

Abstract
Background:
Raloxifene, a second-generation selective estrogen receptor modulator (SERM) used to prevent osteoporosis in postmenopausal women is administered orally in the form of a tablet. The absolute bioavailability of the drug is only 2% because of extensive hepatic first-pass metabolism. Lipid-based formulations are reported to reduce the first-pass metabolism by promoting its lymphatic uptake.
Materials and Methods:
In the present investigation, microemulsion and Self-Microemulsifying Drug Delivery System (SMEDDS) formulations of Raloxifene were prepared. The prepared formulations were characterized for drug loading, size, transparency, zeta potential, Transmission Electron Microscopy (TEM) and in vitro intestinal permeability.
Results:
The results indicated that high drug loading, optimum size and desired zeta potential and transparency could be achieved with both SMEDDS and microemulsion. The TEM studies indicated the absence of aggregation with both the systems. The in vitro intestinal permeability results showed that the permeation of the drug from the microemulsion and SMEDDs was significantly higher than that obtained from the drug dispersion and marketed formulation.
Conclusion:
Lipid based formulations such as microemulsion and Self Microemulsifying drug delivery systems are expected to increase the oral bioavailability as evidenced by the increased intestinal permeation.
Keywords: Bioavailability enhancement, microemulsion, raloxifene, Self Microemulsifying Drug Delivery System, SMEDDS
Delivery of active pharmaceutical ingredients through the oral route is the most preferred and convenient mode of drug administration. However, it has certain limitations, the most important being the poor bioavailability of certain drugs. In spite of showing excellent pharmacological action in vitro, certain drugs show poor absorption from the gastrointestinal tract into the systemic circulation resulting in low bioavailability and hence poor pharmacological effect in vivo. There are various physiological as well as pharmaceutical factors which affect the oral absorption as well as bioavailability of the drugs. The reasons which contribute to poor oral bioavailability include low aqueous solubility, inappropriate partition coefficient, degradation of the drug in the gastrointestinal tract, first-pass metabolism and P-Glycoprotein (Pgp) efflux of certain drugs. High first-pass hepatic metabolism is the major contributory factor responsible for inactivation of the drug before it reaches the systemic circulation. Drugs administered orally are absorbed in the systemic circulation either via portal circulation or lymphatic transport. Drugs which are transported via portal circulation and which are susceptible to the enzymes present in the liver undergo first pass metabolism. Drugs having low solubility (falling in Biopharmaceutical Classification System (BCS) Class II and IV) are particularly susceptible to first-pass metabolism.[1] (For example, carvedilol, cyclosporine, glypizide, indinavir, tacrolimus, amphotericin).[1] This kind of drugs have to be administered by alternative routes such as parenteral which have very less patient compliance. Thus, oral bioavailability improvement is the most rational approach in efficient and effective drug delivery.
Reduction in the first-pass metabolism can lead to enhancement of oral bioavailability of a drug. This can be accomplished by co-administration of the drug with enzyme inhibitors, pro-drug approach, and use of a novel lipid-based drug delivery system such as microemulsion,[2] SMEDDS,[3] and lipid nanoparticles.[4] Although the exact mechanisms responsible for this enhanced absorption are not fully known, the possible reasons for the enhancement of bioavailability are as follows:[2,5]
Enhanced dissolution/solubilization- Increase in pancreatic and gall bladder secretions which helps in solubilization of drugs. Moreover, the exogenous surface active agent used in the preparation of the formulation facilitates the solubilization.
Prolongation of gastric residence time- Lipid in the gastric tract leads to a delay in gastric emptying.
Stimulation of lymphatic transport- Due to the triglyceride core of chylomicrons.[4]
Increased intestinal permeability- Lipid formulations will convert into fine emulsion droplets having better permeability. Moreover, exogenous surfactants facilitate absorption.
Reduced metabolism due to lymphatic transport.
Microemulsion is ‘a system of water, oil and amphiphile which is a single optically isotropic and thermodynamically stable liquid solution.’[6] Advantage of microemulsion over other formulations is that it is easy to prepare and scale up, gives almost 100% entrapment of drug, and because of thermodynamic stability remains stable over a long period of time.
SMEDDS is an isotropic mixture of oil, surfactant and co-surfactant, and drug substance; microemulsion can be generated rapidly upon gentle mixing with water or aqueous media.[7] Generally for oral preparation microemulsion preconcentrate (SMEDDS) is preferred over the regular microemulsion system because it will decrease the volume so it will become easy to fill in the soft gelatin capsule.[7] Examples of commercialized SMEDDS formulations include cyclosporine (Neoral®), ritonavir (Norvir®), and saquinavir (Fortovase®).
Raloxifene is a second-generation SERM used to prevent osteoporosis in postmenopausal women. It is administered in the form of a tablet containing 60 mg, administered as a single daily dose. Raloxifene is rapidly absorbed from the gastrointestinal tract and approximately 60% of an oral dose is absorbed, however, because of extensive presystemic glucuronide conjugation, absolute bioavailability is only 2%[8] and the metabolite is excreted primarily in the feces. Enhancement of the bioavailability of Raloxifene can reduce the dose required to elicit the same pharmacological action and hence reduce the side-effects associated with the drug. Thus, in the present investigation, microemulsion and SMEDDs of Raloxifene were prepared and characterized.
Materials and Methods
Materials
Raloxifene was obtained as a gift sample from Aarti Drugs Limited (Mumbai, India). Capmul MCM C8, Capmul MCM, Acconon CC6 was gifted by Abitec Corporation Limited (Columbus, Ohio). Labrafil M 1944 CS, Labrafac CC, Transcutol HP were gifted by Colorcon Asia Private Ltd. (Goa, India). Akrysol K-140 was gifted by Corel Pharma Chem. (Ahmedabad, India). Tween-20, Tween-60, Tween-80, Polyethylene glycol (PEG)- 200 and 400 were purchased from Hi-media Ltd. (India).
Methods
Determination of solubility of drug in Oil, Surfactant and Co-Surfactant: Selection of components was done based on solubility of drug as well as USFDA approval status of components.
Solubility of Raloxifene was determined in different oils (such as Capmul MCM, Capmul MCM C8, Labrafac CC, Labrafil M1944 CS, Oleic acid, Isopropyl myristate, soya bean oil, castor oil), surfactants (such as Tween-20, Tween-60, Tween-80, Acconon cc6, Akrysol K-140) and co-surfactants (such as PEG-200, PEG-400, propylene glycol, Transcutol HP). Raloxifene is added in excess to different oils, surfactants and co-surfactants and stirred for 24 h on magnetic stirrer. After stirring for 24 h, samples were centrifuged at 7500 rpm for 10 min and the drug in the supernatant was determined after proper dilution with methanol by Ultraviolet-visible spectrophotometric method
Optimization of surfactant: Co-surfactant ratio by phase diagram preparation
For construction of pseudoternary phase diagrams water titration method was used because this method is easy and scalable. In this study, microemulsions were prepared to find the area of a particular component system.
In this method surfactant was blended with co-surfactant in fixed weight ratios (1:1, 2:1, 3:1, and 4:1), as from a review of the literature it was found that at SA/Cos (0.5/1) stable microemulsion formation is not possible. Aliquots of each surfactant and co-surfactant mixture (Smix) were then mixed with oil at ambient temperature. For each phase diagram, the ratio of oil to the Smix was varied as 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9(v/v). Water was added drop-wise to each oil-Smix mixture under vigorous stirring. After equilibrium, the samples were visually checked and determined as being clear microemulsions or emulsions or gels. No heating is conducted during the preparation. Phase diagrams were prepared using chemix software (Chemix Version 3.51).
Optimization of concentration of oil and surfactant-co-surfactant mixture (Smix)
To optimize the concentration of oil and Smix, a total of 16 batches were prepared for the microemulsion system and 10 batches for SMEDDS, the composition of which is shown in Tables 1 and 2.
Table 1.
Optimization of formulation variables for microemulsion
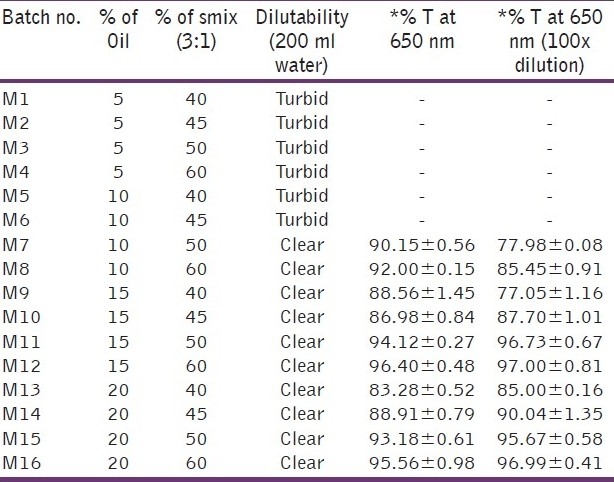
Table 2.
Optimization of formulation variables for SMEDDS
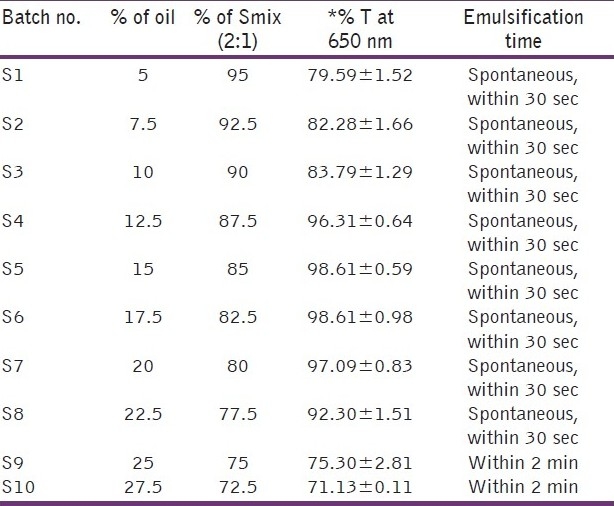
For the microemulsion system, four different oil concentrations i.e. 5%, 10%, 15% and 20% and four different concentrations of Smix i.e. 40%, 45%, 50% and 60% were used as shown in Table 1. For SMEDDS, ten different concentrations of oil ranging from 5-27.5% were used. The concentration of Smix also varied accordingly as SMEDDS consisted of only oil and Smix. Thus, the concentration of Smix varied from 95–72.5% as shown in Table 2.
The parameters assessed were dilutability, clarity (% transmittance) after 100 times dilution, particle size analysis and zeta potential using Malvern zeta sizer nano zs. For particles size and zeta potential analysis sample was diluted 10 times with distilled water
Preparation of microemulsion
Fixed amount of oil, surfactant, co-surfactant and drug were taken in completely dry beaker. The drug was dissolved completely by stirring the mixture using a magnetic stirrer. Required quantity of water was added drop-wise with stirring until a clear and transparent liquid was obtained.
Characterization of microemulsion/SMEDDs
Dilution test
Microemulsion system was diluted to 100 times of its volume and checked for its transparency by visual assessment and effect on globule size and zeta potential.[6,7]
Clarity
Percentage transmittance was checked against distilled water using UV One important consideration when formulating a microemulsion/SMEDDS formulation for the oral route is avoiding precipitation of the drug on dilution in the gut lumen in vivo and for that optimization of system was done using dilutability and transparency. Visible spectrophotometer at 650 nm (UV, 1601, 220X shimadzu, Japan) by dilution with distilled water (100 times) and as such. The wavelength selected was 650 nm as found in the literature and kept constant for measurement of all batches of microemulsions.
Centrifugation test
In this, the samples were centrifuged at 7500 rpm for 10 min at room temperature (25°C) and then were examined for phase separation.
Assay
Accurately measured volume (100 μl) of the optimized formulation was diluted with methanol up to 100 ml and absorbance was measured at 287 nm using methanol as blank. The assay was performed in triplicate.[9]
Globule size determination
To determine globule size, the microemulsion system was diluted with distilled water 10 times and then measured with Malvern zetasizer nanozs using clear disposable zeta cell (model Nano ZS, Malvern Instruments, Worcestershire, UK). The principle was based on the particle diffusion due to Brownian motion, which is related to particle size. Particle size is then calculated from the translational diffusion coefficient using the Stokes-Einstein equation by inbuilt software.
Zeta potential determination
To determine zeta potential, the microemulsion system was diluted with distilled water 10 times and then measured with Malvern zetasizer nano zs using clear disposable zeta cell (model Nano ZS, Malvern Instruments, Worcestershire, UK). The measurement was based on the electrophoretic mobility (μm/s) of the particles, which was converted to the zeta potential by inbuilt software based on the Helmholtz-Smoluchowski equation.
pH
pH of the microemulsion was measured using calibrated pH meter.
Viscosity measurement
The viscosity was determined using rotational viscosity measuring device coupled with concentric cylinders (Brookfield viscometer DVII plus pro, Brookfield engineering laboratory, USA). Viscosity measurement was done at 60 rpm using spindle no. 61 at 25°C.
Dye solubility test
It is also known as the stain test. Staining tests, in which a dye is sprinkled onto the surface of the emulsion. These tests essentially identify the continuous phase. Two to three drops of water-soluble dye was added to the optimized formulation and after 5 mins visual observation was done.
Stability study
The formulations were subjected to stability study for a period of three months at room temperature and refrigeration conditions. After three months of storage the microemulsion is subjected to test for physical stability, accelerated centrifugation cycle, drug content and particle size and zeta potential determination.
In vitro intestinal permeability
All experiments and protocols described in this study were approved by the Institutional Animal Ethics Committee of The MS University of Baroda and are in accordance with the Committee for the purpose of control and supervision of experiments on animals, Ministry of Social Justice and Empowerment, Government of India. The method employed was modified from experimental procedures well described in the literature.[10–12] The experiment was conducted with male Wistar rats (250-300 g).The rats were sacrificed, to study the in vitro intestinal permeability study from duodenum. This tissue was thoroughly washed with cold Ringer's solution to remove the mucous and lumen contents. Each formulation was added in a piece of rat intestine and it was tightly closed on both sides to check diffusion study. This tissue portion with formulation was put into 100 ml diffusion medium (phosphate buffer saline pH 7.4 with 1% Tween-80) into a beaker with continuous carbogen supply and gentle stirring condition at 37±2°C. Sample (5 ml) was collected at different time intervals and replaced with the same volume of diffusion media. The collected samples were analyzed by UV-visible spectroscopy method for drug content.[11]
Results and Discussion
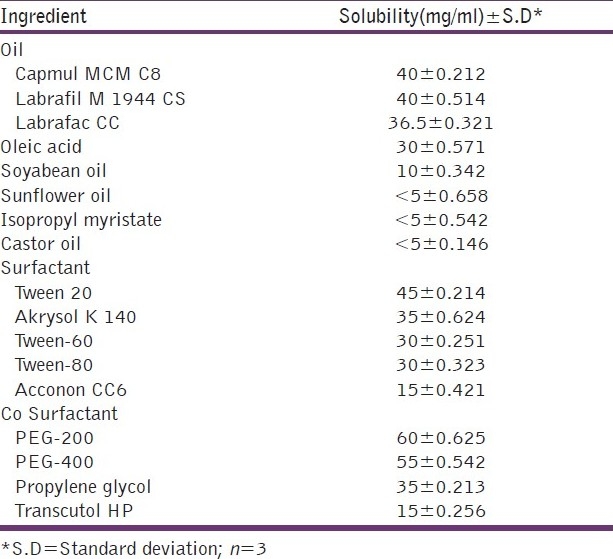
Microemulsions/SMEDDS are essentially clear systems and hence the solubility of the drug in all the components is necessary. One important consideration when formulating a microemulsion/SMEDDS formulation for the oral route is avoiding precipitation of the drug on dilution in the gut lumen in vivo. Therefore, the components used in the system should have high solubilization capacity for the drug, ensuring the solubilization of the drug in the resultant dispersion. Table 3 shows the solubility of Raloxifene in various oils, surfactants and co-surfactants. It can be observed that the highest solubility of Raloxifene was found in Capmul MCM C8, Tween-20 and Akrysol K140 and PEG-200, and hence these components were selected for the preparation of the microemulsion system.
Table 3.
Solubility of drug in oils, surfactants and cosurfactants
In the microemulsion system surfactant and co-surfactant get preferentially adsorbed at the interface, reducing the interfacial energy as well as providing a mechanical barrier to coalescence. The decrease in the free energy required for the emulsion formation consequently improves the thermodynamic stability of the microemulsion formulation. Therefore, the selection of oil and surfactant, and the mixing ratio of oil to S/CoS, play an important role in the formation of the microemulsion. This can be ascertained by the pseudoternary phase diagram as it differentiates the microemulsion region from the macroemulsion region. One can select the microemulsion region from the pseudoternary phase diagram. As seen in Figures 1 and 2, the microemulsion existence area increased as the S/CoS ratio increased. For the Tween-20: PEG-200 system the highest region was found in the 3:1 ratio and hence selected as further preparation. For Akrysol K 140: PEG-200 system the highest region was found in the 2:1 ratio and hence selected for further studies.
Figure 1.
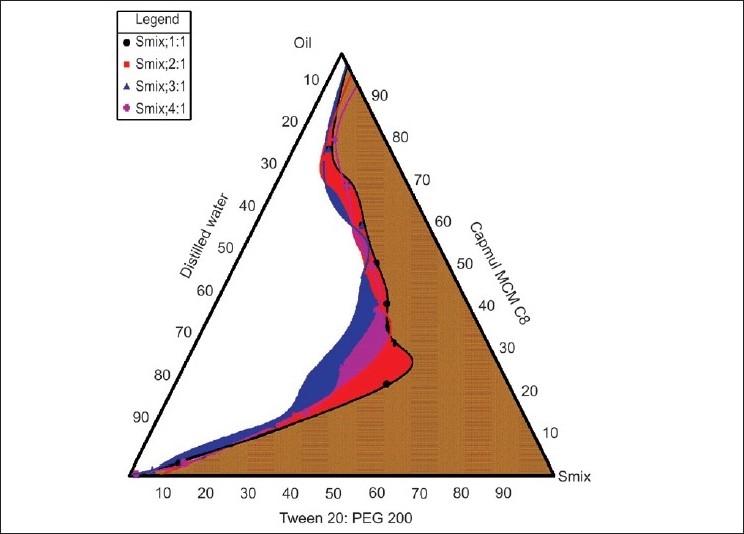
Pseudoternary phase diagrams of Capmul MCM C8 (oil), Tween-20: PEG-400 (S:CoS) and Water system
Figure 2.
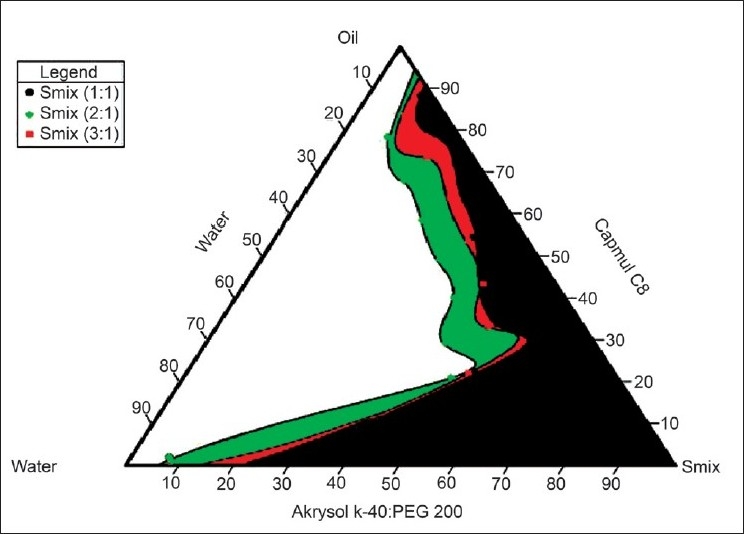
Pseudoternary phase diagrams of Capmul MCM C8 (oil), Akrysol k-140: PEG 400 (S: CoS) and Water system
Based on preliminary studies, various batches of the microemulsion preparation were taken using surfactant to co-surfactant ratio of 3:1. Optimization of the microemulsion system is shown in Table 1. As seen in Table 1, at low concentration of oil batches (M1 to M8), upon dilution the system became turbid which suggests that the system converts into macroemulsion. Batches M9, M10, M13 and M14 of the system remained clear upon dilution but when clarity was checked percentage transmittance was found to be less than 90%. M11, M12, M15 and M16 remained clear upon dilution and percentage transmittance for all batches was more than 90%, which shows that the system remained as microemulsion. Globule size and zeta potential values for all four batches are shown in Table 4, and from that batch M12 was selected as the final formulation as it had low globule size and higher zeta potential compared to the others which indicated that the system would remain stable compared to others. The optimized formulation is shown in Table 5.
Table 4.
Globule size and zeta potential of the optimized batches of microemulsion and SMEDDS
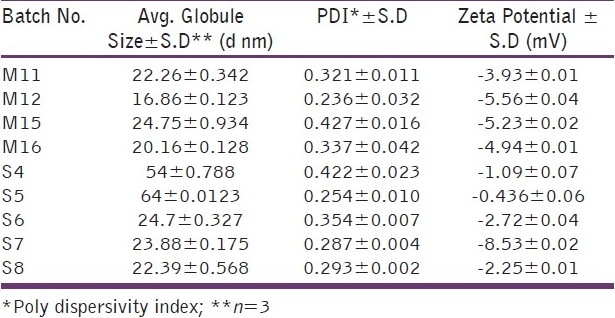
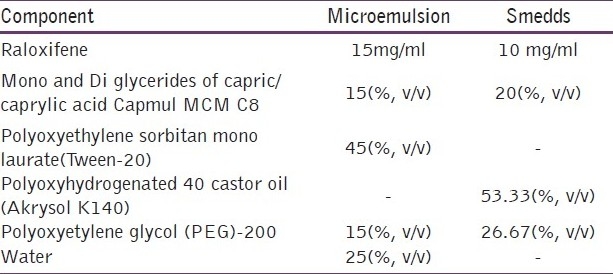
Table 5.
Optimized formula for raloxifene microemulsion and SMEDDS
Based on preliminary studies, various batches of the SMEDDS preparation were taken.one additional parameter checked was emulsification time.
Optimization of SMEDDS is shown in Table 2. Upon dilution of the batches: S1, S2, S3, S9 and S10, the clarity was found to be less than 90% transmittance and S10 become turbid upon visual inspection. Batches S4, S5, S6, S7, S8 remained clear upon dilution and percentage transmittance for all batches was more than 90%, which shows that the system remained as microemulsion. Globule size and zeta potential values for all batches are shown in Table 4, and from that batch S7 was selected as final formulation as it had low globule size and higher zeta potential compared to others which indicated that the system would remain stable compared to others.The optimised formulation is shown in Table 5.
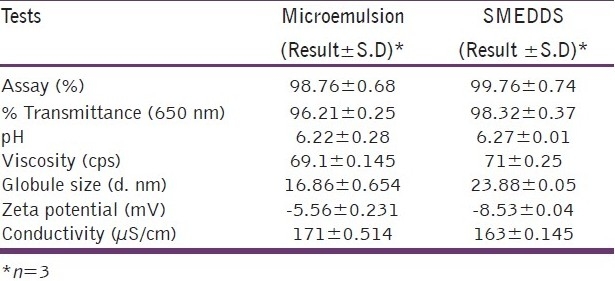
The amount of the drug content in the optimized formulation is around 98% of the added amount indicating the suitability of the system for high entrapment in the internal phase. The high value of percentage transmittance (96.21%) indicates that the system is optically clear which a prerequisite for microemulsions. The size of the globule is also found to be in the desirable size range (10-100 nm) indicating the possibility of enhanced permeation through the biological membrane. The zeta potential of the formulation was found to be slightly negative in spite of the non-ionic nature of the surfactant, co-surfactant and the oil. This indicated that the formulation would remain physically stable on storage, which was further confirmed by stability study. The formulation showed conductivity of 171 μS/cm which indicated that the type of the emulsion formed was oil in water (o/w).
The prepared microemulsion on oral administration gets diluted with the gastrointestinal fluids. Upon dilution, there should be no precipitation or increase in the globule size as this may affect the product performance in vivo. Hence, the dilutability of the microemulsion was ascertained by determining the clarity, size and zeta potential after 100 times dilution with distilled water. The percentage transmission of undiluted microemulsion was 96.21±0.25 and after dilution up to 10 times and 100 times the percentage transmission was 97.85±0.64 and 97.15±0.28 respectively. Therefore upon dilution no significant change in percentage transmission was observed, which indicated that there was no drug precipitation on dilution and the microemulsion exhibited desirable dilutability. This is also supported by the visual observation that there was no precipitation and the system remained clear. Upon 100 times dilution the globule size of microemulsion was 34.27 which indicated that the nanometric size range of the globule was retained even after 100 times dilution with water, which proves the system's compatibility with excess water. The increase in the globule size can be explained by the partition capacity of the co-surfactant. Initially, the interfacial film of the surfactant-co-surfactant mixture is tightly packed and hence the size of the globules is very less. As the volume of the aqueous dispersion medium increases, there is a migration of the co-surfactant in this phase resulting in reduced flexibility of the interface leading to enlargement of the globules. There was no significant change in the zeta potential indicating that the microemulsion was physically stable upon dilution.
The dye used was water-soluble and it was found to be distributed uniformly throughout the microemulsion, which suggests the distribution of the dye throughout the continuous phase. It can thus be concluded that the type of microemulsion is oil in water (o/w) type. This is also supported by the conductivity values as shown in Table 6.
Table 6.
Results of the characterization of raloxifene-loaded microemulsion and SMEDDS
Centrifugation of a biphasic system leads to the formation of two phases if the system is not stable physically. The prepared formulation was able to withstand the centrifugation indicating the long-term stability at the ambient conditions.
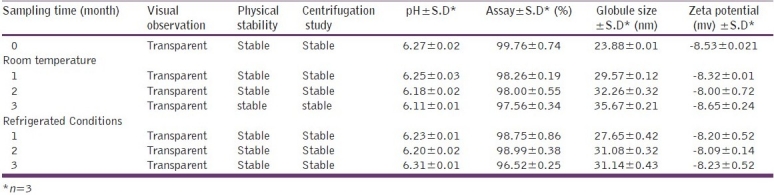
Stability study data reveal that the formulation remained stable over a period of three months both at room temperature and under refrigerated conditions. As shown in Table 7 and 8, there is no significant change in the pH and the assay of the formulation indicating the chemical stability of the formulation. The microemulsion was physically stable as evidenced by the visual observation, and the absence of signs of instability such as phase separation, flocculation or precipitation. As shown in Table 7 and 8, after three months’ storage there was a negligible increase in the globule size of the system stored at both room temperature as well as under refrigerated condition. Based on zeta potential measurement after a three-month interval, change in the zeta potential was found to be non-significant. All these results suggest that the microemulsion was physically and chemically stable on storage.
Table 7.
Results of stability study of raloxifene microemulsion
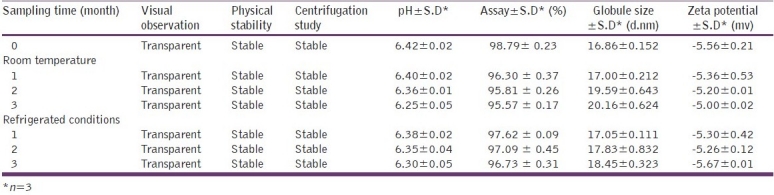
Table 8.
Results of stability study of raloxifene SMEDDS
The amount of the drug content in the optimized formulation was around 99% of the added amount indicating the suitability of the system for high entrapment in the internal phase. The high value of percentage transmittance (98.321%) indicated that the system was optically clear which is a prerequisite for microemulsions. The size of the globule was also found to be in the desirable size range (10-100 nm) indicating the possibility of enhanced permeation through the biological membrane. The zeta potential of the formulation was found to be negative. This indicated that the formulation would remain physically stable on storage, which was further confirmed by the stability study. The formulation showed conductivity of 163 μS/cm which indicated that the type of the microemulsion formed is o/w.
In vitro intestinal permeability data are shown in Figure 3. The total percentage diffusion was much higher for the microemulsion system and SMEDDS than for the raloxifene plain dispersion. After 5.5 h of diffusion, more than 90% of the drug was diffused, as compared to 41.06% diffused from the plain dispersion. The drug diffused at a faster rate from the microemulsion system than from the plain dispersion.
Figure 3.
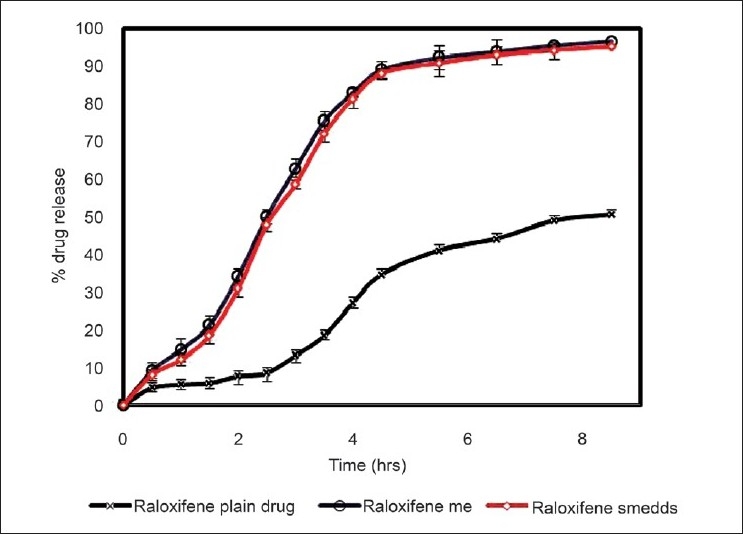
In vitro intestinal permeability study
The enhanced absorption may be explained in terms of (1) the huge specific surface area of the lipid nanoemulsion droplets (mean droplet size ~20 nm), (2) improved permeation of Raloxifene because of the presence of surfactant, which reduces the interfacial tension of formulation. There was no permeation of the drug from the stomach during the period of 1 h indicating that the site of absorption is the intestine. This may be due to the large surface area of the intestine and also the presence of microvilli which may aid in the absorption.
Conclusion
Raloxifene microemulsion/SMEDDS were prepared by the water titration method. This method of manufacturing was found to be simple, did not require specialized equipments. The prepared formulations exhibited all the desirable attributes of an ideal microemulsion and were also found to be stable over a period of three months both at room temperature and under refrigerated conditions. The in vitro intestinal permeability studies demonstrated that the microemulsion exhibited significantly higher permeation compared to the plain drug suspension. This increase in the bioavailability can lead to reduction in the dose and frequency of administration of a drug, fewer side-effects and overall cost of therapy. However, these assumptions can be confirmed only after animal experimentation on at least two or more animal models followed by an extensive clinical evaluation.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: Transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 2.Khoo SM, Shackleford DM, Porter CJ, Edwards GA, Charman WN. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm Res. 2003;20:1460–5. doi: 10.1023/a:1025718513246. [DOI] [PubMed] [Google Scholar]
- 3.Mei-Ling Chen. AAPS workshop- Effective utilization of lipid-based systems to enhance the delivery of poorly soluble drugs, lipid excipients and delivery systems for pharmaceutical development – An FDA Perspective. 2007 [Google Scholar]
- 4.Gande S, Manjunath K, Vobalaboina V, Vemula S. Preparation, characterization and in vitro and In vivo Evaluation of Lovastatin Solid Lipid Nanoparticles. AAPS Pharm Sci Tech. 2007;8(1):E1–E9. doi: 10.1208/pt0801024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Driscoll CM, Griffin BT. Biopharmaceutical challenges associated with drugs with low aqueous solubility—The potential impact of lipid-based formulations. Adv Drug Deliv Rev. 2008;60:617–24. doi: 10.1016/j.addr.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh PK, Majithiya RJ, Umrethia ML, Murthy RS. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech. 2006;7:77. doi: 10.1208/pt070377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355:269–27. doi: 10.1016/j.ijpharm.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 8. [Last accessed on 2010 Oct 28]. Available from http://www.drugbank.ca/Drugs/Db00481/Fda_Labels/652 .
- 9.Pavithra DC, Sivasubramanium L. New spectrophotometric determination of Raloxifene HCl in tablets. Indian J Pharm Sci. 2006;68:375–6. [Google Scholar]
- 10.Smith PL. Methods for evaluating intestinal permeability and metabolism in vitro. Pharm Biotechnol. 1996;8:13–34. doi: 10.1007/978-1-4899-1863-5_2. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh PK, Majithiya RJ, Umrethia ML, Murthy RS. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech. 2006;7:3–16. doi: 10.1208/pt070377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abrol S, Trehan A, Katare OP. Comparative study of different silymarin formulations: Formulation, characterisation and In vitro/in vivo evaluation. Curr Drug Deliv. 2005;2:45–51. doi: 10.2174/1567201052772870. [DOI] [PubMed] [Google Scholar]