

Abstract
Background:
Neurocutaneous syndromes (NCS) are a group of genetic disorders that produce a variety of developmental abnormalities of the skin along with an increased risk of neurological complications. Cutaneous manifestations usually appear early in life and progress with time, but neurological features generally present at a later age. There is a paucity of data regarding the evolution of skin lesions and their correlation with the central nervous system involvement in children.
Aim:
The primary objective was to track the course of skin lesions in various forms of NCS in the pediatric age group. Our secondary aim was to assess whether there was any predictive value of the lesions in relation to the neurological manifestations.
Materials and Methods:
This prospective longitudinal study was conducted at a tertiary care pediatric dermatology referral clinic of the Institute of Child Health, Kolkata, West Bengal. Children between the age group 0 and 12 years were included in the study on the basis of standard diagnostic criteria for different NCS, during the period from March, 2000 to February, 2004, and each of the enrolled cases were followed up for a duration of six years.
Results:
The study population comprised of 67 children (35 boys, 32 girls).The mean age of presentation was 33.8±27.8 months (range 10 days to 111 months). The various forms of NCS observed was neurofibromatosis 1(NF1) (n=33), tuberous sclerosis complex (TSC) (n=23), Sturge Weber syndrome (n=6), ataxia telangiectasia (n=2), PHACE syndrome (n=1), incontinentia pigmenti (n=1), and hypomelanosis of Ito (n=1). The presentations were varied, ranging from predominantly cutaneous to primarily neurological, depending on the disease entity and age group concerned. There was a significant increase in the number of café au lait macules (CALMs) with time (P=0.0002) in NF1, unlike that of hypopigmented macules of TSC (P=0.15). Statistically, no relation was documented between the evolution of skin lesions and neurological manifestations in the major groups.
Conclusion:
As NCS is not an uncommon disease in children, it is always necessary to find out the subtle neurological signs, whenever we observe any case with cutaneous markers suggestive of NCS. In addition, it is a must to do a detailed dermatological examination in a child with central nervous system involvement, in the pediatric population. However, the neurological course cannot be predicted from skin lesions.
Keywords: Neurocutaneous, neurofibromatosis, tuberous sclerosis
Introduction
Neurocutaneous syndromes (NCS), also known as phakomatosis, include a heterogeneous group of disorders characterized by abnormalities of both the integument and central nervous system (CNS). Most disorders are inherited as autosomal dominant conditions with variable penetrance, and are believed to originate from a defect in differentiation of the primitive ectoderm.[1] Disorders classified as NCS include neurofibromatosis 1 (NF1), tuberous sclerosis (TS), Sturge-Weber syndrome (SWS), von Hippel-Lindau (VHL) disease, PHACE syndrome, ataxia telangiectasia (AT), linear nevus syndrome (LNS), hypomelanosis of Ito (HOI), and incontinentia pigmenti (IP).[2] Being the tertiary care referral pediatric dermatology center, we came across quite a number of such cases in our institute. Therefore, we planned to conduct a study to monitor the varied presentations in children. The study was designed to evaluate the progression of skin lesions in various forms of NCS. Our secondary objective was to identify whether the characteristics of these lesions had any predictive value for neurological manifestations.
Materials and Methods
This study was carried out on a pediatric population at a tertiary care pediatric dermatology referral clinic of the Institute of Child Health, Kolkata, West Bengal. After approval from the Institutional Ethical Committee, subjects between ages of 0 and 12 years, who fulfilled the inclusion criteria, were enrolled, after obtaining an informed consent from the parents, during the period from March, 2000 to February, 2004. The diagnosis of neurofibromatosis1 (NF1) was established when the children presented with two or more of the following features: six or more café au lait macules (CALMs) over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals; two or more neurofibromas of any type or 1 plexiform neurofibroma; freckling in the axillary or inguinal region; optic glioma; two or more Lisch nodules; a distinctive osseous lesion; and a first-degree relative with NF1.[3] Definite tuberous sclerosis complex (TSC) was confirmed when at least two major or one major plus two minor features were present. Major features included skin lesions, brain and eye lesions, and tumors in the heart, kidneys, or lungs. Minor features comprised of bone cysts, rectal polyps, dental enamel pits, CNS white matter migrational abnormalities, gingival fibromas, non-renal hamartomas, retinal achromic patches, ‘confetti’ skin lesions, and multiple renal cysts.[4]
Sturge-Weber syndrome, PHACE syndrome, ataxia telangiectasia, hypomelanosis of Ito, and incontinentia pigmenti, were diagnosed on the basis of standard clinical features and relevant investigations, including neuroimaging, genetic analysis, and skin biopsy, wherever applicable.
All the patients were initially admitted in the Inpatient Department of the institute for a detailed workup. A complete evaluation, including history, physical, and neurological examinations were performed. In addition to dermatologists, pediatricians and neurologists were also involved in managing the subjects. Each of the cases was followed up on a half-yearly basis for the next six years. Patients were referred for quarterly ophthalmological evaluation, with a slit lamp examination as a part of the routine care, wherever applicable. Follow-up data, including chronology of the developing additional features, were collected in our clinic through the time of the last follow-up examination. Those subjects who were lost to follow-up were excluded from the study population. Statistical analysis was done by SPSS ver. 12, with a P value less than 0.05 taken as significant.
Results
A total of 67 children were included in the study of which 35 were boys and 32 were girls (M : F ratio 1.09 : 1). They presented with the age range of 10 days to 111 months with a mean age of 33.8±27.8 months. Among 67 patients of NCS, 33 cases were NF1, 23 cases were TS, six cases were SWS, and two cases were AT. Only one case each of PHACE syndrome, HOI, and IP was diagnosed in the stipulated period.
Out of a total of 33 children (18 male and 15 female) who met the inclusion criteria of NF1, 27 (81.8%) children were diagnosed before the age of 72 months, with the mean age of presentation being 51.1±23.9 months. All the cases of NF1 presented with one consistent finding, that is, prerequisite number and size of CALM. They were more pronounced on the trunk (n=29) and extremities (n=20). Initially in 21 subjects the number of CALM was restricted to 10 or less. The average number of CALMs at the time of presentation was 9.6±3.2. During the course of the study the number and size increased in most of the cases (n=27 and n=29). However, the increase in the number of CALMs was mostly restricted within the first decade of life. Table 1 shows the change in the number of CALMs in different age groups. At the end of follow-up, the mean number of CALMs was 11.5±2.3. Figure 1 shows the CALMs in the trunk of an 11-year-old child with NF1.
Table 1.
Shows the change in the number of café au lait macules (CALMs) in different age groups
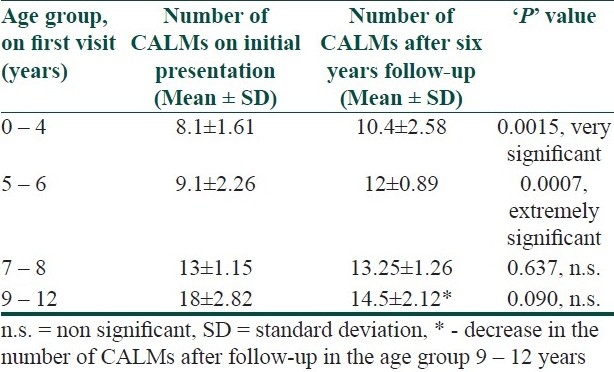
Figure 1.
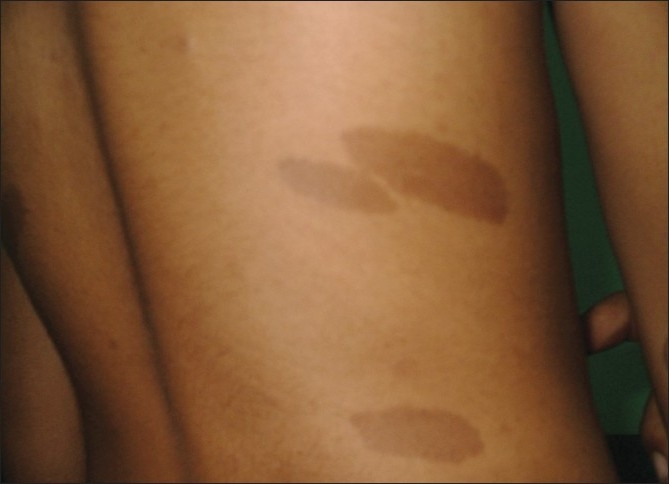
Showing café au lait macules (CALMs) in the trunk of an 11-year-old child with NF1
At the time of diagnosis, the most common second feature in patients having NF1 was axillary or inguinal freckling, occurring in 24 (72.7%) patients. Other signs that occurred, included, the following: Lisch nodules in 15 children, plexiform neurofibromas in three cases, and optic gliomas in three children. Following a diagnosis of NF1, subjects in this clinic population developed the following additional features of NF1 during the course of our study: axillary or inguinal freckling, Lisch nodules, and neurofibromas, respectively, in eight cases and optic gliomas in four patients. Skeletal anomalies (tibial pseudoarthrosis) developed in three children, plexiform neurofibromas, and precocious puberty in one case each. None developed malignant peripheral nerve sheath tumor or pheochromocytoma; 11 of 18 patients who underwent neuroimaging were noted to have ‘unidentified bright objects (UBOs)’ on magnetic resonance imaging.
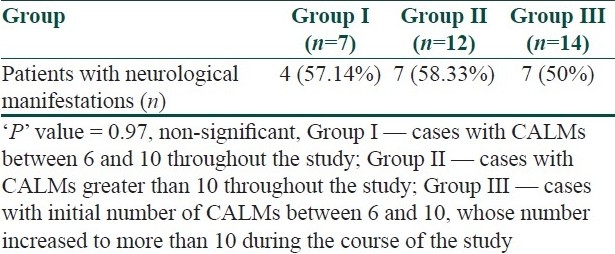
Eight subjects presented with neurological manifestations on the first visit only. Most of them (n=6) presented with seizures. However, CNS complications were noted in another 10 patients after detailed history taking and clinical examination during enrolment and subsequent follow-up. Cognitive dysfunction was found in 11 cases, seizures in nine patients, and hemiparesis in two cases. Table 2 shows the relative frequency of neurological complications in different age groups. Table 3 shows the relationship between the number of CALMs and neurological features.
Table 2.
shows the proportion of neurological presentation in different age groups at the time of enrolment

Table 3.
showing the correlation between the numbers of café au lait macules (CALMs) and neurological manifestations
Twenty-three children (13 boys and 10 girls) met the diagnostic criteria of the TS complex. The mean age of presentation was 11.8±8.4 months. At the time of inclusion in the study, the maximum number (n=11) of patients was in the age group of 7–12 months. Nine of them had seizure as their presenting complaint. In contrast, in four of the five cases, who were diagnosed before the age of six months, the only presenting feature was hypopigmented macules (ash leaf spots). Five subjects were diagnosed between the age of 13 and 24 months and the remaining children beyond that.
An adequate number of hypopigmented macules were the most frequent cutaneous finding seen in 21 of 23 children (91.3%) at the time of enrolment, and in all cases, after a six-year follow-up. They were predominantly distributed over the trunk (n=14), face (n=13), and extremities (n=10), and varied in size (1–3 cm in length). There was no significant increase in the number of hypopigmented macules at the end of the study period compared to the number of macules noted at the point of entry into the study (mean,6.04±1.9 vs. 5.4±2.03, P=0.15). The next most common dermatological marker observed in TSC was adenoma sebaceum (n=16), most (n=14, 87.5%) being evident by six years of age. Shagreen patch was noted in 11 cases (47.8%). Figure 2 shows hypopigmented macule and adenoma sebaceum in a 12-year-old child.
Figure 2.
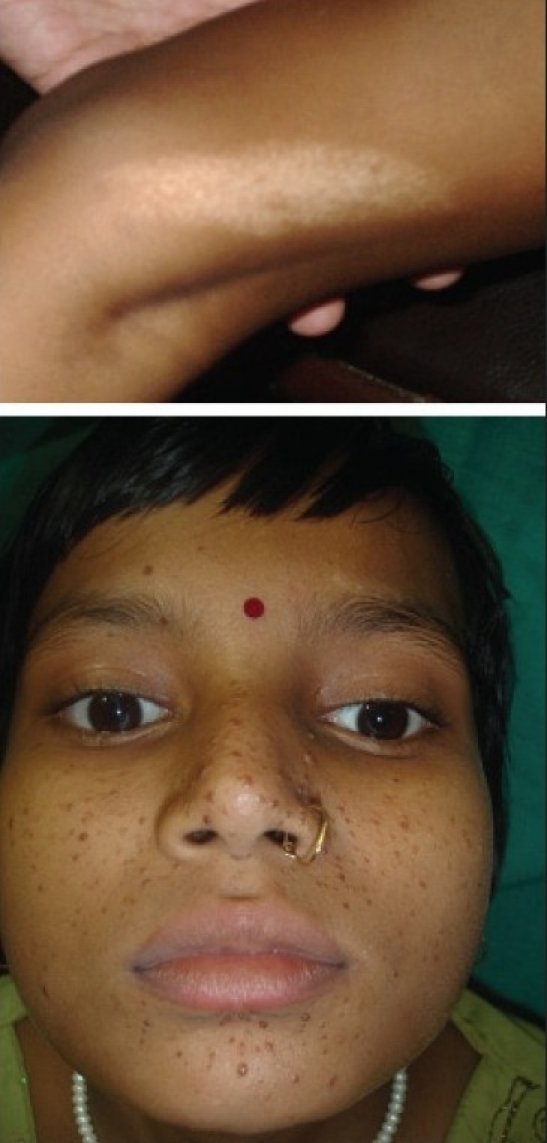
Showing hypopigmented macule and adenoma sebaceum in a 12-year-old child with tuberous sclerosis complex
Convulsion was seen in 15 of 23 children (78.2%) initially, with 10 (66.6%) occurring during the first year of life; and eventually most cases (n=20; 86.95%) developed seizures. Convulsions typical of infantile spasms and corroborated by a chaotic pattern in the electroencephalogram (EEG) were observed in seven subjects. Six patients developed refractory myoclonic epilepsy in due course. Three of these cases eventually died. Mental retardation, behavioral abnormalities, and cognitive dysfunction were detected in 18, 16, and 11 children, respectively. Serial CT scan brain ultimately showed typical calcified tubers in 21 children, most (n=13) being evident after four years of age. However, there was very poor correlation between the number of tubers in neuroimaging and that of the hypopigmented macules (r=0.08).
The following additional features of TS were detected during the course of our study: Rhabdomyomas of the heart, mulberry tumors of the optic nerve head, and angiomyolipomas of the kidney in seven, six, and three cases, respectively. None of them developed lymphangiomyomatosis of the lung. Out of seven children with rhabdomyomas of the heart, two developed congestive heart failure with arrhythmia.
Six children (three male and three female) were diagnosed with SWS, and their mean age of presentation was 10±4.6 months (range 4–16 months). History revealed that all had typical facial nevus (port-wine stain) since birth, but seizure developed later (four cases within the first year of life). Serial ophthalmological check-up revealed increased intra-ocular tension in four cases (three cases within 12 months and one case at the age of six years). All subjects developed developmental delay and mental retardation subsequently, whereas, two of them developed hemiparesis. Two patients had died of intractable seizures. Figure 3 shows a large hemangioma in the right side of the face of a four-month-old infant with SWS.
Figure 3.
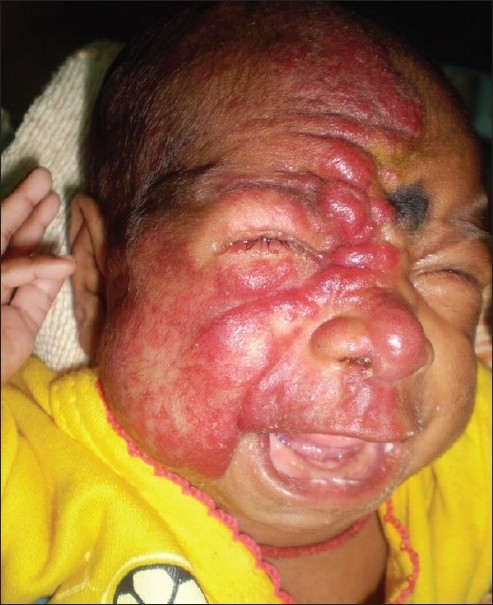
Showing a large hemangioma on the right side of the face of a four-month-old infant with SWS
Two cases of AT were diagnosed; one at six years and another at eight years of age. Past history revealed ataxia, which became evident in both the cases when they began to walk, whereas, bulbar telangiectasia developed at the time they were diagnosed. They obviously had frequent episodes of recurrent chest infections, prior to the diagnosis.
Both the patients of HOI and IP presented with frank generalized tonic–clonic convulsions at three years of age, but definitive diagnosis was made on the basis of typical skin lesions. HOI was manifested by appearance of depigmented, whorled areas over the limbs and trunk, which persisted throughout. The cutaneous finding in IP was noted in the pigmentary stage (brown), mainly over the trunk, which gradually faded over time.
We also documented the PHACE syndrome in a 10-day-old neonate who presented with a large facial hemangioma and Dandy-Walker cyst. This case was unique because the cyst was detected antenatally and there was associated congenital hypothyroidism.
Discussion
There have been studies on the various forms of NCS, but a collective study on them, along with their midterm follow-up, exclusively in the pediatric population, is virtually nonexistent.[5,6]
There have been quite a number of studies on the clinical profile of NF1 both in children as well as in adults.[5,7–9] As found by Nunley KS et al.,[10] most of our study population met the criteria of NF1 before 72 months of age. However, at the time of enrolment, the average number of CALMs in our cases was less than that of their patients (9.6 vs. 11.8). A significant increase in the number of CALMs occurred only in the first decade, which has been established both by our study (vide Table 1) as well as by Huson SM et al.[5] We noted that there was absolutely no relation between the number of CALMs and neurological manifestations (p=0.97).
In our series, TSC was the second most common group (69.6%). Similar to what was documented by Jozwiak S et al.,[6] we found that hypopigmented macules were the most common cutaneous finding. We noted that most of the children had an onset of neurological presentation when they were below one year of age, which was also corroborated by the previous study. Our observation on the mean age of presentation in TSC was considerably lower than the study done by Brigid AS et al.,[11] probably because their series was a retrospective one and included both children and adults. We could not find any relation between the magnitude of skin manifestation and tubers in neuroimaging. However, larger studies are needed to validate our observation.
Sturge-Weber syndrome is the only form of NCS that is not genetically determined and involves a facial port-wine stain reaching the first branch of the trigeminal nerve (V1), ophthalmological abnormalities (especially congenital glaucoma), and neurological signs (seizure, mental retardation, hemiparesis).[1,12] The relatively less reporting of the syndrome in our study is perhaps due to the less incidence of the disease (one in 50,000 live births).[2] Ataxia-telangiectasia (AT) is an autosomal recessive inherited disease caused by the mutational inactivation of the ATM gene. It is a multisystemic disease, characterized by progressive neurological dysfunction, especially in the cerebellum, oculocutaneous telangiectasia, immunodeficiency, and recurrent sinopulmonary infections.[13] Literatures on HOI and IP are mostly limited to case reports.[14–16]
PHACE is an acronym coined to describe a neurocutaneous syndrome. It was proposed by Frieden, et al.,[17] in 1996, encompassing the following features: Posterior fossa malformations, Hemangiomas, Arterial anomalies, Coarctation of the aorta and other cardiac defects, and Eye abnormalities. Association of congenital hypothyroid in our case has been described previously in very few articles.[18]
As it is not unusual to encounter NCS in the pediatric population, it is imperative to look for neurological manifestations, especially things that are often missed, like cognitive dysfunction in a child with neurocutaneous markers. Opinion from colleagues of the Pediatric Neurology Department must be taken, as and when it is felt necessary. However, at the same time the neurologists must also be sensitized about performing a dermatological examination in a child with central nervous system involvement.
Acknowledgments
We profusely thank Prof. Apurba Ghosh, Director, Institute of Child Health, Kolkata and Prof. Swapan Mukhopadhay, Head, Department of Pediatric Neurology, Institute of Child Health for their support and guidance.
Footnotes
Source of support: Nil
Conflict of Interest: Nil.
References
- 1.Dahan D, Fenichel GM, El-Said R. Neurocutaneous syndromes. Adolesc Med. 2002;13:495–509. [PubMed] [Google Scholar]
- 2.Haslam Robert HA. Neurocutaneous Syndromes. In: Kliegman RA, Jenson HB, Behrman RE, Stanton BF, editors. Nelson Textbook of Pediatrics. 18th ed. Vol. 2. Philadelphia: Elsevier; 2008. pp. 2483–8. [Google Scholar]
- 3.National Institutes of Health Consensus Development Conference. Neurofibromatosis: Conference Statement. Arch Neurol. 1988;45:575–8. [PubMed] [Google Scholar]
- 4.Hyman MH, Whittemore VH. National Institutes of Health consensus conference: Tuberous sclerosis complex. Arch Neurol. 2000;57:662–5. doi: 10.1001/archneur.57.5.662. [DOI] [PubMed] [Google Scholar]
- 5.Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis: A clinical and population study in South East Wales. Brain. 1988;111:1355–81. doi: 10.1093/brain/111.6.1355. [DOI] [PubMed] [Google Scholar]
- 6.Jozwiak S, Schwartz RA, Janniger CK, Michalowicz R, Chmielik J. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol. 1998;37:911–7. doi: 10.1046/j.1365-4362.1998.00495.x. [DOI] [PubMed] [Google Scholar]
- 7.Obringer AC, Meadows AT, Zackai EH. The diagnosis of neurofibromatosis 1 in the child under the age of 6 years. Am J Dis Child. 1989;143:717–9. doi: 10.1001/archpedi.1989.02150180099028. [DOI] [PubMed] [Google Scholar]
- 8.Créange A, Zeller J, Rostaing-Rigattieri S, Brugières P, Degos JD, Revuz J, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999;122:473–81. doi: 10.1093/brain/122.3.473. [DOI] [PubMed] [Google Scholar]
- 9.North K. Neurofibromatosis type 1: Review of the first 200 patients in an Australian clinic. J Child Neurol. 1993;8:395–402. doi: 10.1177/088307389300800421. [DOI] [PubMed] [Google Scholar]
- 10.Nunley KS, Gao F, Albers AC, Bayliss SJ, Gutmann DH. Predictive value of café au lait macules at initial consultation in the diagnosis of neurofibromatosis type 1. Arch Dermatol. 2009;145:883–7. doi: 10.1001/archdermatol.2009.169. [DOI] [PubMed] [Google Scholar]
- 11.Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127:e117–25. doi: 10.1542/peds.2010-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruani A. Sturge-Weber syndrome. Presse Med. 2010;39:482–6. doi: 10.1016/j.lpm.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 13.Bott L, Thumerelle C, Cuvellier JC, Deschildre A, Vallée L, Sardet A. Ataxia-telangiectasia: A review. Arch Pediatr. 2006;13:293–8. doi: 10.1016/j.arcped.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Chen YB, Hao YP, Liang D. Hypomelanosis of Ito and brain abscess in a boy. Zhongguo Dang Dai Er Ke Za Zhi. 2011;13:440–1. [PubMed] [Google Scholar]
- 15.Lee Y, Kim S, Kim K, Chang M. Incontinentia pigmenti in a newborn with NEMO mutation. J Korean Med Sci. 2011;26:308–11. doi: 10.3346/jkms.2011.26.2.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Florin TA, Shah KN. Picture of the month. Incontinentia pigmenti. Arch Pediatr Adolesc Med. 2011;165:367–8. doi: 10.1001/archpediatrics.2011.23-a. [DOI] [PubMed] [Google Scholar]
- 17.Frieden IJ, Reese V, Cohen D. PHACE syndrome: The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132:307–11. doi: 10.1001/archderm.132.3.307. [DOI] [PubMed] [Google Scholar]
- 18.Metry DW, Dowd CF, Barkovich AJ, Frieden IJ. The many faces of PHACE Syndrome. J Pediatr. 2001;139:117–23. doi: 10.1067/mpd.2001.114880. [DOI] [PubMed] [Google Scholar]