of a constant number of MGSCs by probably stimulating asymmetric cell division[66] (Figure 2A). Loss of the tumor suppressor protein shifts proliferation to the symmetrical division of MGSCs, resulting in the production of only stem cells that all retain pluripotency and self-renewal capacity. This symmetric division of MGSCs results in significant enlargement of the stem cell pool, which favors tumorigenesis of the mammary gland[66,67].
Figure 2.

p53 suppresses self-renewal and promotes differentiation of adult stem cells. A: In mammary gland stem cells, p53 promotes asymmetric cell division, resulting in an increased number of differentiated cells and a reduction in the stem cell pool; B: In mesenchymal stem cells (MSCs), p53 suppresses proliferation and self-renewal and reduces differentiation by suppressing runx2, osterix and pparγ; C: p53 supports quiescence of hematopoietic stem cells and reduces proliferation. This effect is mediated by transcriptional activation of gf1, necdin and p21 (Top). In response to DNA damage, p53 enhances the death of severely damaged cells by cell competition (below).
In addition to controlling proliferation and symmetry of MGSC division, p53 regulates epithelial-mesenchymal transition (EMT) in the mammary gland. EMT and the reverse process, mesenchymal-epithelial transition (MET), are key processes for the regulation of embryogenesis. EMT and MET are a series of events during which epithelial or mesenchymal cells lose many of their characteristics and take on properties that are typical of the other cell type. This transition occurs as a result of a number of intercellular and intracellular adjustments[68]. p53 suppresses EMT by binding to the promoter of the microRNA miR-200c and activating its expression[69] (Table 1). miR-200c is a microRNA that regulates EMT by inhibiting ZEB1/2, a transcriptional repressor of E-cadherin[70]. Re-expression of miR-200c in stem cells with deleted p53 reduces formation of mammospheres, indicating that the control of proliferation and asymmetric cell division of MGSCs by p53 may primarily occur by regulating this microRNA[69].
Mesenchymal stem cells
Mesenchymal stem cells (MSCs) reside in the bone marrow and can differentiate into different types of cells of mesodermal origin, such as osteoblasts, adipocytes and chondrocytes[71]. Multipotent bone marrow MSCs express low levels of adipogenic and osteogenic factors. When this balance of adipogenic and osteogenic factors is tipped, cells become committed toward one of these lineages, and lineage-specific transcription factors that promote one particular cell fate are transcribed. Lineage-specific transcription factor activation usually occurs concomitantly with repression of the other cells’ fate[72]. Loss of p53 in MSCs results in severe alterations of tissue homeostasis. The complete absence of p53 promotes a higher proliferation rate of bone-marrow-derived MSCs, which acquire the typical MSC surface phenotype earlier than wild-type MSCs do. In addition, more precursors are generated that are able to form colonies.
MSCs can be isolated and expanded in vitro, which predestines these cells for tissue engineering and therapeutic applications[73-75]. However, MSCs that have been extensively propagated in vitro frequently acquire mutations that may lead to spontaneous transformation[57,76,77]. Therefore, extensively propagated MSCs should be taken with care with regard to patient therapy. Moreover, the expression profiles and mutation spectra of p53 of these extensively propagated MSCs are similar to those found in human tumors. Therefore, these findings raise the conjecture that mesenchymal tumors may originate from aged MSCs[78]. The coincidence of p53 mutations and the development of tumors in aged MSCs suggests that p53 activity might be required for maintaining genomic stability in this cell type, and for suppression of spontaneous transformation in long-term MSC cultures[77]. This notion is further supported by the observation that MSCs from mice with a genetic deletion of both p53 alleles are capable of forming tumors when they are injected into immunodeficient mice[79]. Conversely, when both alleles of the p21 gene, a target gene of p53 that is mainly responsible for p53-mediated cell cycle arrest, are deleted, MSCs do not show any signs of tumoral transformation[80,81]. Only when at least one allele of p53 is mutated in addition to p21 does p53 expression become lost after long-term culture, and a significant increase in growth rate and genomic instability is observed[82]. Loss of p53 expression is further accompanied by the loss of expression of the cdk inhibitor p16, upregulation of p19Arf and c-myc, and by the complete loss of any senescence phenotype[82].
In addition to controlling proliferation and transformation, p53 also influences the differentiation of MSCs (Figure 2B). MSCs that lack p53 generally differentiate into adipocytes or osteocytes more rapidly than wild-type MSCs. This increased differentiation occurs along with enhanced expression of osteoblast differentiation factors osterix and runx2, which are normally repressed by p53, and by increased expression of runx2 and ppary[83-85] (Table 1). Conversely, osteoblast progenitor cells with elevated p53 activity show reduced proliferation and reduced differentiation, indicating that p53 may negatively regulate the differentiation process[84]. Accelerated differentiation in combination with an increase in macrophage colony-stimulating factor is thought to be the reason for the high bone mass that is observed in the absence of p53[83].
Neural stem cells
The nervous system develops from neural stem cells (NSCs), which have the capacity to self-renew and differentiate into neurons, oligodendrocytes and astrocytes[86,87]. Proliferation and differentiation of the nervous system of mammals is limited after birth, although certain areas in the brain retain multipotent precursor cells with the ability to self-renew and differentiate along neural lineages. Neurogenesis in the adult brain has been particularly observed in the subventricular zone (SVZ) of the lateral ventricle and in the subgranular zone of the hippocampus[88].
p53 has emerged as an important regulator of cell division both in the developing and in the adult brain[89-93]. The tumor suppressor protein has been shown to be expressed in the SVZ as well as the lateral ventricle wall, where most of the glial-fibrillary-acidic-protein-positive astrocytes and Musashi1-positive progenitor cells of the SVZ reveal p53 immunoreactivity[90]. The major function of p53 in the brain is to protect neurons from DNA damage and transformation. Accordingly, p53 induces cell-cycle arrest, DNA repair and cell death in the brain in response to genotoxic insults. In addition, p53 regulates proliferation, differentiation and development of the brain. It promotes neuronal maturation, axon outgrowth and regeneration after injury, and has a crucial role in eliciting neuronal cell death during development[94].
The first indication that p53 might regulate NSC proliferation came about 15 years ago when the p53 gene was deleted in mice. Of those animals that lacked both alleles of p53, more than 15% showed cellular overgrowth in the mid-brain[95]. Comparisons of wild-type and p53 knockout mice have revealed that the number of proliferating cells in the SVZ from p53-null mice is significantly higher than in p53 wild-type mice. This hyperproliferation of cells of the SVZ results in areas of increased cell density that are distributed along the walls of the lateral ventricle[90,91]. The hyperproliferation of cells of the SVZ is probably caused by expansion of the stem cell/progenitor compartment, along with rapid differentiation towards neuronal and glial lineages[90,91]. An increased proliferation of p53-negative NSCs becomes apparent when NSCs from p53 knockout mice are taken into culture. These NSCs form an increased number of clonal aggregates called neurospheres in comparison to those from wild-type mice. In addition, neurospheres from p53-null NSCs are larger in size, due to increased cell content[90,91]. When NSCs are recovered from these neurospheres, a greater proportion of cells are capable of initiating new neurospheres compared to NSCs from wild-type mice[90,91]. These observations strongly suggest that p53 is a negative regulator of NSC self-renewal.
Proliferation of NSCs lacking p53 is further enhanced when cells are also null for phosphatase and tensin homolog (PTEN). Moreover, although NSCs that lack p53 still respond to differentiation cues, this property is not the case when PTEN and p53 are both genetically deleted. Instead, the cells retain their stem-cell-like morphology and continue to express NSC lineage markers, even after experiencing differentiation cues[96]. The reason for this increased proliferation and inhibition of differentiation is probably a substantial increase in c-myc expression in the absence of p53 and PTEN, whereas c-myc expression is only marginally elevated in p53-/- or PTEN-/- NSCs. Enforced expression of c-myc in p53-/- NSCs also enhances proliferation and represses differentiation in the absence of p53 alone, whereas genetic deletion of c-myc in p53/PTEN-deficient NSCs restores their differentiation potential[96]. Thus, p53 and PTEN may both suppress c-myc expression in NSCs and allow differentiation to take place. Loss of one of these proteins may be compensated by the other factor or other components in the cell, while loss of both leads to snapping through of the differentiation-blocked phenotype.
Alterations in cell number can be achieved by modulating proliferation or by enhancing or reducing cell death. p53 can affect both possibilities; it influences cell proliferation by controlling expression of the cdk-inhibitor p21, and cell death by activating transcription of proapoptotic genes such as bax, puma or noxa, as well as by binding to pro- and antiapoptotic members of the Bcl-2 family[3,4]. Yet, the ability of p53 to induce cell death appears to be negligible for constraining proliferation of NSCs. The smaller neurospheres that are formed in the presence of p53 show no indication of cell death. Also, overexpression of an isoform of p53 (ΔNp53) that is shorter and more stable, and therefore, more active, reduces the size of neurospheres but does not increase the level of apoptosis[97-99]. Consistent with these in vitro results, mice overexpressing ΔNp53 show a clear age-dependent decline in the number of proliferating cells in the SVZ, and a reduction in the supply of new olfactory bulb neurons compared to mice expressing normal p53. Olfactory bulb neuron exhaustion is not apparent in younger mice, indicating that it is caused by premature NSC exhaustion[99]. In line with the reduction in NSC proliferation and the absence of increased cell death, overexpression of ΔNp53 leads to constitutive expression of the cdk-inhibitor p21, compromises the re-entry of dormant NSCs into the cell cycle and to extended durations of cell-cycle passages[99].
Although there is unanimity about the regulatory role of p53 in NSC proliferation, there are differing reports regarding the impact of p53 for differentiation. Jonas Meletis et al[90] have reported that neurospheres from p53-null and wild-type NSCs contain a similar number of neurons, astrocytes and oligodendrocytes, whereas Armesilla-Diaz et al[89] and Masato Nagao et al[100] have found that differentiation of neurospheres from p53-null mice is biased towards neuronal precursors. Regardless of the differing reports regarding the role of p53 in NSC differentiation, p53 activity increases during development and is at the time when neuronal differentiation takes place at its maximum[101,102]. Also in cultured NSCs, p53 expression increases during differentiation[103], which might be caused by a gradual upregulation of p19Arf. p19Arf binds to the central domain of the Mdm2 , which is the most important negative regulator of p53, and inhibits Mdm2-mediated degradation of the tumor suppressor protein[5]. As a result of this inhibition, p53 accumulates in the cell in an Arf-dose-dependent manner. The amount of p19Arf increases up to 20-fold from E13.5 to postnatal day 2, which could be responsible for the increase in p53 expression[100]. Consistent with a regulation of p53 by p19Arf and a putative role of p53 in suppressing self-renewal of NSCs and supporting differentiation towards glia cells, early-stage NSCs, which express little p19Arf, retain a high self-renewal capacity and differentiate towards the neurogenic lineage, whereas late-stage NSCs, which possess more p19Arf, have a lower self-renewal capacity and predominantly generate glia cells[100]. In line with this notion, the enhanced downregulation of p19Arf or genetic deletion of p53 enhances the self-renewal potential of NSCs as well as the production of neurons, whereas overexpression of p19Arf reduces NSC proliferation and promotes differentiation into glia cells[100] (Figure 3). Furthermore, induction of NSC differentiation leads to phosphorylation of p53 at serine 15 and enhances its DNA binding activity. It is, therefore, not surprising that differentiation of NSCs is concurrent with alterations in the expression of p53 target genes[104]. Two possible target genes of p53 during neuronal differentiation are dual oxidase (DUOX1) and its maturation factor DOUXA1 (Table 1). Expression of p53 in P19 pluripotent embryonal carcinoma cells increases the level of both DUOX1 and DUOXA1 and allows their physical interaction[103]. Both proteins, DUOX1 and DUOXA1, play an important role in the regulation of neuronal differentiation through DOUX1-mediated reactive oxygen species production and modulation of intermediate filaments[105]. Transcriptional activation of these two proteins may, therefore, contribute to the differentiation-inducing activity of p53 in NSCs.
Figure 3.
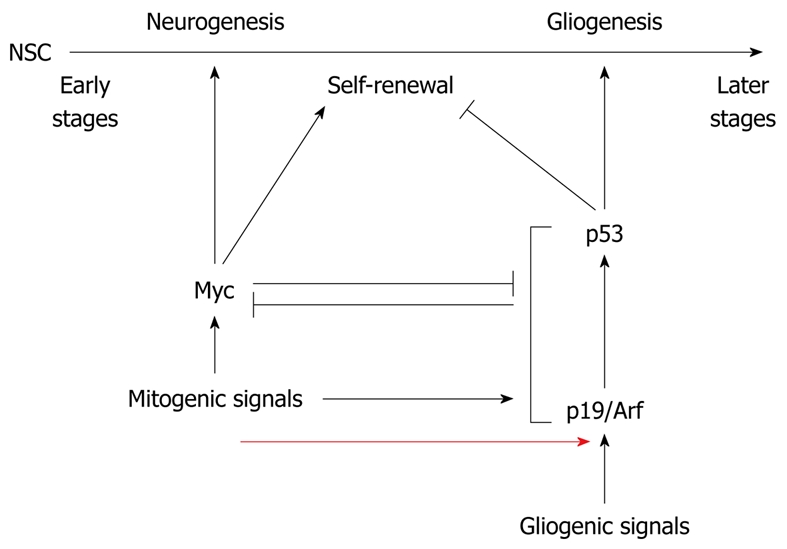
Antagonistic regulation of differentiation of neural stem cells by Arf/p53 and Myc. Myc regulates self-renewal of neural stem cells (NSCs) and enhances their differentiation into the neuronal lineage. At early stages of embryogenesis, myc expression is high and p19Arf expression is low. Later in development, mitogenic signals upregulate p19Arf expression, resulting in enhanced abundance of the p53 tumor suppressor protein, which suppresses self-renewal of NSCs and differentiation towards the neuronal lineage while it supports gliogenesis [100].
About 50% of human tumors express a mutated p53 gene, including brain tumors, where p53 mutations are found frequently. Mutations of p53 are associated with both tumor initiation and expansion in the brain, supporting the importance of this tumor suppressor protein for maintaining tissue homeostasis[106]. In differentiated cells, the activity of p53 comes mainly into play when the DNA of a cell has been damaged, at which point p53 initiates cell cycle arrest and cell death[107]. More recently, it has been shown that when cell cycle regulation or DNA integrity is perturbed in neural precursors, p53 is a major proapoptotic protein[108]. Telencephalic cells grown to neurospheres are arrested at the G1/S boundary of the cell cycle after irradiation, and show enhanced induction of apoptosis along with enhanced abundance and nuclear localization of p53 and its downstream targets p21 and Bax. At the same time, Rad51 and Rrm2 are transcriptionally repressed by p53, demonstrating that in response to genotoxic stress, p53 regulates transcription and repression of its target genes in neural progenitors[89,109,110]. Cultures from p53-/- neural progenitors are not arrested in G1/S phase and exhibit a significantly lower level of spontaneous and DNA-damage-induced apoptosis than their wild-type counterparts, thereby showing that DNA damage-induced G1 arrest and apoptosis in neural precursor cells are critically dependent on p53[89,109,110]. Also, immortalized human NSCs show upregulation and phosphorylation of p53 in response to ionizing radiation[111], indicating that this is a general response of NSCs to DNA damage. Importantly, although neurospheres derived from wild-type animals exhibit only slight karyotypic modifications, neurospheres derived from p53-null animals show major structural chromosomal aberrations and aneuploidy, particularly with higher passage numbers[89], demonstrating that p53 is required for the maintenance of chromosomal stability in NSCs.
Hematopoietic stem cells
Most adult hematopoietic stem cells (HSCs) exist in a relatively quiescent state in the microenvironment of the bone marrow. Once activated, they start to proliferate and differentiate along the different hematopoietic cell lineages[112,113].
As shown for stem cells in other tissues, p53 activity also affects cell number, proliferation potential and differentiation of HSCs. Accordingly, HSCs of mice with a hyperactive mutant form of p53[64] show a reduced number of proliferating HSCs. Conversely, a reduction in p53 increases proliferation and self-renewal of HSCs[114-116]. Absence of the p53 target gene p21[117] (Table 1) results in an increased number of HSCs, whereas high levels of p21 mRNA in the quiescent stem cell fraction restrict stem cells from entry into the cell cycle[118]. Nevertheless, it should be noted that p21 abundance is also regulated by mechanisms independent of p53[119].
In addition to controlling proliferation of HSCs, p53 is an essential component for maintaining quiescence of HSCs[120]. This activity of p53 may also be regulated by p21[118], although others have found that the role of p21 in maintaining HSC function is limited[115,121]. Other p53 target genes that might be involved in mediating quiescence are gfi-1 and necdin (Figure 2C). Both genes have been identified as direct p53 targets by transcript profiling[115] (Table 1). Gfi-1 is a zinc-finger-containing repressor protein that has been shown in the past to restrict HSC proliferation[122,123]. Necdin is a growth-inhibitory protein that interacts with multiple proliferation-promoting proteins, such as simian virus 40 large T antigen or E2F-1[124-126]. Downregulation of Necdin diminishes HSC quiescence, whereas quiescence is increased when necdin is overexpressed[115].
Furthermore, p53 is an important component of the DNA damage response of HSCs. HSCs from mice lacking p53 are more resistant to irradiation-induced apoptosis compared to HSCs from wild-type mice, indicating that p53 induces cell death in HSCs in response to DNA damage[115]. p53 also controls a process called cell competition in HSCs in response to DNA damage (Figure 2C). Cell competition is the active elimination of suboptimal cells, basically enabling the selection of the fittest. This process has initially been described for Drosophila, but it probably plays a wider role in tissue homeostasis of all metazoans[127]. Cell competition is activated in HSCs after DNA damage and selects for the least-damaged cells. Despite being controlled by p53, cell competition is distinct from the classical p53-mediated DNA damage response. It persists for several months, appears to be specific for HSCs and progenitor cells, and depends on relative rather than absolute p53 levels in competing cells. Cell competition in response to DNA damage is probably mediated by a non-cell-autonomous induction of proliferation arrest and senescence-related gene expression in those cells that possess higher p53 activity[128].
p53 IN INDUCED PLURIPOTENT STEM CELLS
Somatic cells can be reprogrammed into pluripotent stem cell-like cells by overexpression of combinations of different pluripotency factors. The reprogramming of differentiated cells is a possible source for autologous pluripotent cells that can be transplanted into patients.
Since 2006, when the first induced pluripotent stem cells (iPSCs) were generated by overexpression of oct4, sox2, klf4 and c-myc in adult and embryonic fibroblasts[129], iPSCs have been generated from cells of multiple origins, including embryonic fibroblasts, adult fibroblasts, neural progenitor cells, hepatocytes, epithelial cells and keratinocytes, by combinations of different reprogramming factors[130,131]. However, the low frequency and the tendency to induce malignant transformation by overexpression of proto-oncogenes raises doubts about the clinical applicability of this approach.
Reprogramming only occurs in a very small percentage of transfected cells, suggesting the existence of reprogramming barriers. Since decreased protein levels of p53 and p21 were observed in iPSCs derived from MEFs[129], it was assumed that p53 might play a role in the reprogramming process of somatic cells. One of the first such reports came from Zhao et al[132] who have observed that downregulation of p53 by siRNA dramatically enhances the efficiency of iPSC generation from human adult fibroblasts. This result has been confirmed by Kawamura et al[133] and Li et al[134], who have observed that reducing p53 levels by p53 shRNA or by using p53-null MEFs increases the efficiency of reprogramming, whereas re-expression of p53 in p53-null MEFs markedly reduces it. Downregulation of p19Arf, a prominent regulator of p53 activity[5], also enhances reprogramming efficiency of MEFs and murine keratinocytes[134]. The reprogramming efficiency of mouse fibroblasts lacking p19Arf is similar to that of MEFs lacking p53, suggesting that the reprogramming barrier that is controlled by p19Arf is mediated by p53[134] (Figure 4). However, despite the leading role of p53 for reprogramming of human cells, p19Arf appears to be more important for murine cells[132,134]. It is presently unclear how and why p53 imposes a reprogramming barrier. One possible explanation is that the reprogramming factors c-Myc, Oct4 and Sox2 induce p53 activity, thus, eliciting a stress response that finally leads to cell cycle arrest and/or cell death (Figure 4). This assumption is further supported by the finding that overexpression of the antiapoptotic gene bcl-2 increases the efficiency of reprogramming, indicating that p53 implements the reprogramming barrier by eliminating cells that express the reprogramming factors by apoptosis[133]. Most interestingly, when p53 levels are reduced, MEFs can be reprogrammed to pluripotency in the absence of oncogenes, such as c-myc or klf4, simply by the overexpression of oct4 and sox2[133]. Moreover, in the absence of p53, up to 10% of transduced cells became iPSCs, and iPSCs can even be generated from terminally differentiated lymphocytes[135]. p53 may also implement a barrier for reprogramming by inducing senescence. Expression of the four reprogramming factors oct4, sox2, klf4 and c-myc triggers a senescence-like phenotype in human fibroblasts[136] (Figure 4). Senescence is an irreversible cell cycle arrest in the G1 phase of the cell cycle caused, for example, in response to cellular stress, such as DNA damage, treatment with chemotherapeutic drugs, or aberrant expression of oncogenes. This arrest occurs mainly through the activation of p53 and upregulation of p16INK4 and p21[137]. In fact, p21 has been shown to be an important target of p53 in the context of implementing a reprogramming barrier[135]. Nevertheless, other target genes of p53 also appear to contribute, as the absence of p21 enhances reprogramming efficiency by only about fourfold, whereas the absence of p53 does so by 7-10-fold[134,135]. Transfection of somatic cells with the four reprogramming factors significantly increases the percentage of cells arrested in G1 phase without inducing apoptosis, indicating a possible involvement of p21. Moreover, cells expressing the reprogramming factors display an enlarged cytoplasm, a senescence-associated β-galactosidase activity and heterochromatic foci. Ablation of the senescence effectors, p16INK4a, p53 and p21 improves the efficiency of reprogramming significantly[136].
Figure 4.
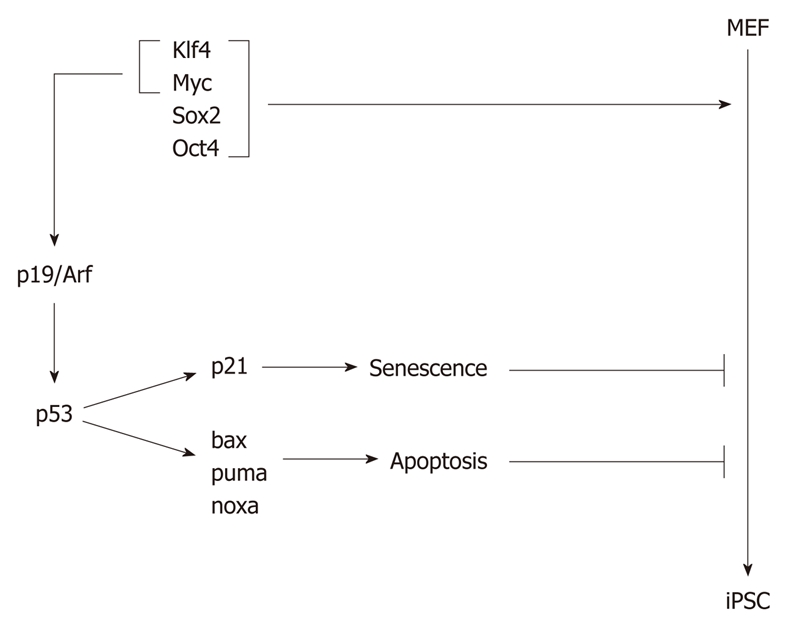
p53 provides a barrier for the reprogramming of somatic cells to induced pluripotent stem cells. Somatic cells, such as mouse embryonic fibroblasts (MEFs), can be reprogrammed to induced pluripotent stem cells (iPSCs) after overexpression of a combination of reprogramming factors, such as Klf4, Sox2, Myc and Oct4. Klf4 and Myc induce p19Arf expression, which prevents p53 degradation, resulting in its accumulation and expression of its target genes. Among the target genes of p53 is p21, which causes senescence, and bax, puma and noxa, which lead to apoptosis.
Although suppression of p53 appears to be a feasible way of improving reprogramming efficiency, permanent suppression of p53 may lower the quality of iPSCs, by allowing the outgrowth of iPSCs with permanent DNA lesions and chromosomal aberrations. p53 appears to be critically involved in preventing the reprogramming of cells with damaged DNA, short telomeres or reduced DNA repair deficiency. MEFs with critically short telomeres or MEFs treated with low doses of γ-irradiation or UV light show low reprogramming efficiency. Abrogation of p53 in these cells restores reprogramming efficiency to the level of undamaged p53-null cells[138].
CONCLUSION
Although mice with genetic deletions of both p53 alleles develop normally, there is increasing evidence for a role of p53 in the regulation of stem cell proliferation, differentiation and the maintenance of stem cell genetic stability. At present, we are only beginning to understand the different actions that p53 exerts in stem cells, and the underlying mechanisms are even less clear. It is of particular interest to establish how p53 avoids the acquisition of mutations and genomic alterations after prolonged propagation of ASCs in vitro. Another important question is why p53 implements a barrier for the generation of iPSCs and how to circumvent this barrier for the generation of isotypic stem cells from patients without instigating the risk of expanding stem cells with genetic alterations. Transient suppression of p53 might be a useful compromise, however, it is presently unclear whether cells that survive the reprogramming process are truly free of mutations and aberrations when p53 has been inactivated for some duration. Therefore, more research is required to allow the safe use of stem cells for therapy.
Footnotes
Peer reviewer: Carlo Ventura, MD, PhD, Full Professor of Molecular Biology, Chief, Laboratory of Molecular Biology and Stem Cell Engineering-National Institute of Biostructures and Biosystems, University of Bologna, S. Orsola-Malpighi Hospital, Cardiovascular Department, Pavilion 21 Via Massarenti 9 Bologna 40138, Italy
S- Editor Cheng JX L- Editor Kerr C E- Editor Zheng XM
References
- 1.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 2.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 4.Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol. 2005;17:631–636. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Boehme KA, Blattner C. Regulation of p53--insights into a complex process. Crit Rev Biochem Mol Biol. 2009;44:367–392. doi: 10.3109/10409230903401507. [DOI] [PubMed] [Google Scholar]
- 6.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulikov R, Letienne J, Kaur M, Grossman SR, Arts J, Blattner C. Mdm2 facilitates the association of p53 with the proteasome. Proc Natl Acad Sci USA. 2010;107:10038–10043. doi: 10.1073/pnas.0911716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machado-Silva A, Perrier S, Bourdon JC. p53 family members in cancer diagnosis and treatment. Semin Cancer Biol. 2010;20:57–62. doi: 10.1016/j.semcancer.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 10.Mora PT, Chandrasekaran K, McFarland VW. An embryo protein induced by SV40 virus transformation of mouse cells. Nature. 1980;288:722–724. doi: 10.1038/288722a0. [DOI] [PubMed] [Google Scholar]
- 11.Chandrasekaran K, McFarland VW, Simmons DT, Dziadek M, Gurney EG, Mora PT. Quantitation and characterization of a species-specific and embryo stage-dependent 55-kilodalton phosphoprotein also present in cells transformed by simian virus 40. Proc Natl Acad Sci USA. 1981;78:6953–6957. doi: 10.1073/pnas.78.11.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogel A, Popliker M, Webb CG, Oren M. p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol Cell Biol. 1985;5:2851–2855. doi: 10.1128/mcb.5.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid P, Lorenz A, Hameister H, Montenarh M. Expression of p53 during mouse embryogenesis. Development. 1991;113:857–865. doi: 10.1242/dev.113.3.857. [DOI] [PubMed] [Google Scholar]
- 14.Sabapathy K, Klemm M, Jaenisch R, Wagner EF. Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 1997;16:6217–6229. doi: 10.1093/emboj/16.20.6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solozobova V, Blattner C. Regulation of p53 in embryonic stem cells. Exp Cell Res. 2010;316:2434–2446. doi: 10.1016/j.yexcr.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, Wahl GM. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol. 1998;8:145–155. doi: 10.1016/s0960-9822(98)70061-2. [DOI] [PubMed] [Google Scholar]
- 17.Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, Li J, Song Z, Qu X, Zhou P, et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842–5852. doi: 10.1074/jbc.M610464200. [DOI] [PubMed] [Google Scholar]
- 18.Solozobova V, Rolletschek A, Blattner C. Nuclear accumulation and activation of p53 in embryonic stem cells after DNA damage. BMC Cell Biol. 2009;10:46. doi: 10.1186/1471-2121-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grandela C, Pera MF, Grimmond SM, Kolle G, Wolvetang EJ. p53 is required for etoposide-induced apoptosis of human embryonic stem cells. Stem Cell Res. 2007;1:116–128. doi: 10.1016/j.scr.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Liu G, Lozano G. p21 stability: linking chaperones to a cell cycle checkpoint. Cancer Cell. 2005;7:113–114. doi: 10.1016/j.ccr.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Chowdhury I, Tharakan B, Bhat GK. Current concepts in apoptosis: the physiological suicide program revisited. Cell Mol Biol Lett. 2006;11:506–525. doi: 10.2478/s11658-006-0041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 24.Maimets T, Neganova I, Armstrong L, Lako M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene. 2008;27:5277–5287. doi: 10.1038/onc.2008.166. [DOI] [PubMed] [Google Scholar]
- 25.Lee MK, Hande MP, Sabapathy K. Ectopic mTERT expression in mouse embryonic stem cells does not affect differentiation but confers resistance to differentiation- and stress-induced p53-dependent apoptosis. J Cell Sci. 2005;118:819–829. doi: 10.1242/jcs.01673. [DOI] [PubMed] [Google Scholar]
- 26.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 27.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 28.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 29.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 30.Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Schöler H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 31.Ogawa K, Nishinakamura R, Iwamatsu Y, Shimosato D, Niwa H. Synergistic action of Wnt and LIF in maintaining pluripotency of mouse ES cells. Biochem Biophys Res Commun. 2006;343:159–166. doi: 10.1016/j.bbrc.2006.02.127. [DOI] [PubMed] [Google Scholar]
- 32.Dravid G, Ye Z, Hammond H, Chen G, Pyle A, Donovan P, Yu X, Cheng L. Defining the role of Wnt/beta-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells. 2005;23:1489–1501. doi: 10.1634/stemcells.2005-0034. [DOI] [PubMed] [Google Scholar]
- 33.Hendrickx M, Leyns L. Non-conventional Frizzled ligands and Wnt receptors. Dev Growth Differ. 2008;50:229–243. doi: 10.1111/j.1440-169X.2008.01016.x. [DOI] [PubMed] [Google Scholar]
- 34.Lee KH, Li M, Michalowski AM, Zhang X, Liao H, Chen L, Xu Y, Wu X, Huang J. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci USA. 2010;107:69–74. doi: 10.1073/pnas.0909734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuykin IA, Lianguzova MS, Pospelova TV, Pospelov VA. Activation of DNA damage response signaling in mouse embryonic stem cells. Cell Cycle. 2008;7:2922–2928. doi: 10.4161/cc.7.18.6699. [DOI] [PubMed] [Google Scholar]
- 37.Hong Y, Stambrook PJ. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci USA. 2004;101:14443–14448. doi: 10.1073/pnas.0401346101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malashicheva AB, Kisliakova TV, Savatier P, Pospelov VA. [Embryonal stem cells do not undergo cell cycle arrest upon exposure to damaging factors] Tsitologiia. 2002;44:643–648. [PubMed] [Google Scholar]
- 39.Filion TM, Qiao M, Ghule PN, Mandeville M, van Wijnen AJ, Stein JL, Lian JB, Altieri DC, Stein GS. Survival responses of human embryonic stem cells to DNA damage. J Cell Physiol. 2009;220:586–592. doi: 10.1002/jcp.21735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farnebo M, Bykov VJ, Wiman KG. The p53 tumor suppressor: a master regulator of diverse cellular processes and therapeutic target in cancer. Biochem Biophys Res Commun. 2010;396:85–89. doi: 10.1016/j.bbrc.2010.02.152. [DOI] [PubMed] [Google Scholar]
- 41.Corbet SW, Clarke AR, Gledhill S, Wyllie AH. P53-dependent and -independent links between DNA-damage, apoptosis and mutation frequency in ES cells. Oncogene. 1999;18:1537–1544. doi: 10.1038/sj.onc.1202436. [DOI] [PubMed] [Google Scholar]
- 42.Milne DM, Palmer RH, Campbell DG, Meek DW. Phosphorylation of the p53 tumour-suppressor protein at three N-terminal sites by a novel casein kinase I-like enzyme. Oncogene. 1992;7:1361–1369. [PubMed] [Google Scholar]
- 43.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 44.Pinto D, Clevers H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res. 2005;306:357–363. doi: 10.1016/j.yexcr.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 45.Taylor RB. Pluripotential stem cells in mouse embryo liver. Br J Exp Pathol. 1965;46:376–383. [PMC free article] [PubMed] [Google Scholar]
- 46.Ghins E, Colson-van Schoor M, Maréchal G. The origin of muscle stem cells in rat triceps surae regenerating after mincing. J Muscle Res Cell Motil. 1984;5:711–722. doi: 10.1007/BF00713929. [DOI] [PubMed] [Google Scholar]
- 47.Dulbecco R, Allen WR, Bologna M, Bowman M. Marker evolution during the development of the rat mammary gland: stem cells identified by markers and the role of myoepithelial cells. Cancer Res. 1986;46:2449–2456. [PubMed] [Google Scholar]
- 48.McKay R, Renfranz P, Cunningham M. Immortalized stem cells from the central nervous system. C R Acad Sci III. 1993;316:1452–1457. [PubMed] [Google Scholar]
- 49.Reddi AH. Bone morphogenetic proteins, bone marrow stromal cells, and mesenchymal stem cells. Maureen Owen revisited. Clin Orthop Relat Res. 1995:115–119. [PubMed] [Google Scholar]
- 50.Jensen UB, Lowell S, Watt FM. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: a new view based on whole-mount labelling and lineage analysis. Development. 1999;126:2409–2418. doi: 10.1242/dev.126.11.2409. [DOI] [PubMed] [Google Scholar]
- 51.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 52.Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- 53.Levi BP, Morrison SJ. Stem cells use distinct self-renewal programs at different ages. Cold Spring Harb Symp Quant Biol. 2008;73:539–553. doi: 10.1101/sqb.2008.73.049. [DOI] [PubMed] [Google Scholar]
- 54.Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769–778. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- 55.Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 56.Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, Biernaskie JA, Sinha S, Prives C, Pevny LH, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64–75. doi: 10.1016/j.stem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miura M, Miura Y, Padilla-Nash HM, Molinolo AA, Fu B, Patel V, Seo BM, Sonoyama W, Zheng JJ, Baker CC, et al. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells. 2006;24:1095–1103. doi: 10.1634/stemcells.2005-0403. [DOI] [PubMed] [Google Scholar]
- 58.Warburton D, Perin L, Defilippo R, Bellusci S, Shi W, Driscoll B. Stem/progenitor cells in lung development, injury repair, and regeneration. Proc Am Thorac Soc. 2008;5:703–706. doi: 10.1513/pats.200801-012AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rubio D, Garcia S, Paz MF, De la Cueva T, Lopez-Fernandez LA, Lloyd AC, Garcia-Castro J, Bernad A. Molecular characterization of spontaneous mesenchymal stem cell transformation. PLoS One. 2008;3:e1398. doi: 10.1371/journal.pone.0001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woodward WA, Chen MS, Behbod F, Rosen JM. On mammary stem cells. J Cell Sci. 2005;118:3585–3594. doi: 10.1242/jcs.02532. [DOI] [PubMed] [Google Scholar]
- 61.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 62.Vaillant F, Asselin-Labat ML, Shackleton M, Lindeman GJ, Visvader JE. The emerging picture of the mouse mammary stem cell. Stem Cell Rev. 2007;3:114–123. doi: 10.1007/s12015-007-0018-2. [DOI] [PubMed] [Google Scholar]
- 63.Gatza CE, Dumble M, Kittrell F, Edwards DG, Dearth RK, Lee AV, Xu J, Medina D, Donehower LA. Altered mammary gland development in the p53+/m mouse, a model of accelerated aging. Dev Biol. 2008;313:130–141. doi: 10.1016/j.ydbio.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 65.Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, Butel JS, Medina D. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–1058. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 66.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 67.Tao L, Roberts AL, Dunphy KA, Bigelow C, Yan H, Jerry DJ. Repression of mammary stem/progenitor cells by p53 is mediated by Notch and separable from apoptotic activity. Stem Cells. 2011;29:119–127. doi: 10.1002/stem.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 69.Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13:R45. doi: 10.1186/bcr2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–192. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 72.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 73.Tropel P, Noël D, Platet N, Legrand P, Benabid AL, Berger F. Isolation and characterisation of mesenchymal stem cells from adult mouse bone marrow. Exp Cell Res. 2004;295:395–406. doi: 10.1016/j.yexcr.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 74.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 75.Phinney DG. Biochemical heterogeneity of mesenchymal stem cell populations: clues to their therapeutic efficacy. Cell Cycle. 2007;6:2884–2889. doi: 10.4161/cc.6.23.5095. [DOI] [PubMed] [Google Scholar]
- 76.Rubio D, Garcia-Castro J, Martín MC, de la Fuente R, Cigudosa JC, Lloyd AC, Bernad A. Spontaneous human adult stem cell transformation. Cancer Res. 2005;65:3035–3039. doi: 10.1158/0008-5472.CAN-04-4194. [DOI] [PubMed] [Google Scholar]
- 77.Armesilla-Diaz A, Elvira G, Silva A. p53 regulates the proliferation, differentiation and spontaneous transformation of mesenchymal stem cells. Exp Cell Res. 2009;315:3598–3610. doi: 10.1016/j.yexcr.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 78.Li H, Fan X, Kovi RC, Jo Y, Moquin B, Konz R, Stoicov C, Kurt-Jones E, Grossman SR, Lyle S, et al. Spontaneous expression of embryonic factors and p53 point mutations in aged mesenchymal stem cells: a model of age-related tumorigenesis in mice. Cancer Res. 2007;67:10889–10898. doi: 10.1158/0008-5472.CAN-07-2665. [DOI] [PubMed] [Google Scholar]
- 79.Rubio R, García-Castro J, Gutiérrez-Aranda I, Paramio J, Santos M, Catalina P, Leone PE, Menendez P, Rodríguez R. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer Res. 2010;70:4185–4194. doi: 10.1158/0008-5472.CAN-09-4640. [DOI] [PubMed] [Google Scholar]
- 80.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 81.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 82.Rodriguez R, Rubio R, Masip M, Catalina P, Nieto A, de la Cueva T, Arriero M, San Martin N, de la Cueva E, Balomenos D, et al. Loss of p53 induces tumorigenesis in p21-deficient mesenchymal stem cells. Neoplasia. 2009;11:397–407. doi: 10.1593/neo.81620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang X, Kua HY, Hu Y, Guo K, Zeng Q, Wu Q, Ng HH, Karsenty G, de Crombrugghe B, Yeh J, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J Cell Biol. 2006;172:115–125. doi: 10.1083/jcb.200507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, Stein GS, Lian JB, Jones SN. Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J Cell Biol. 2006;172:909–921. doi: 10.1083/jcb.200508130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Molchadsky A, Shats I, Goldfinger N, Pevsner-Fischer M, Olson M, Rinon A, Tzahor E, Lozano G, Zipori D, Sarig R, et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS One. 2008;3:e3707. doi: 10.1371/journal.pone.0003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cayre M, Malaterre J, Scotto-Lomassese S, Strambi C, Strambi A. The common properties of neurogenesis in the adult brain: from invertebrates to vertebrates. Comp Biochem Physiol B Biochem Mol Biol. 2002;132:1–15. doi: 10.1016/s1096-4959(01)00525-5. [DOI] [PubMed] [Google Scholar]
- 87.Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- 88.Galvan V, Jin K. Neurogenesis in the aging brain. Clin Interv Aging. 2007;2:605–610. doi: 10.2147/cia.s1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Armesilla-Diaz A, Bragado P, Del Valle I, Cuevas E, Lazaro I, Martin C, Cigudosa JC, Silva A. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience. 2009;158:1378–1389. doi: 10.1016/j.neuroscience.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 90.Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 91.Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Medrano S, Scrable H. Maintaining appearances--the role of p53 in adult neurogenesis. Biochem Biophys Res Commun. 2005;331:828–833. doi: 10.1016/j.bbrc.2005.03.194. [DOI] [PubMed] [Google Scholar]
- 93.Eizenberg O, Faber-Elman A, Gottlieb E, Oren M, Rotter V, Schwartz M. p53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol Cell Biol. 1996;16:5178–5185. doi: 10.1128/mcb.16.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tedeschi A, Di Giovanni S. The non-apoptotic role of p53 in neuronal biology: enlightening the dark side of the moon. EMBO Rep. 2009;10:576–583. doi: 10.1038/embor.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- 96.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–6728. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
- 98.Yin Y, Stephen CW, Luciani MG, Fåhraeus R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462–467. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]
- 99.Medrano S, Burns-Cusato M, Atienza MB, Rahimi D, Scrable H. Regenerative capacity of neural precursors in the adult mammalian brain is under the control of p53. Neurobiol Aging. 2009;30:483–497. doi: 10.1016/j.neurobiolaging.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nagao M, Campbell K, Burns K, Kuan CY, Trumpp A, Nakafuku M. Coordinated control of self-renewal and differentiation of neural stem cells by Myc and the p19ARF-p53 pathway. J Cell Biol. 2008;183:1243–1257. doi: 10.1083/jcb.200807130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Komarova EA, Chernov MV, Franks R, Wang K, Armin G, Zelnick CR, Chin DM, Bacus SS, Stark GR, Gudkov AV. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J. 1997;16:1391–1400. doi: 10.1093/emboj/16.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M. Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J. 1997;16:1381–1390. doi: 10.1093/emboj/16.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ostrakhovitch EA, Semenikhin OA. p53-mediated regulation of neuronal differentiation via regulation of dual oxidase maturation factor 1. Neurosci Lett. 2011;494:80–85. doi: 10.1016/j.neulet.2011.02.061. [DOI] [PubMed] [Google Scholar]
- 104.Aranha MM, Solá S, Low WC, Steer CJ, Rodrigues CM. Caspases and p53 modulate FOXO3A/Id1 signaling during mouse neural stem cell differentiation. J Cell Biochem. 2009;107:748–758. doi: 10.1002/jcb.22172. [DOI] [PubMed] [Google Scholar]
- 105.Kennedy KA, Ostrakhovitch EA, Sandiford SD, Dayarathna T, Xie X, Waese EY, Chang WY, Feng Q, Skerjanc IS, Stanford WL, et al. Mammalian numb-interacting protein 1/dual oxidase maturation factor 1 directs neuronal fate in stem cells. J Biol Chem. 2010;285:17974–17985. doi: 10.1074/jbc.M109.084616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature. 1992;355:846–847. doi: 10.1038/355846a0. [DOI] [PubMed] [Google Scholar]
- 107.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 108.Akhtar RS, Geng Y, Klocke BJ, Roth KA. Neural precursor cells possess multiple p53-dependent apoptotic pathways. Cell Death Differ. 2006;13:1727–1739. doi: 10.1038/sj.cdd.4401879. [DOI] [PubMed] [Google Scholar]
- 109.Semont A, Nowak EB, Silva Lages C, Mathieu C, Mouthon MA, May E, Allemand I, Millet P, Boussin FD. Involvement of p53 and Fas/CD95 in murine neural progenitor cell response to ionizing irradiation. Oncogene. 2004;23:8497–8508. doi: 10.1038/sj.onc.1207821. [DOI] [PubMed] [Google Scholar]
- 110.D’Sa-Eipper C, Leonard JR, Putcha G, Zheng TS, Flavell RA, Rakic P, Kuida K, Roth KA. DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development. 2001;128:137–146. doi: 10.1242/dev.128.1.137. [DOI] [PubMed] [Google Scholar]
- 111.Carlessi L, De Filippis L, Lecis D, Vescovi A, Delia D. DNA-damage response, survival and differentiation in vitro of a human neural stem cell line in relation to ATM expression. Cell Death Differ. 2009;16:795–806. doi: 10.1038/cdd.2009.10. [DOI] [PubMed] [Google Scholar]
- 112.Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol. 1997;25:445–453. [PubMed] [Google Scholar]
- 113.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol. 2003;31:521–527. doi: 10.1016/s0301-472x(03)00072-9. [DOI] [PubMed] [Google Scholar]
- 117.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 118.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 119.Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Givol D. Induction of WAF1/CIP1 by a p53-independent pathway. Cancer Res. 1994;54:3391–3395. [PubMed] [Google Scholar]
- 120.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, Huang G, Di Giandomenico S, Koff A, Nimer SD. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–3124. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, Dontje B, de Haan G. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007;25:836–843. doi: 10.1634/stemcells.2006-0631. [DOI] [PubMed] [Google Scholar]
- 122.Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, Orkin SH. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004;431:1002–1007. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- 123.Zeng H, Yücel R, Kosan C, Klein-Hitpass L, Möröy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 2004;23:4116–4125. doi: 10.1038/sj.emboj.7600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hu B, Wang S, Zhang Y, Feghali CA, Dingman JR, Wright TM. A nuclear target for interleukin-1alpha: interaction with the growth suppressor necdin modulates proliferation and collagen expression. Proc Natl Acad Sci USA. 2003;100:10008–10013. doi: 10.1073/pnas.1737765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Taniura H, Kobayashi M, Yoshikawa K. Functional domains of necdin for protein-protein interaction, nuclear matrix targeting, and cell growth suppression. J Cell Biochem. 2005;94:804–815. doi: 10.1002/jcb.20345. [DOI] [PubMed] [Google Scholar]
- 126.Taniura H, Matsumoto K, Yoshikawa K. Physical and functional interactions of neuronal growth suppressor necdin with p53. J Biol Chem. 1999;274:16242–16248. doi: 10.1074/jbc.274.23.16242. [DOI] [PubMed] [Google Scholar]
- 127.Morata G, Martín FA. Cell competition: the embrace of death. Dev Cell. 2007;13:1–2. doi: 10.1016/j.devcel.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 128.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 130.Cox JL, Rizzino A. Induced pluripotent stem cells: what lies beyond the paradigm shift. Exp Biol Med (Maywood) 2010;235:148–158. doi: 10.1258/ebm.2009.009267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhao Y, Yin X, Qin H, Zhu F, Liu H, Yang W, Zhang Q, Xiang C, Hou P, Song Z, et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell. 2008;3:475–479. doi: 10.1016/j.stem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 133.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Belmonte JC. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, Blasco MA, Serrano M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–2139. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 138.Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]