

Abstract
Previous genome-wide association studies (GWAS) have shown several risk alleles to be associated with breast cancer. However, the variants identified so far contribute to only a small proportion of disease risk. The objective of our GWAS was to identify additional novel breast cancer susceptibility variants and to replicate these findings in an independent cohort. We performed a two-stage association study in a cohort of 3,064 women from Alberta, Canada. In Stage I, we interrogated 906,600 single nucleotide polymorphisms (SNPs) on Affymetrix SNP 6.0 arrays using 348 breast cancer cases and 348 controls. We used single-locus association tests to determine statistical significance for the observed differences in allele frequencies between cases and controls. In Stage II, we attempted to replicate 35 significant markers identified in Stage I in an independent study of 1,153 cases and 1,215 controls. Genotyping of Stage II samples was done using Sequenom Mass-ARRAY iPlex platform. Six loci from four different gene regions (chromosomes 4, 5, 16 and 19) showed statistically significant differences between cases and controls in both Stage I and Stage II testing, and also in joint analysis. The identified variants were from EDNRA, ROPN1L, C16orf61 and ZNF577 gene regions. The presented joint analyses from the two-stage study design were not significant after genome-wide correction. The SNPs identified in this study may serve as potential candidate loci for breast cancer risk in a further replication study in Stage III from Alberta population or independent validation in Caucasian cohorts elsewhere.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-011-0973-1) contains supplementary material, which is available to authorized users.
Introduction
Breast cancer is a heterogeneous disease strongly influenced by genetic, environmental and life-style factors. Mutations in BRCA1 (Hall et al. 1990) and BRCA2 (Wooster et al. 1995) tumor suppressor genes confer familial breast cancer risk and account for the high penetrance alleles characterized thus far. Subsequently, certain genes of moderate penetrance such as ATM (Ahmed and Rahman 2006; Renwick et al. 2006), CHEK2 (Meijers-Heijboer et al. 2002) and PALB2 (Rahman et al. 2007) were shown to predispose to breast cancer susceptibility. However, these genes account only for a small proportion of genetic risk. Intensive research efforts to identify BRCA-like genes to explain the breast cancer risk in populations were not successful, thus invoking a polygenic model of disease susceptibility to explain the remaining genetic risk in non-familial or sporadic breast cancer cases (Pharoah et al. 2002; 2008; Smith et al. 2006). The polygenic model enables identification of several common genetic variants that each individually confers only modest risk effect to the disease.
Subsequent genome-wide association studies (GWAS) have identified several new risk alleles to be associated with breast cancer (Ahmed et al. 2009; Easton et al. 2007; Gold et al. 2008; Hunter et al. 2007; Murabito et al. 2007; Stacey et al. 2007, 2008; Thomas et al. 2009; Zheng et al. 2009), thus lending support to the polygenic model of disease susceptibility. Most of these studies were conducted in women of European ancestry with the exception of two studies which investigated the risk alleles in Chinese (Zheng et al. 2009) and Ashkenazi Jewish (Gold et al. 2008) populations. Nonetheless, it is of continuing importance to conduct GWAS over ethnically diverse populations including European ancestry to uncover the full spectrum of breast cancer susceptibility variants. Such studies are expected to show both unique variants and confirm previously reported variants.
We performed a two-stage association study on cohorts from Alberta, Canada to identify novel loci potentially associated with breast cancer susceptibility. A prior study from our group successfully validated several previously reported low-penetrance alleles in the same study population (data not shown). The object of our present GWAS was to identify novel risk loci, i.e., ones not previously reported in the literature (Ahmed et al. 2009; Easton et al. 2007; Gold et al. 2008; Hunter et al. 2007; Murabito et al. 2007; Stacey et al. 2007, 2008; Thomas et al. 2009; Zheng et al. 2009) and to replicate these findings in an independent cohort. Herein, we report six previously unreported loci significantly associated with breast cancer, replicated in one independent cohort.
Materials and methods
Study population
We accessed information about women in Alberta with confirmed diagnosis of breast cancer (cases) with no documented family history in the first and second degree relatives and clinicopathological information from the PolyomX project (PolyomX 2001) and Canadian Breast Cancer Foundation (CBCF) tumor bank (CBCF 2005), located at the Cross Cancer Institute, Edmonton, Alberta, Canada. The province of Alberta has centralized cancer registry, and all cancer patients receive treatment within the provincial health care services (Alberta Health Services, AHS). Patients with banked tissues gave prospective written approval for treatment information and life-long follow-up subject to ethics approval for the research studies and strict adherence to the health agency guidelines on privacy, confidentiality, security of patient personal information and other identifiers associated with banked specimens in the project database. The PolyomX project accrued tumor and matching buffy coat samples for breast and other cancer types during the years 2001–2005 from four regional hospitals in Edmonton. The CBCF tumor bank was initiated in 2005 in Edmonton and Calgary to bank tumor specimens and to serve as an open source bank to provide access to samples and associated clinical information to cancer researchers in Canada.
The histological subtypes of breast cancer subjects were predominantly invasive ductal carcinomas, non-metastatic at presentation, with a median age at diagnosis of 53 years. Control subjects were age- and gender-matched healthy women selected from the same geographic region (Alberta, Canada) and were free of cancer at the time of recruitment for the study, with no documented family history of breast cancer in the first and second degree relatives. Previous studies addressed Stage I of the whole genome polymorphisms scans with emphasis to positive family history of breast cancer, ethnic diversity (Chinese or Ashkenazi Jewish populations) or cases with breast cancer in post-menopausal women (Easton et al. 2007; Gold et al. 2008; Hunter et al. 2007; Zheng et al. 2009). Controls were obtained from an AHS cohort study recruiting up to 50,000 Albertans (age group 35–69 years) willing to provide extensive health and life-style questionnaires and DNA as part of the “Tomorrow Project” (Tomorrow Project 2001). We accessed a subset of these individuals (samples banked between 2002 and 2008) who consented to participate in the Tomorrow Project, donated blood and provided detailed family history of breast cancer. Informed consent was obtained from each participant included in the research project, and the study was approved by the institutional research ethics board. Stage I of this study evaluated 348 breast cancer cases and 348 controls; and Stage II studied a completely independent group comprising 1,153 cases and 1,215 controls. Cases and controls were predominantly of Caucasian origin, determined based on the self-completed ethnicity questionnaires. The blood or buffy coat samples were retrieved from the PolyomX and Tomorrow projects, and genomic DNA was extracted for each sample using commercially available Qiagen™ (Mississauga, Ontario, Canada) DNA isolation kits. We quantified isolated genomic DNA using NanoDrop 2000 spectrophotometer (Wilmington, DE, USA), and we adhered to the good practices for sample quality as recommended by Affymetrix genotyping protocols.
Genotyping and quality control measures
For Stage I, 348 breast cancer cases and 348 controls were genotyped using the Affymetrix genome-wide Human SNP Array 6.0, which features 906,600 SNP probes with each probe represented 4–6 times on the array. Sample processing was performed following the protocols provided by Affymetrix. After labeling of DNA, hybridization and washing steps, the arrays were scanned using the GeneChip® Scanner 3000 7G (Santa Clara, CA, USA). Genotype data were acquired by genotype calling of samples in batches of 96 using a default genotyping algorithm (Birdseed v2) provided by Affymetrix, as per the recommended guidelines. For Stage II, 1,153 breast cancer cases and 1,215 controls were genotyped using Sequenom Mass-ARRAY iPlex technology (Gabriel et al. 2009). Genotyping services were provided by Genome Quebec Innovation Centre (Montreal, QC, Canada). The Stage I samples were then re-genotyped on the Sequenom platform to evaluate the genotype concordance between the platforms, prior to replication of select markers in an independent cohort (Stage II).
We applied the following filters to the genotype data prior to the association analysis to minimize false-positive associations, which helps to increase the overall power of the study:
Individual chip call rate Affymetrix recommends the chip call rate threshold of >86% for SNP 6.0 arrays. The sample call rates for the 696 samples were: two samples in the interval (89, 90], 39 in the interval (90, 95], 480 in the interval (95, 98], and 175 in the interval (98, 100]. In addition, quality for each sample was determined by contrast quality control (CQC) as recommended for the SNP Array 6.0 by the manufacturers. CQC is a cluster-based algorithm that is a good predictor of sample genotyping performance. Intensity of each spot on the array following hybridization, washing and scanning along with clustering of data (based on genotype calls of homozygous wild type, heterozygotes and variant homozygous) was assessed. Default average CQC for a sample to be included in further analysis was set at ≥1.7. Most of our samples used in this study had a CQC of >2.0.
Deviation from Hardy–Weinberg equilibrium (HWE) We assessed for deviations from HWE using χ2 test, with 1 degree of freedom (df). Significant deviations were observed for 30,636 SNPs (3.38% of the 906,600 SNPs) in controls at p < 0.001 (user-defined stringent cut-off). We excluded these SNPs from our association analysis.
SNP call rate Failure to assign genotypes for certain SNPs in a sample affects the completeness of the data, the association test results and the replication of the findings. Different studies to date had adopted different cut-off points ranging from 80 to 99.7% (Ahmed et al. 2009; Easton et al. 2007; Gold et al. 2008; Hunter et al. 2007; Stacey et al. 2007, 2008; Thomas et al. 2009; Turnbull et al. 2010; Zheng et al. 2009). We adopted a stringent call rate cut-off of ≥99%, which meant eliminating a total of 93,126 SNPs (10.3% of the 906,600 SNPs).
Concordance of genotype calls (a) Within Affymetrix Batch effects of the Affymetrix Birdseed v2 genotype calling algorithm were assessed by grouping the samples (348 cases and 348 controls) into batches (7 × 96 plus 24 in the eighth), with 96 samples in each batch. To assess genotype call concordance across batches, we included randomly selected raw data from the first seven batches (47 cases and 25 controls) to the batch eight to bring the sample size to 96. The mean genotype concordance rate for the samples achieved in this analysis was very high (>99.9%). (b) Affymetrix versus Sequenom The SNPs that were statistically significant (p < 0.001) in Stage I on Affymetrix platform and those that were selected for replication were re-genotyped in 647 samples (326 cases and 321 controls) on Sequenom Mass-ARRAY iPlex platform. We consistently observed high mean genotype concordance rate of >95% between these two genotyping platforms. (c) Within Sequenom To assess the genotype concordance within Sequenom platform, 132 replicate samples (67 cases and 65 controls) were randomly distributed in each of the 96-well plate assay. The mean genotype concordance rate of the replicates was again high at >98.6%. We have also analyzed the sample call rates for the 2,368 Stage II samples (replication study) within Sequenom and found that 26 samples were in the interval (80, 90], 67 in the interval (90, 95], 394 in the interval (95, 98] and 1,881 in the interval (98, 100].
Assessment of population stratification We explored the genetic relatedness of our case–control cohorts prior to independent validation of loci from GWAS reported here. Detection and removal of outliers (population stratification) is important to minimize false-positive findings. To assess the substructure, we applied the principal component analysis (PCA)-based EIGENSTRAT method (Price et al. 2006) embedded in our statistical software (HelixTree™) used for the data analysis reported here. Using a conservative threshold of ≥3 standard deviations away from the mean on one of the two principal components, we detected a total of 73 samples (46 cases and 27 controls) as outliers. The detected outliers were removed from our dataset leaving with 302 cases and 321 controls for further scrutiny.
Statistical analysis
We calculated the power assuming an additive model of genetic inheritance, a minor allele frequency (MAF) of 0.1, genotype relative risk of 1.2 (based on odds ratio (OR) and allele frequency estimates from previous GWAS for Caucasian population), alpha of 0.05 and determined that our study cohort has more than 80% power to detect associations (Klein 2007).
The statistical analysis was conducted using commercially available software, HelixTree™. Association analysis was carried out using the case versus control status as the binary variable. After filtering out SNPs and samples (PCA outliers, deviations from HWE and missing values), a total of 782,838 SNPs and 302 cases/321 controls were included in the analysis for Stage I of the study. A χ2 test with 1 df was carried out to determine the allele frequency differences between cases and controls for all the datasets reported here. Unconditional logistic regression analysis was used to estimate the OR and 95% confidence intervals (CI). A genome-wide correction to correct for multiple marker testing was applied (Bonferroni method: p < 6.4 × 10−8 calculated from nominal p value of 0.05/total number of markers as 782,838).
Whole genome association analysis from Stage I enabled us to identify subset of markers with nominal p values (<0.05) that showed association with the disease trait. As this subset of markers may also include several false associations, we attempted to replicate these primary findings by testing them on an independent cohort with higher sample sizes than in Stage I. We selected the markers for replication from Stage I in a systematic manner proposed by Zheng et al. (2009). Of the 35,859 markers from Stage I that showed association with breast cancer risk (p < 0.05) and showed conformity with the HWE criteria, we selected the ones with p < 0.001, MAF > 0.1, distinct genotype clusters and novel markers that were not previously identified in other GWAS. We identified haplotype blocks using a feature available in the software, using the default parameters—maximum length of 160 kb and maximum of 30 markers per block. A haplotype association analysis was performed in a case–control setting to improve statistical power. We selected the markers that showed statistical significance (p < 0.001) in both (1) allelic and haplotype association analyses, with each identified block containing more than two SNPs and (2) chose representative SNPs with r 2 ≥ 0.8. After applying these selection criteria, we selected 35 SNPs for replication in Stage II with a completely independent series of 1,153 cases and 1,215 controls. Association analysis and tests of significance were carried out independently for Stage II and in combined samples from Stages I and II (potential combined sample size of 2,991 from cases and controls left for association analysis following data filtering criteria described above).
Results
GWAS in 348 cases and 348 controls (Stage I)
Allelic association analysis with 782,838 SNPs showed statistically significant (p < 0.05) differences between the cases and the controls at multiple genomic locations (35,859 SNPs) scattered across all chromosomes (Fig. 1). We explored multi-dimensional scaling using PCA-based method. Case and control samples of this study showed significant overlap with the Central European population cluster of HapMap samples on PCA plots (Supplementary Figure S1). It indicates that our predominantly Caucasian population has (1) high genetic similarity with the European population when compared with the Asian or Yoruba Indians (African) and (2) the cases and the controls from the Alberta region showed near genetic homogeneity, i.e., both appear to be of eastern European ancestry. Quantile–quantile plot showed that most of the observed associations lie along the line of best fit (expected distribution) conforming to the null hypothesis of no association for majority of the SNPs (Supplementary Figure S2).
Fig. 1.
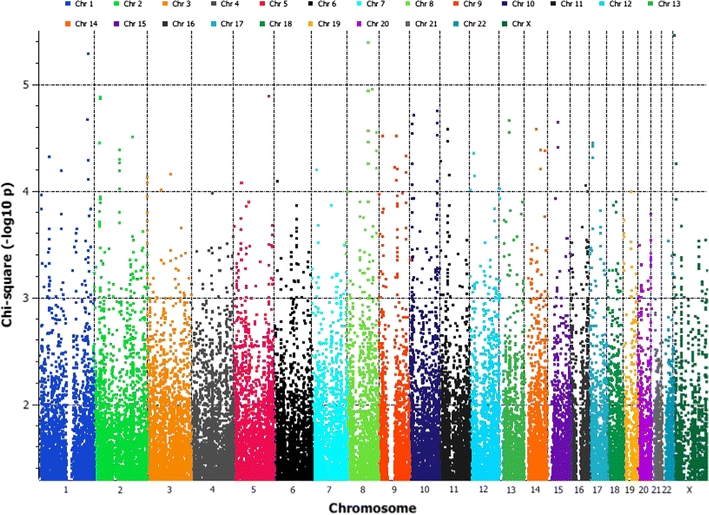
Manhattan plot for Stage I association study showing 35,589 markers (p < 0.05) distributed across chromosomes. This graph is plotted against allelic χ2 p values on –log10 scale to indicate polygenic nature of breast cancer susceptibility
Replication of markers from Stage I in independent study (Stage II)
In Stage II, we genotyped 35 SNPs using Sequenom Mass-ARRAY iPlex technology in independent case and control subjects (1,153 cases and 1,215 controls). We also performed a joint analysis which is considered the best way to confer power and confidence in the results, as well as to address the possible sampling bias and inherent heterogeneity of breast cancer as a phenotype (Skol et al. 2006). The joint analysis consisted of a total of 1,455 breast cancer cases and 1,536 controls obtained by combining the samples from the two stages of the study. Of the 35 SNPs considered for replication, 6 SNPs showed statistical significance in all stages (Stages I and II) and in joint analysis (Table 1). The data from the remaining 29 SNPs are summarized in Supplementary Table S1. Data summarized in Table 1 also lists the OR, 95% CI, false discovery rate (FDR), SNP call rate and MAF for the six significant polymorphisms described above.
Table 1.
Six novel loci showing consistent association with breast cancer in both stages of the study
| dbSNP rs# | Polymorphism | Chr | Associated gene | Relative location | Stage I χ 2 p value | Stage II χ2 p value | Joint analysis | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| χ 2 p value | χ2 FDR | OR (95% CI) | CR | MA | MAF | Cases | Controls | |||||||||||||
| AA | AB | BB | n | AA | AB | BB | n | |||||||||||||
| rs1092913 | C>T | 5p15.2 | ROPN1L |
2.5 kb Downstream |
7.00E−04 | 2.17E−04 | 1.89E−06 | 3.30E−05 |
1.45 (1.24, 1.69) |
0.99 | T | 0.13 | 1,054 | 333 | 48 | 1,435 | 1,228 | 275 | 28 | 1,531 |
| rs10411161 | C>T | 19q13.33 | ZNF577 | 3′ UTR | 1.08E−03 | 6.16E−04 | 7.09E−06 | 8.27E−05 |
1.42 (1.22, 1.65) |
0.99 | T | 0.13 | 1,068 | 306 | 64 | 1,438 | 1,232 | 251 | 45 | 1,528 |
| rs3848562 | C>T | 19q13.33 | ZNF577 | Intron | 8.01E−04 | 9.78E−04 | 9.23E−06 | 8.08E−05 |
1.42 (1.22, 1.66) |
1.00 | C | 0.12 | 1,078 | 336 | 38 | 1,452 | 1,238 | 274 | 23 | 1,535 |
| rs11878583 | C>T | 19q13.33 | ZNF577 | Intron | 1.25E−03 | 7.59E−03 | 1.35E−04 | 9.45E−04 |
1.35 (1.16, 1.57) |
1.00 | C | 0.13 | 1,077 | 332 | 42 | 1,451 | 1,214 | 301 | 19 | 1,534 |
| rs1429142 | C>T | 4q31.23 | EDNRA |
112.5 kb Upstream |
2.80E−03 | 1.28E−02 | 3.59E−04 | 2.10E−03 |
1.27 (1.11, 1.45) |
1.00 | C | 0.18 | 926 | 466 | 57 | 1,449 | 1,074 | 417 | 44 | 1,535 |
| rs1981867 | A>G | 16q23.2 | C16orf61 |
85.9 kb Downstream |
3.70E−04 | 3.17E−02 | 4.32E−04 | 2.16E−03 |
1.22 (1.09, 1.36) |
1.00 | A | 0.31 | 664 | 611 | 171 | 1,446 | 769 | 651 | 116 | 1,536 |
Chr chromosome, UTR untranslated region, χ 2 chi-square, FDR false discovery rate, OR odds ratio, CI confidence interval, CR call rate, MA minor allele, MAF minor allele frequency; genotypes: AA wild-type homozygous, AB heterozygous, BB variant homozygous, n sample size
The polymorphisms with high significance (10−6–10−4) in joint analysis and conferring risk for breast cancer are in chromosomes 4, 5, 16 and 19. Of these, rs1092913 is located on chromosome 5p15.2 [p value 1.89 × 10−6, FDR 3.30 × 10−5, OR (95% CI) 1.45 (1.24–1.69)], with the ropporin-1-like (ROPN1L) gene present 2.5 kb downstream of the polymorphism; the three SNPs present on chromosome 19q13.33, ZNF577 (zinc finger protein 577) gene are (1) rs10411161 (p value 7.09 × 10−6, FDR 8.27 × 10−5) located in the 3′ untranslated region (UTR; 2.8 kb downstream of the stop codon, Goldenpath-hg 18/db SNP build 130), (2) rs3848562 (p value 9.23 × 10−6, FDR 8.08 × 10−5) and (3) rs11878583 (p value 1.35 × 10−4, FDR 9.45 × 10−4) located in the introns 6 and 2, respectively. We observed ORs (95% CI) of 1.42 (1.22–1.65), 1.42 (1.22–1.66) and 1.35 (1.16–1.57), respectively, for the three ZNF577 SNPs. However, rs10411161 showed deviation from HWE in the Stage II sample set and in the joint analysis for both cases and controls. We ruled out the obvious possibility of genotyping errors, and both Stage I and Stage II samples (with several replicates) showed good concordance within and across the genotyping platforms (see “Materials and methods”). The fifth SNP, rs1429142, is located on chromosome 4q31.23 (p value 3.59 × 10−4, FDR 2.10 × 10−3), with EDNRA (endothelin receptor type A) gene present approximately 112.5 kb downstream of the polymorphism. An OR of 1.27 (95% CI 1.11–1.45) was noted for the minor allele C. Finally, rs1981867 located on chromosome 16q23.2 showed satisfactory statistical significance in Stage I (p value 3.7 × 10−4) and in joint analysis (p value 4.32 × 10−4, FDR 2.16 × 10−3) but showed only marginal significance in Stage II (p value 0.03). An OR of 1.22 (95% CI 1.09–1.36) for the minor allele A was noted.
Discussion
Increasingly, assessing genetic risk in complex traits requires identification of multiple loci conferring risk and/or mining of the data in an integrated manner to identify potential gene–gene interactions that together may explain a higher proportion of risk than the single-locus analysis (Park et al. 2010; Yang et al. 2010). This approach calls for identification of potential novel risk-associated SNPs and their subsequent validation to improve the accuracy of genetic risk assessment models. We performed a whole genome analysis to identify novel markers (single-locus analysis) associated with breast cancer and confirmed several new loci in a larger, independent replication set of cases and controls. None of these six SNPs were significant after genome-wide correction in individual stages or in the joint analysis from the two-stage association study presented here.
The conduct of our study was appropriate, in that the sample size used in our Stage I GWAS (348 cases and 348 controls) closely matched those used in Stage I of earlier studies, e.g., 390 familial breast cancer cases and 364 controls used by Easton et al. (2007) and 249 Ashkenazi Jew familial cases and 299 controls used by Gold et al. (2008). Furthermore, this was only the second study, after Zheng et al. (2009), to use high-density Affymetrix SNP arrays (906,600 SNPs/array), which provides a vast physical coverage of the genome in an unbiased manner, to identify the markers associated with disease susceptibility. A precedent exists for using high-density SNP arrays to identify breast cancer susceptibility loci and to subsequently replicate those identified variants in large cohorts (Ahmed et al. 2009; Easton et al. 2007; Gold et al. 2008; Hunter et al. 2007; Murabito et al. 2007; Stacey et al. 2007, 2008; Thomas et al. 2009; Turnbull et al. 2010; Zheng et al. 2009). The SNPs identified, so far, in GWAS including our study were largely surrogate markers; fine mapping and independent validation studies are underway to identify causal variants.
Using the cohorts described here, we successfully validated the FGFR2 polymorphisms (data not shown), which were also highly reproducible in several cohorts and disease models (Raskin et al. 2008; Rebbeck et al. 2009; Thomas et al. 2009; Turnbull et al. 2010; Zheng et al. 2009). In this study, we report six putative candidate loci, and further large-scale studies are required to confirm their association with breast cancer. These include SNP rs1981867 in the open reading frame on chromosome 16 (C16orf61), a gene that has been shown to be associated with multi-drug resistance (Campone et al. 2008). Easton et al. (2007) and Stacey et al. (2007) previously reported that rs3803662 positioned on chromosome 16q is associated with breast cancer risk in two independent GWAS. The identified SNP rs1981867 in this study from chromosome 16 further emphasizes the importance of this region in breast cancer. While these results and interpretations require large-scale studies and independent confirmation, repeated and independent observations by several research groups suggesting breast cancer risk related to these open reading frames underscores the importance of this region. Functional characterization of these variants is warranted.
Zinc finger proteins are commonly involved in transcriptional regulation of genes. Tan et al. (2004b) have shown that C-terminal transcriptional repression domain of zinc finger protein ZBRK1 interacts with BRCA1 tumor suppressor gene to repress transcription. Previous linkage studies have shown that mutations in BRCA1 gene are a common event in early-onset, multiple-case breast cancer families (Hall et al. 1990). The C-terminal extension of ZNF577 shares sequence homology with ZBRK1 (Tan et al. 2004a). It remains to be determined whether ZNF577 identified in our study also plays a role in transcriptional repression by binding to the BRCA1 protein. We found three SNPs from ZNF577 gene region to be associated with disease susceptibility: rs10411161 (only SNP in the replication stage that showed deviation from HWE) found 2.8 kb downstream of the gene in the 3′ UTR, and the other two markers rs3848562 and rs11878583 are present in the introns 2 and 6, respectively. The reasons for the deviation of the 3′ UTR SNP in ZNF577 gene require further scrutiny and validation from independent studies. Similarly, a recent GWAS has shown a polymorphism rs10995190 located within the intron 4 of zinc finger protein 365 (ZNF365) to be associated with breast cancer susceptibility (Turnbull et al. 2010). Further studies are required to understand the functional role of ZNF577.
The endothelin receptor type A (EDNRA) gene is located 112.5 kb upstream of the polymorphism rs1981867, which we identified in our association study. Its role in breast cancer susceptibility requires further attention. Interestingly, constitutive co-expression of endothelin-1 growth factor and EDNRA often results in ovarian carcinoma (Salani et al. 2000) and also contributes to bone metastases in different primary tumors (Medinger et al. 2003).
The ROPN1L gene on chromosome 5p15.2 is present 2.5 kb downstream of the polymorphism rs1092913. There is no previous evidence on the association of the gene to breast cancer susceptibility. The ROPN1L gene encodes for a sperm protein known to interact with A-kinase anchoring protein (GeneCards 2010). It is evident from previous GWAS that the p arm of chromosome 5 harbors several polymorphisms implicated in breast cancer susceptibility (Stacey et al. 2008; Thomas et al. 2009). Moreover, Lowe et al. (2007) showed that ROPN1L gene is highly expressed in pancreatic cancer when compared to the normal pancreatic tissues and other tumors in their dataset. Again, our results motivate further investigation in this gene.
Conclusion
We report six candidate polymorphisms that were not previously associated with breast cancer risk in Caucasian population from Alberta. These findings merit further replication and, if confirmed, warrant fine mapping and functional validation studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Funding support for this project was provided by Alberta Cancer Research Institute (ACRI), Alberta Cancer Board (ACB) operating grants to SD, and an operating grant from Canadian Breast Cancer Foundation (CBCF) to SD and JM. Support from Canadian Partnership Against Cancer (PR), Natural Sciences and Engineering Research Council (NSERC) and the Alberta Ingenuity Centre for Machine Learning is acknowledged (RG). We thank Yadav Sapkota, Kathryn Calder, Adrian Driga, Jennifer Dufour, Diana Carandang, and Lillian Cook for assistance and technical help. PolyomX project and CBCF tumor bank received funding from Alberta Cancer Foundation and CBCF, respectively. We also extend our sincere thanks to the anonymous reviewers for their suggestions.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
B. Sehrawat and M. Sridharan contributed equally to the work.
References
- Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25:5906–5911. doi: 10.1038/sj.onc.1209873. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, Platte R, Morrison J, Maranian M, Pooley KA, Luben R, Eccles D, Evans DG, Fletcher O, Johnson N, dos Santos Silva I, Peto J, Stratton MR, Rahman N, Jacobs K, Prentice R, Anderson GL, Rajkovic A, Curb JD, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Diver WR, Bojesen S, Nordestgaard BG, Flyger H, Dork T, Schurmann P, Hillemanns P, Karstens JH, Bogdanova NV, Antonenkova NN, Zalutsky IV, Bermisheva M, Fedorova S, Khusnutdinova E, Kang D, Yoo KY, Noh DY, Ahn SH, Devilee P, van Asperen CJ, Tollenaar RA, Seynaeve C, Garcia-Closas M, Lissowska J, Brinton L, Peplonska B, Nevanlinna H, Heikkinen T, Aittomaki K, Blomqvist C, Hopper JL, Southey MC, Smith L, Spurdle AB, Schmidt MK, Broeks A, van Hien RR, Cornelissen S, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Schmutzler RK, Burwinkel B, Bartram CR, Meindl A, Brauch H, Justenhoven C, Hamann U, Chang-Claude J, Hein R, Wang-Gohrke S, Lindblom A, Margolin S, Mannermaa A, Kosma VM, Kataja V, Olson JE, Wang X, Fredericksen Z, Giles GG, Severi G, Baglietto L, English DR, Hankinson SE, Cox DG, Kraft P, Vatten LJ, Hveem K, Kumle M, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet. 2009;41:585–590. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campone M, Campion L, Roche H, Gouraud W, Charbonnel C, Magrangeas F, Minvielle S, Geneve J, Martin AL, Bataille R, Jezequel P. Prediction of metastatic relapse in node-positive breast cancer: establishment of a clinicogenomic model after FEC100 adjuvant regimen. Breast Cancer Res Treat. 2008;109:491–501. doi: 10.1007/s10549-007-9673-x. [DOI] [PubMed] [Google Scholar]
- CBCF (2005) http://www.abtumorbank.com/?about
- Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel S, Ziaugra L, Tabbaa D (2009) SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet Chapter 2:Unit 2.12 [DOI] [PubMed]
- GeneCards (2010) http://www.genecards.org/. Accessed 5 May 2010
- Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB, Gregersen P, Kosarin K, Olsh A, Bergeron J, Ellis NA, Klein RJ, Clark AG, Norton L, Dean M, Boyd J, Offit K. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci USA. 2008;105:4340–4345. doi: 10.1073/pnas.0800441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ. Power analysis for genome-wide association studies. BMC Genet. 2007;8:58. doi: 10.1186/1471-2156-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe AW, Olsen M, Hao Y, Lee SP, Taek Lee K, Chen X, van de Rijn M, Brown PO. Gene expression patterns in pancreatic tumors, cells and tissues. PLoS One. 2007;2:e323. doi: 10.1371/journal.pone.0000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medinger M, Adler CP, Schmidt-Gersbach C, Soltau J, Droll A, Unger C, Drevs J. Angiogenesis and the ET-1/ETA receptor system: immunohistochemical expression analysis in bone metastases from patients with different primary tumors. Angiogenesis. 2003;6:225–231. doi: 10.1023/B:AGEN.0000021395.43438.44. [DOI] [PubMed] [Google Scholar]
- Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- Murabito JM, Rosenberg CL, Finger D, Kreger BE, Levy D, Splansky GL, Antman K, Hwang SJ (2007) A genome-wide association study of breast and prostate cancer in the NHLBI’s Framingham Heart Study. BMC Med Genet 8(Suppl 1):S6 [DOI] [PMC free article] [PubMed]
- Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet. 2002;31:33–36. doi: 10.1038/ng853. [DOI] [PubMed] [Google Scholar]
- Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med. 2008;358:2796–2803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- PolyomX (2001) http://www.abtumorbank.com/?about
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin L, Pinchev M, Arad C, Lejbkowicz F, Tamir A, Rennert HS, Rennert G, Gruber SB. FGFR2 is a breast cancer susceptibility gene in Jewish and Arab Israeli populations. Cancer Epidemiol Biomarkers Prev. 2008;17:1060–1065. doi: 10.1158/1055-9965.EPI-08-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck TR, DeMichele A, Tran TV, Panossian S, Bunin GR, Troxel AB, Strom BL. Hormone-dependent effects of FGFR2 and MAP3K1 in breast cancer susceptibility in a population-based sample of post-menopausal African-American and European-American women. Carcinogenesis. 2009;30:269–274. doi: 10.1093/carcin/bgn247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- Salani D, Di Castro V, Nicotra MR, Rosano L, Tecce R, Venuti A, Natali PG, Bagnato A. Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol. 2000;157:1537–1547. doi: 10.1016/S0002-9440(10)64791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- Smith P, McGuffog L, Easton DF, Mann GJ, Pupo GM, Newman B, Chenevix-Trench G, Szabo C, Southey M, Renard H, Odefrey F, Lynch H, Stoppa-Lyonnet D, Couch F, Hopper JL, Giles GG, McCredie MR, Buys S, Andrulis I, Senie R, Goldgar DE, Oldenburg R, Kroeze-Jansema K, Kraan J, Meijers-Heijboer H, Klijn JG, van Asperen C, van Leeuwen I, Vasen HF, Cornelisse CJ, Devilee P, Baskcomb L, Seal S, Barfoot R, Mangion J, Hall A, Edkins S, Rapley E, Wooster R, Chang-Claude J, Eccles D, Evans DG, Futreal PA, Nathanson KL, Weber BL, Rahman N, Stratton MR. A genome wide linkage search for breast cancer susceptibility genes. Genes Chromosomes Cancer. 2006;45:646–655. doi: 10.1002/gcc.20354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Tres A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F, Gudbjartsson D, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Holst S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39:865–869. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, Jakobsdottir M, Bergthorsson JT, Gudmundsson J, Aben KK, Strobbe LJ, Swinkels DW, van Engelenburg KC, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Saez B, Lambea J, Godino J, Polo E, Tres A, Picelli S, Rantala J, Margolin S, Jonsson T, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Sveinsdottir SG, Alexiusdottir K, Saemundsdottir J, Sigurdsson A, Kostic J, Gudmundsson L, Kristjansson K, Masson G, Fackenthal JD, Adebamowo C, Ogundiran T, Olopade OI, Haiman CA, Lindblom A, Mayordomo JI, Kiemeney LA, Gulcher JR, Rafnar T, Thorsteinsdottir U, Johannsson OT, Kong A, Stefansson K. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2008;40:703–706. doi: 10.1038/ng.131. [DOI] [PubMed] [Google Scholar]
- Tan W, Kim S, Boyer TG. Tetrameric oligomerization mediates transcriptional repression by the BRCA1-dependent Kruppel-associated box-zinc finger protein ZBRK1. J Biol Chem. 2004;279:55153–55160. doi: 10.1074/jbc.M410926200. [DOI] [PubMed] [Google Scholar]
- Tan W, Zheng L, Lee WH, Boyer TG. Functional dissection of transcription factor ZBRK1 reveals zinc fingers with dual roles in DNA-binding and BRCA1-dependent transcriptional repression. J Biol Chem. 2004;279:6576–6587. doi: 10.1074/jbc.M312270200. [DOI] [PubMed] [Google Scholar]
- Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K, Chatterjee N, Garcia-Closas M, Gonzalez-Bosquet J, Prokunina-Olsson L, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Diver R, Prentice R, Jackson R, Kooperberg C, Chlebowski R, Lissowska J, Peplonska B, Brinton LA, Sigurdson A, Doody M, Bhatti P, Alexander BH, Buring J, Lee IM, Vatten LJ, Hveem K, Kumle M, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Chanock SJ, Hunter DJ. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomorrow Project (2001) http://www.albertahealthservices.ca/1476.asp
- Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, Seal S, Ghoussaini M, Hines S, Healey CS, Hughes D, Warren-Perry M, Tapper W, Eccles D, Evans DG, Hooning M, Schutte M, van den Ouweland A, Houlston R, Ross G, Langford C, Pharoah PD, Stratton MR, Dunning AM, Rahman N, Easton DF. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, Goddard ME, Visscher PM. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Long J, Gao YT, Li C, Zheng Y, Xiang YB, Wen W, Levy S, Deming SL, Haines JL, Gu K, Fair AM, Cai Q, Lu W, Shu XO. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–328. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.