

Abstract
The continuous emergence of drug-resistant viruses is a major obstacle for the successful treatment of viral infections, thus representing a persistent spur to the search for new therapeutic strategies. Among them, multidrug treatments are currently at the forefront of pharmaceutical, clinical, and computational investigation. Still, there are many unknowns in the way that different drugs interact among themselves and with the pathogen that they aim to control. Inspired by experimental studies with picornavirus, here, we discuss the performance of sequential vs. combination therapies involving two dissimilar drugs: the mutagen ribavirin and an inhibitor of viral replication, guanidine. Because a systematic analysis of viral response to drug doses demands a precious amount of time and resources, we present and analyze an in silico model describing the dynamics of the viral population under the action of the two drugs. The model predicts the response of the viral population to any dose combination, the optimal therapy to be used in each case, and the way to minimize the probability of appearance of resistant mutants. In agreement with the theoretical predictions, in vitro experiments with foot-and-mouth disease virus confirm that the suitability of simultaneous or sequential administration depends on the drug doses. In addition, intrinsic replicative characteristics of the virus (e.g., replication through RNA only or a DNA intermediate) play a key role to determine the appropriateness of a sequential or combination therapy. Knowledge of several model parameters can be derived by means of few, simple experiments, such that the model and its predictions can be extended to other viral systems.
Keywords: population dynamics, antiviral design, viral quasispecies, mutation, viral adaptation
The ability of diverse and rapidly multiplying parasites to adapt to environmental changes is a major obstacle for the design of successful therapeutic strategies able to control their proliferation and propagation. RNA viruses count among the most plastic organisms on Earth and are an iconic example of populations persistently escaping the action of antiviral drugs (1, 2). Drugs exert constant selection pressures on viral populations, and as such, the question is not whether a resistant form of the virus will appear but when it will occur. Alternative therapies are continuously sought with the aim of impeding the appearance and fixation of resistance mutants (3–5). Among them, multidrug treatments have been established as an efficient way of delaying the appearance of resistant forms. However, to understand the mode of action of combined treatments, it is urgent to clarify the degree of interaction between the different drugs used (6) and the quantitative impact that they have on the virus that they are affecting. The joint action of two drugs can rarely be reduced to the simple addition of their independent effects (7), and the same drug might elicit different responses in different viral systems, including the appearance of compensatory mutations able to induce resistance (8). Thus, the complete characterization of a multidrug treatment applied to a particular viral system might require in the last term a systematic assay with a large number of drug doses delivered under different administration protocols. A full characterization of the response of the viral system in vitro is needed before any in vivo assay of the treatment. This procedure demands a costly amount of time and resources that can be significantly reduced through the guide offered by formal approaches to therapies.
Knowledge derived from in vitro studies of the response of pathogens to the action of one or a number of drugs might be combined with the information yielded by well-designed mathematical models involving the relevant mechanisms of action of and interaction among the drugs (9, 10), virus–host interactions (11), or role of the immune response (12). An important feature, rarely taken into account, is the intrinsic replicative ability of the pathogen. Many different ways to encode genomic information (ds- and ssRNA and different polarities) and a repertoire of replicating strategies have been selected by different RNA viruses. Hence, the knowledge gained for a particular virus may not be extrapolated to other systems, although suitable modifications of dynamical models can likely account for these different replication modes.
The dynamics of the picornavirus foot-and-mouth disease virus (FMDV) have been explored under different experimental regimens with the aim of disclosing protocols able to cause its extinction during replication in cell culture. It has been shown that a combination of mutagenic agents and antiviral inhibitors is an efficient way to drive FMDV to extinction (13, 14). The mutagen succeeds in raising the fraction of defective and lethal mutants in the population, thus decreasing its overall fitness. Defectors may also interfere with infectivity, as shown with specific mutants (15) and preextinction viral populations (16), and thus, accelerate extinction. For its part, the inhibitor contributes to extinction by reducing the viral load. However, the joint effect of a mutagen and an inhibitor cannot be reduced to the addition of their individual effects (9). Since the dawn of quasispecies theory (17), the use of mutagens has been proposed as a plausible strategy to induce viral extinction (18, 19). A significant increase in the mutation rate has been, indeed, successful to cause the extinction of infectivity in many different viral systems (20, 21), although the mechanisms through which extinction supervenes are diverse and related to a variety of molecular and population responses (22). In particular, increased mutagenesis can also bear beneficial effects for viral populations through an enhancement of their diversity, which promotes adaptation of low-fitness viruses (23) and facilitates the appearance of resistance mutants when an inhibitor of viral replication is present (24).
The increased likelihood of developing resistance to the inhibitor when dispensed simultaneously with a mutagen is what advocates the sequential administration of the two drugs, with the inhibitor followed by the mutagen. In a study with FMDV subjected to the action of the mutagen ribavirin (R) and the inhibitor of viral replication guanidinium hydrochloride (GU), it was shown that, under the experimental conditions assayed, the sequential therapy performs better than the simultaneous administration of both drugs (24). This finding is an interesting result that immediately queries the range of applicability and relative success of combined or sequential administration of those two dissimilar drugs. In this contribution, we face the characterization of the response of a general viral system to the action of an inhibitor of its replication and a mutagenic agent through a model for the dynamics of two different viral classes in the viral population. We obtain several exact results that predict the preferred therapy as a function of intrinsic viral characteristics and the administered drug doses. Our predictions are tested and confirmed by experiments with FMDV at different doses of inhibitor.
Results
Mathematical Model.
The present model is intended to reproduce the dynamics of a viral population after the addition of mutagens and/or replication inhibitors using the protocol represented in Fig. 1. Mutagens mostly promote the appearance of deleterious variants, although some particular mutations may, however, confer resistance to the inhibitor, thus favoring its survival. To capture this double role of the mutagen, the model considers that the dynamics are dominated by two types of individuals: viable individuals that are susceptible to the inhibitor (v) and viable individuals that are resistant to the inhibitor (V). Viable individuals are able to infect cells and replicate by themselves.
Fig. 1.

Schematic outline of the experimental protocol in cell culture. (A) Combined therapy. A dose dGU of guanidine and a dose dR of ribavirin are simultaneously added to the initial population. The viral titer 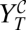 is calculated after a single passage. (B) Sequential therapy. A dose dGU is added to the initial population. After 24 h, the inhibitor is removed, and a dose dR of ribavirin is added. The viral titer
is calculated after a single passage. (B) Sequential therapy. A dose dGU is added to the initial population. After 24 h, the inhibitor is removed, and a dose dR of ribavirin is added. The viral titer 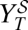 is obtained after this second passage. (C) Experimental results in a case example. The initial population has a titer of 3.7 × 106 pfu/mL. The applied doses are dGU = 18 mM and dR = 5 mM. With these drug doses, the viral titers after applying the protocols described in A and B are
is obtained after this second passage. (C) Experimental results in a case example. The initial population has a titer of 3.7 × 106 pfu/mL. The applied doses are dGU = 18 mM and dR = 5 mM. With these drug doses, the viral titers after applying the protocols described in A and B are  pfu/mL and
pfu/mL and 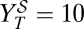 pfu/mL, respectively. Passages are performed and quantified as described in Materials and Methods.
pfu/mL, respectively. Passages are performed and quantified as described in Materials and Methods.
Let w0 be the rate at which viable individuals produce, under replication, a nonviable class, including lethal variants, defective interfering forms, or any other mutant unable to complete an infection cycle on its own [(1 − w0) is the intrinsic copying fidelity of the virus].† Resistant forms appear at a rate of μ0 = kw0. Because mutations providing resistance to the inhibitor are rare compared with deleterious ones, k ≪ 1.‡ Addition of a mutagen is implemented by increasing the mutation rate from its natural value w0 to a higher w > w0. As a consequence, the rate of appearance of resistant mutants, μ = kw, increases when the mutagen is added. Each time a viral genome replicates inside the cell, m copies are produced, and therefore, m is the replicative ability per genome and replication cycle. The effect of an inhibitor is to slow down replication of the susceptible type by multiplying parameter m by a factor 0 < i ≤ 1.
Let v(g) and V(g) denote the number of individuals of each type at replication cycle g. Populations after one replication cycle in the presence of a mutagen and an inhibitor will be (Eqs. 1 and 2)
To study the dynamics of the model and compare the sequential vs. the combined administration of the drugs, we will mimic the protocol described in Fig. 1. At cycle 0 (initial condition), we will assume that the population is formed by S0 individuals of type v, which because of mutations, will populate the class or resistant mutants in successive replication cycles. A full solution of the model is given in Materials and Methods. The dynamics of the combined therapy correspond to applying Eqs. 1 and 2 iteratively for g = 1, … , G cycles with parameters i < 1 and w > w0; the dynamics of the sequential therapy imply replication for G cycles in absence of the mutagen (i < 1 and w = w0) and use of the generated population as initial condition to replicate for G additional cycles in absence of the inhibitor and presence of the mutagen (i = 1, w > w0).
Model Analysis.
Viral particles are released from the cell after G replication cycles. This release finishes the process for the combined therapy, whereas there is a second passage to undergo in the sequential therapy. The size of the infective population [let us call it YT(G, w, i) to make explicit the experimental parameters involved] is obtained by adding the population of individuals in the two relevant classes after the first passage, YT(G, w, i) = v(G) + V(G) (Materials and Methods), and therefore (Eq. 3),
 |
which for i = 1 (absence of inhibition), yields YT(G, w, 1) = S0mG(1 − w)G. The size of a viable viral population after one passage with a mutagen and/or a replication inhibitor is, thus, given by Eq. 3 with the corresponding parameters w and i. In the absence of drugs, the total population obtained is S0mG(1 − w0)G, such that the initial population is simply multiplied by m(1 − w0) at each replication cycle, which is the basic reproductive ratio of the population.
Comparison between treatments: Viral titer.
Let us call 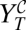 the viral titer after the application for one passage of the combined treatment. According to the experimental protocol, the mutagen and the inhibitor have been simultaneously administered, and therefore, the viral yield is immediately given by Eq. 3:
the viral titer after the application for one passage of the combined treatment. According to the experimental protocol, the mutagen and the inhibitor have been simultaneously administered, and therefore, the viral yield is immediately given by Eq. 3: 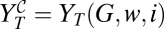 . Analogously,
. Analogously, 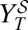 is defined as the viral yield after the sequential administration of the drugs: first, the inhibitor for one passage and then, the mutagen for a second passage (Eq. 4),
is defined as the viral yield after the sequential administration of the drugs: first, the inhibitor for one passage and then, the mutagen for a second passage (Eq. 4),
where the initial condition for the second passage requires S0 to be substituted by the yield obtained, YT(G, w0, i). The comparison of the yields obtained using either treatment determines which of them is more efficient for each dose of mutagen and inhibitor. The curve C
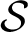 , defined by
, defined by 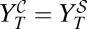 , separates two regions in the space of parameters {i, w}, where one therapy or the other is better suited to obtain a low viral titer (Materials and Methods has an approximate expression of curve C and its asymptotic value at large values of the inhibitor). Fig. 2 A, C, and E shows the viral titer corresponding to the numerical solution of the equations above for the combined and sequential treatments.
, separates two regions in the space of parameters {i, w}, where one therapy or the other is better suited to obtain a low viral titer (Materials and Methods has an approximate expression of curve C and its asymptotic value at large values of the inhibitor). Fig. 2 A, C, and E shows the viral titer corresponding to the numerical solution of the equations above for the combined and sequential treatments.
Fig. 2.

Theoretical phase space representation of the model. (A) Viral titer 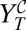 after application of the combination treatment. (B) Doses that fall on the gray area cause viral extinction under application of the combination treatment. The frontier with the blue region corresponds to values {i, w} yielding
after application of the combination treatment. (B) Doses that fall on the gray area cause viral extinction under application of the combination treatment. The frontier with the blue region corresponds to values {i, w} yielding  , which defines curve
, which defines curve  . (C) Viral titer
. (C) Viral titer 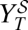 after application of the sequential treatment. (D) Doses that fall on the gray area cause viral extinction under application of the sequential treatment. The frontier with the red region corresponds to values {i, w} yielding
after application of the sequential treatment. (D) Doses that fall on the gray area cause viral extinction under application of the sequential treatment. The frontier with the red region corresponds to values {i, w} yielding 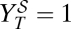 , which defines curve
, which defines curve 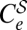 . (E) Comparison of the titers produced by combination and sequential therapies. In the z axis, we represent the minimum value of the viral yield for each pair of {i, w} values. (F) In the gray area, extinction occurs if the appropriate therapy is used. Blue and red lines correspond to extinction with the combination or sequential treatment, respectively, and are as above. Black curve is C. It signals the pairs of {i, w} values where both therapies produce the same titer. Violet curve CR indicates when both treatments produce the same amount of resistants. In the violet region between CR and C curves, the combined treatment yields the lower titer, but the sequential treatment is less prone to produce resistance. In the limit of large amounts of inhibitor (i → 0), both therapies perform equally well in regard to titer and resistant production. The expression for the limit value of mutagen wi → 0(Materials and Methods) is indicated with an arrow in the plot. Finally, the dotted line stands for curve Ci, which indicates the values of i (below the curve) that cause an efficient decrease of the population. Curve Cw, representing the values of the mutagen above which the population of viable individuals decreases from passage to passage, is out of the range shown in the plots. Parameters for all plots are m = 50, G = 2.5, w0 = 0.9, and k = 0.01.
. (E) Comparison of the titers produced by combination and sequential therapies. In the z axis, we represent the minimum value of the viral yield for each pair of {i, w} values. (F) In the gray area, extinction occurs if the appropriate therapy is used. Blue and red lines correspond to extinction with the combination or sequential treatment, respectively, and are as above. Black curve is C. It signals the pairs of {i, w} values where both therapies produce the same titer. Violet curve CR indicates when both treatments produce the same amount of resistants. In the violet region between CR and C curves, the combined treatment yields the lower titer, but the sequential treatment is less prone to produce resistance. In the limit of large amounts of inhibitor (i → 0), both therapies perform equally well in regard to titer and resistant production. The expression for the limit value of mutagen wi → 0(Materials and Methods) is indicated with an arrow in the plot. Finally, the dotted line stands for curve Ci, which indicates the values of i (below the curve) that cause an efficient decrease of the population. Curve Cw, representing the values of the mutagen above which the population of viable individuals decreases from passage to passage, is out of the range shown in the plots. Parameters for all plots are m = 50, G = 2.5, w0 = 0.9, and k = 0.01.
Comparison between treatments: Appearance of resistant mutants.
In addition to the viral titer produced after application of one or another therapy, it is important to know the fraction of resistant mutants that each treatment produces on average. This quantity corresponds to the final populations of type V(G) after one passage with both the mutagen and the inhibitor (combination therapy) or one passage with the inhibitor plus a second passage with the mutagen (sequential therapy). Let us call R and R the two final populations of resistants. The exact expressions for the two quantities are obtained as R = V(G, w, i) and 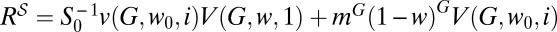 , analogous to the way in which the titers were obtained. The amount of resistants produced with either treatment will be equal at values of w and i that fulfill R = R. This finding defines curve CR. A convenient way of calculating some of the properties of that curve is to express the difference in the number of resistants generated for a pair of values {w, i} as ΔR(w, i) = ΔY(w, i) − ΔS(w, i), where
, analogous to the way in which the titers were obtained. The amount of resistants produced with either treatment will be equal at values of w and i that fulfill R = R. This finding defines curve CR. A convenient way of calculating some of the properties of that curve is to express the difference in the number of resistants generated for a pair of values {w, i} as ΔR(w, i) = ΔY(w, i) − ΔS(w, i), where 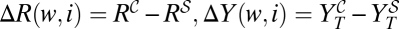 ; ΔS(w, i) = S0(im(1 − μ − w))G(1 − mG(1 − μ0 − w0)G) is defined as the difference in the amount of susceptible virus produced by the combined and sequential treatments, with v(G, w, i) as calculated in Materials and Methods.
; ΔS(w, i) = S0(im(1 − μ − w))G(1 − mG(1 − μ0 − w0)G) is defined as the difference in the amount of susceptible virus produced by the combined and sequential treatments, with v(G, w, i) as calculated in Materials and Methods.
The values of ΔR(w, i), when the yields of the two treatments are equal [that is, on curve C, where ΔY(w, i) = 0], are always positive, because ΔS(w, i) < 0 because of the biological fact that the replication rate of the susceptible type is larger than one in the absence of drugs: m(1 − μ0 − w0) > 1 and 1 ≥ μ + w. The production of resistant mutants is, thus, lower through the sequential treatment when both therapies are equally efficient in terms of total titer. This finding also implies that curve CR is always above curve C (Fig. 2F). Hence, there is a region of doses of mutagen and inhibitor between curves CR and C where the combination treatment leads to lower titers but at the same time, the sequential treatment causes a lower population of resistants.
In the limit of high doses of the inhibitor, i → 0, and because limi → 0Δv(w, i) → 0 (the population of susceptibles is completely suppressed), the total population coincides with the population of resistants and ΔR(w, i) → ΔY(w, i). As a result, the limit value of w on curve CR coincides with the limit value of w on curve C (Materials and Methods).
Effect of treatment and virus parameters.
Not all possible drug doses are experimentally meaningful. First, there is a minimum amount of mutagen required for the therapy to be effective, and it corresponds to those values of w large enough to cause a decrease of viral yield in the absence of the inhibitor. This condition takes the formal expression m(1 − w) < 1. We, thus, define curve Cw as those points where m(1 − w) = 1, separating the regions of increase and decrease of the population size under the action of the mutagen. A similar reasoning leads to curve Ci, which marks the values of i below which the inhibitor is able to cause a decrease of the viable population before resistance appears, (im)G(1 − w0)G = 1.
There is a limit under high enough values of the drug dose where either treatment may cause the extinction of the virus with the applied protocol. Curve 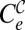 is determined by those combinations of {i, w} where the population size of the combined treatment falls below one. The exact solution for curve
is determined by those combinations of {i, w} where the population size of the combined treatment falls below one. The exact solution for curve 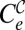 can only be given in implicit form as those values solution of YT(G, w, i) = 1. To first order in μ, the curve fulfils the approximate expression mGS0(1 − w)G − 1(iG(1 − w) + kwγ) ≅ 1. Analogously, curve
can only be given in implicit form as those values solution of YT(G, w, i) = 1. To first order in μ, the curve fulfils the approximate expression mGS0(1 − w)G − 1(iG(1 − w) + kwγ) ≅ 1. Analogously, curve 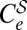 is defined by those pairs {i, w} at which S0−1YT(G, w0, i)YT(G, w, 1) = 1. Fig. 2 illustrates, in a representative case, the response of a viral population to the action of a combined or sequential treatment. All curves defined above (except for Cw, which occurs at values of w quite far from the domains shown) are represented in Fig. 2 B, D, and F.
is defined by those pairs {i, w} at which S0−1YT(G, w0, i)YT(G, w, 1) = 1. Fig. 2 illustrates, in a representative case, the response of a viral population to the action of a combined or sequential treatment. All curves defined above (except for Cw, which occurs at values of w quite far from the domains shown) are represented in Fig. 2 B, D, and F.
Parameters m and G depend significantly on the mode of replication of the virus inside an infected cell. ssRNA viruses are characterized by large values of m and probably, quite low values of G. In double-stranded genomes with semiconservative replication, one would expect a value of m ∼ 2 and a larger value of G. In both cases, the accumulation of mutations proceeds differently, and therefore, w0 would also vary accordingly. The model presented here cannot be directly applied to viruses that use other replication modes, such as retroviruses (with a DNA provirus phase) or DNA viruses such as the herpesviruses that include a latency step. Extension to such systems would require significant modifications of the dynamical rules.
Case of FMDV with R and GU.
The determination of viral productivity for a virus growing in excess of resources (P0) in the presence of a given amount of mutagen (Pw) and inhibitor (Pi) bounds for the values of the model parameters (SI Text). Because FMDV is an ssRNA virus, we take m between 50 and 1,000. As for the intrinsic mutation rate w0, data available put it in the range of 0.4–0.9. With a basal productivity of P0 = 460 ± 35, G is bounded between 1 and 3.8. Estimation of Pw yields the range of possible values of w, which is bounded between 0.992 and 0.999. Similarly, estimation of the productivity in the presence of different doses of the inhibitor permits estimating parameter i as i = (Pi/P0)1/G (SI Text). More accurate measurements of the viral parameters by means of specific experiments directed to quantify m, G, and/or w0 could significantly narrow the intervals compatible with the experimental results.
Results with FMDV subjected to the therapeutic protocols described under fixed doses of R and GU revealed that the sequential treatment could be better suited to cause viral extinction (24). However, in the light of the model described, the adequacy of sequential vs. combination treatment with a mutagen and an inhibitor of the viral replication depends on (i) the administered doses and (ii) the natural productivity of the virus when infecting a given host (i.e., parameters m, w0, and G). In particular, the model predicts that, in most cases at a sufficiently low amount of inhibitor and for a fixed value of the amount of mutagen, the combination treatment should yield a lower titer, the same change holding for a fixed amount of inhibitor and a sufficiently low amount of mutagen. Increase of the doses dGU or dR above a threshold value changes the treatment that produces the lower amount of infectious virions. To test this prediction, we carried out several experiments at increasing doses of inhibitor and compared the viral yield after both therapeutic protocols (Fig. 3A). At low doses of inhibitor, the combined therapy causes lower viral yields than the sequential therapy. However, for a dose of the inhibitor between 6 and 12 mM, it is the sequential treatment that begins to yield lower viral titers. The effect of the therapies is, thus, exchanged as theoretically predicted (Fig. 3B).
Fig. 3.

Experimental protocol results using FMDV infection of cell cultures. Comparison with theoretical predictions. (A) Experimental results. Passages were carried out by infecting 2 × 106 BHK-21 cells with pMT28 strain of FMDV (0.2 mL supernatant from the previous passage), and infectivity levels were determined as detailed in Materials and Methods. The combination or sequential protocol therapies were applied in the presence of a dose of mutagen dR = 5 mM and increasingly high doses of viral inhibitor, dGU = 3, 6, 12, or 20 mM, as indicated. The combination treatment is indicated in brackets (Left), whereas for the sequential treatment, the order of administration of GU and R is separated by a comma (Right). A control measure is shown for reference (first left bar in each panel), and other columns represent the mean ± SD (error bars) from triplicate determinations. The discontinuous lines in the virus titers indicate the limit of detection of infectivity. (B) Comparison of experimental results with the predictions of the model. Viral titers expected after applying the sequential or combination protocol in the mathematical model are shown as continuous lines (blue, combined treatment; red, sequential treatment; color code for data is the same as in A). Curves obtained with parameters that yield the least squares deviation to the logarithm of experimental data are shown as blue (combination) and red (sequential) curves. The obtained parameters are w0 = 0.76, w = 0.998, k = 0.005, and m = 195. The values of the inhibitor i = 4.25 × 10−2, 3.47 × 10−3, 2.10 × 10−4, and 1.41 × 10−4, corresponding to the four doses assayed, are shown in the x axis (SI Text).
Discussion
Ever since the quasispecies dynamics were revealed as general for pathogenic RNA viruses and retroviruses, the problem of treatment failure because of selection of drug-escape viral mutants was recognized (25). Several approaches have been used in medical practice to minimize selection of viral mutants resistant to antiviral agents. The most successful strategy was the implementation of combination therapy involving the simultaneous administration of two or more drugs directed to different viral targets. The advantages of combination therapy over monotherapy stem from basic statistical considerations on the frequency of generation of multidrug-resistant mutants (25). The advantage of combination vs. various regimens of sequential or treatment interruption (structured treatment interruptions or drug holidays) regimens has been supported by results of multiple clinical trials as well as theoretical models of viral dynamics (5, 11, 26–30). However, whenever residual viral replication is allowed, viral rebound and treatment failure often occur. For this reason, new proposals for the administration of drugs have been made. A recent one, yet to be tested in clinical trials, is the so-called proactive treatment in two steps: an induction regimen aimed at reducing the background of viral mutants followed by a maintenance regimen to control the viral load for extensive time periods (31). An alternative strategy is to target cellular factors needed for viral replication either alone or combined with drugs directed to viral functions (32). Provided no toxic effects on the cells intervene, resistance to inhibitors of cellular functions can also develop when the host factor interacts with viral nucleic acids or proteins.
Lethal mutagenesis introduces an important element in antiviral therapy in that a mutagenic agent is involved in treatment. When administered together with a nonmutagenic inhibitor, the mutagen can play a dual and opposite role: to deteriorate viral functions because of the excess mutations that it provokes, and increase the frequency of mutations that confer resistance to the partner, coadministered inhibitor. An advantage of a sequential over combination treatment during lethal mutagenesis protocols was suggested by a study of lethal mutagenesis of FMDV (33), and this advantage prompted the theoretical generalization reported in the present study and has been further supported by additional experiments also reported herein.
In this contribution, we have developed a theory that quantitatively describes the response of a viral population under two different protocols that involve the action of an inhibitor of viral replication and a mutagenic drug. A limited number of simple experiments allows us to estimate the parameters that describe the dominant processes, and knowledge of these few relevant parameters permits us to predict the behavior of the model for other combinations of drugs. Two important outcomes of the treatments have to be considered when choosing between a combined or sequential administration of the drugs: in a region of mutagen and inhibitor doses that can be calculated, the viral titer produced by one or another therapy is minimized. Furthermore, there is a domain of drug doses where combination treatment yields the lower titer, but the probability of appearance of mutants resistant to the inhibitor is lower with a sequential treatment. The use of analogous models can significantly reduce the number of in vitro assays to be performed in other viral systems as well, where different replication strategies should translate into dynamical equations similar to our Eqs. 1 and 2. The simulation of an in vivo situation entails additional difficulties, like development of a large viral population from an infecting seed, interaction with the immune system, or environmental and individual characteristics that may not lead to deterministic equations but should be included as noisy, fluctuating dynamical variables. The predictions of the model after being tested in vitro could be taken only as a rough guide to apply one or another administration protocol and infer minimum drug doses in in vivo assays.
Our predictions acquire more relevance in view of the evidence that lethal mutagenesis can indeed be effective in vivo (34) and that some clinical trials for AIDS patients that involve administration of a mutagenic nucleoside analog have been implemented (35). Our results will be particularly relevant when considering a lethal mutagenesis approach to combat viruses for which a repertoire of nonmutagenic inhibitory agents is already available. Detailed predictions of the viral response tailored to the particular system under study are now possible.
Materials and Methods
Cells and Viruses.
The origin of BHK-21 cells and procedures for cell growth in DMEM and plaque assays in semisolid agar have been previously described (36, 37). FMDV C-S8c1 is a plaque-purified derivative of serotype C isolate C1 Santa Pau-Sp70 (37). An infectious clone of FMDV C-S8c1, termed pMT28, was constructed by recombining into pGEM-1 plasmid subclones that represented the C-S8c1 genome as described (38, 39). Thus, FMDV pMT28 used in the experiments is the progeny of infectious transcripts that express the standard FMDV C-S8c1. To control for the absence of contamination, mock-infected cells were cultured, and their supernatants were titrated in parallel with the infected cultures; no signs of infectivity or cytopathology in the cultures or control plaque assays were observed in any of the experiments.
Treatment with R and GU.
A solution of GU in DMEM was prepared at a concentration of 50 mM, sterilized by filtration, and stored at 4 °C. A solution of R in PBS was prepared at a concentration of 100 mM, sterilized by filtration, and stored at −70 °C. Before use, the stock solutions were diluted in DMEM to reach the desired concentration. For infections of BHK-21 cells with FMDV in the presence of GU, no pretreatment of the cell monolayer with GU was performed. For infections in the presence of R, cell monolayers were pretreated during 7 h with 5 mM R before infection. After addition of FMDV and washing of the cell monolayers, infections were allowed to continue in the presence of a combination of GU + R or sequential passages consisting of a first passage in the presence of GU and a second passage in the presence of R. For the combination treatment, the infections were carried out at an MOI of 0.4 pfu/cell. For the sequential treatment, the initial infection in the presence of increasing concentrations of GU was carried out also at an MOI of 0.4 pfu/cell. The second infection in the presence of 5 mM R was carried out at MOIs of 1.1, 2.0 × 10−2, 2.2 × 10−4, and 1.2 × 10−4 pfu/cell for GU 3, 6, 12, and 20 mM GU, respectively. Infections in the absence of GU, R, or a combination of GU + R and mock-infected cells were maintained in parallel, and no evidence of contamination of cells with virus was observed at any time.
Complete Solution of Eqs. 1 and 2.
The dynamics of the model system can be written in compact form through the vector n(g), whose components are the number of individuals in each of the classes v and V after g replication cycles (Eq. 5):
with A being the transition matrix of the system (Eq. 6),
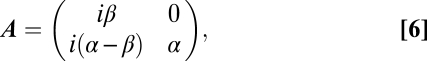 |
and where we have defined α = 1 − w and β = 1 − μ − w. This dynamical system can be exactly solved for the initial condition n(0) = {S0, 0} to yield the population of each viral class after g cycles (Eqs. 7 and 8),
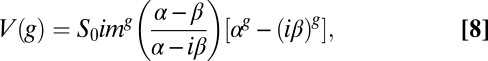 |
from where the exact expression for the total number of viable elements is obtained (Eq. 3).
Approximate Analytic Expression for C.
The expression obtained from Eq. 4 yields the dependence between w and i in an essential nonalgebraic way, and therefore, it can be only numerically solved. However, if we assume that μ ≪ 1, an expansion in powers of μ yields the following approximate dependence for the points on curve C (Eq. 9):
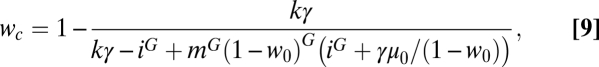 |
where γ = [i + iG(iG − i − G)]/(1 − i). Given the amount of mutagen i, the sequential treatment causes a larger decrease in the viral titer for values of w > wc, whereas the combined treatment is more efficient for w < wc. The preferred therapy changes as well if the value of w is fixed and the amount of inhibitor increases. For values of i close to one, the combined treatment is better, whereas the sequential treatment will be preferred above a certain amount of inhibitor. Curve (Eq. 9) has two important limits, absence of inhibitor (i → 1) and large doses of inhibitor (i → 0). In the former case, both treatments become equivalent for values of w → 1 (Fig. 2F), a situation where all genomes produced under replication would be nonviable. Actually, w = 1 cannot be empirically tested, because the complete extinction of the population occurs at values of w below one, which will be shown. However, there is a saturation effect when the amount of inhibitor is very high in the sense that additional decreases in i diminish the viral titer but do not change the preference for one or another therapy (Eq. 10):
 |
which is independent of i.
Supplementary Material
Acknowledgments
The authors acknowledge the support of Comunidad Autónoma de Madrid through a grant to J.I. and through Project MODELICO (S2009/ESP-1691; to S.C.M. and J.I.). Work at Centro de Astrobiología was supported by Ministerio de Ciencia e Innovación Project FIS2008-05273. Work at Centro de Biología Molecular “Severo Ochoa” was supported by Grant BFU2008-02816/BMC from MICINN. Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas is funded by Instituto de Salud Carlos III.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
†Under low multiplicity of infection (MOI), only viable individuals can infect new cells. Independent of the size of the population of nonviable mutants, they are unable to produce infection in the next passage because of the low MOI (from 10−5 to 10−1 pfu/cell) (Materials and Methods). This is the reason why the dynamic equations for nonviable types are not explicitly considered.
‡For the sake of simplicity, back mutations are not considered in the model. They would represent a small contribution to the population numbers of type v in the form of an additive term of order mμ and do not significantly affect population dynamics, because initially, only susceptible individuals are present. Note that a term of the same order is, however, required to trigger the growth of population V. After resistant individuals have appeared, their dynamics are dominated by the term of order (1 − w)m, in front of which the first term could be then discarded.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110489108/-/DCSupplemental.
References
- 1.Richman DD, editor. Antiviral Drug Resistance. New York: Wiley; 1996. [Google Scholar]
- 2.Domingo E, et al. Viral quasispecies and the problem of vaccine-escape and drug-resistant mutants. Prog Drug Res. 1997;48:99–128. doi: 10.1007/978-3-0348-8861-5_4. [DOI] [PubMed] [Google Scholar]
- 3.Endy D, Yin J. Toward antiviral strategies that resist viral escape. Antimicrob Agents Chemother. 2000;44:1097–1099. doi: 10.1128/aac.44.4.1097-1099.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vignuzzi M, Wendt E, Andino R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat Med. 2008;14:154–161. doi: 10.1038/nm1726. [DOI] [PubMed] [Google Scholar]
- 5.Domingo E, Grande-Pérez A, Martín V. Future prospects for the treatment of rapidly evolving viral pathogens: Insights from evolutionary biology. Expert Opin Biol Ther. 2008;8:1455–1460. doi: 10.1517/14712598.8.10.1455. [DOI] [PubMed] [Google Scholar]
- 6.Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nat Rev Drug Discov. 2005;4:71–78. doi: 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- 7.Torella JP, Chait R, Kishony R. Optimal drug synergy in antimicrobial treatments. PLoS Comput Biol. 2010;6:e1000796. doi: 10.1371/journal.pcbi.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Handel A, Regoes RR, Antia R. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput Biol. 2006;2:e137. doi: 10.1371/journal.pcbi.0020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–466. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 10.Yeh P, Tschumi AI, Kishony R. Functional classification of drugs by properties of their pairwise interactions. Nat Genet. 2006;38:489–494. doi: 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- 11.Bonhoeffer S, May RM, Shaw GM, Nowak MA. Virus dynamics and drug therapy. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komarova NL, Barnes E, Klenerman P, Wodarz D. Boosting immunity by antiviral drug therapy: A simple relationship among timing, efficacy, and success. Proc Natl Acad Sci USA. 2003;100:1855–1860. doi: 10.1073/pnas.0337483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pariente N, Airaksinen A, Domingo E. Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J Virol. 2003;77:7131–7138. doi: 10.1128/JVI.77.12.7131-7138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pariente N, Sierra S, Airaksinen A. Action of mutagenic agents and antiviral inhibitors on foot-and-mouth disease virus. Virus Res. 2005;107:183–193. doi: 10.1016/j.virusres.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Perales C, Mateo R, Mateu MG, Domingo E. Insights into RNA virus mutant spectrum and lethal mutagenesis events: Replicative interference and complementation by multiple point mutants. J Mol Biol. 2007;369:985–1000. doi: 10.1016/j.jmb.2007.03.074. [DOI] [PubMed] [Google Scholar]
- 16.González-López C, Arias A, Pariente N, Gómez-Mariano G, Domingo E. Preextinction viral RNA can interfere with infectivity. J Virol. 2004;78:3319–3324. doi: 10.1128/JVI.78.7.3319-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eigen M, Schuster P. The Hypercycle. A Principle of Natural Self-Organization. Berlin: Springer; 1979. [DOI] [PubMed] [Google Scholar]
- 18.Eigen M. Error catastrophe and antiviral strategy. Proc Natl Acad Sci USA. 2002;99:13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domingo E, Escarmís C, Lázaro E, Manrubia SC. Quasispecies dynamics and RNA virus extinction. Virus Entry into Error Catastrophe as a New Antiviral Strategy, ed E. Domingo. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Lee CH, et al. Negative effects of chemical mutagenesis on the adaptive behavior of vesicular stomatitis virus. J Virol. 1997;71:3636–3640. doi: 10.1128/jvi.71.5.3636-3640.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: Direct molecular test by using ribavirin. Proc Natl Acad Sci USA. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manrubia SC, Domingo E, Lázaro E. Pathways to extinction: Beyond the error threshold. Philos Trans R Soc Lond B Biol Sci. 2010;365:1943–1952. doi: 10.1098/rstb.2010.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cases-González C, Arribas M, Domingo E, Lázaro E. Beneficial effects of population bottlenecks in an RNA virus evolving at increased error rate. J Mol Biol. 2008;384:1120–1129. doi: 10.1016/j.jmb.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 24.Perales C, Agudo R, Tejero H, Manrubia SC, Domingo E. Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS Pathog. 2009;5:e1000658. doi: 10.1371/journal.ppat.1000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domingo E, Holland JJ. Complications of RNA heterogeneity for the engineering of virus vaccines and antiviral agents. In: Setlow JK, editor. Genetic Engineering, Principles and Methods. Vol 14. New York: Plenum; 1992. pp. 13–31. [DOI] [PubMed] [Google Scholar]
- 26.Ho DD. Time to hit HIV, early and hard. N Engl J Med. 1995;333:450–451. doi: 10.1056/NEJM199508173330710. [DOI] [PubMed] [Google Scholar]
- 27.Domingo E. RNA virus evolution and the control of viral disease. Prog Drug Res. 1989;33:93–133. doi: 10.1007/978-3-0348-9146-2_5. [DOI] [PubMed] [Google Scholar]
- 28.Nijhuis M, van Maarseveen NM, Boucher CA. Handbook of Experimental Pharmacology. Berlin: Springer; 2009. pp. 299–320. [DOI] [PubMed] [Google Scholar]
- 29.Ribeiro RM, Bonhoeffer S. Production of resistant HIV mutants during antiretroviral therapy. Proc Natl Acad Sci USA. 2000;97:7681–7686. doi: 10.1073/pnas.97.14.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller V, Bonhoeffer S. In: Origin and Evolution of Viruses. 2nd Ed. Domingo E, Parrish CR, Holland JJ, editors. London: Academic; 2008. pp. 279–302. [Google Scholar]
- 31.von Kleist M, et al. HIV quasispecies dynamics during pro-active treatment switching: Impact on multi-drug resistance and resistance archiving in latent reservoirs. PLoS One. 2011;6:e18204. doi: 10.1371/journal.pone.0018204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geller R, Vignuzzi M, Andino R, Frydman J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007;21:195–205. doi: 10.1101/gad.1505307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perales C, Agudo R, Domingo E. Counteracting quasispecies adaptability: Extinction of a ribavirin-resistant virus mutant by an alternative mutagenic treatment. PLoS One. 2009;4:e5554. doi: 10.1371/journal.pone.0005554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz-Jarabo CM, Ly C, Domingo E, de la Torre JC. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) Virology. 2003;308:37–47. doi: 10.1016/s0042-6822(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 35.Mullins JI, et al. Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS One. 2011;6:e15135. doi: 10.1371/journal.pone.0015135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domingo E, Dávila M, Ortín J. Nucleotide sequence heterogeneity of the RNA from a natural population of foot-and-mouth-disease virus. Gene. 1980;11:333–346. doi: 10.1016/0378-1119(80)90073-6. [DOI] [PubMed] [Google Scholar]
- 37.Sobrino F, Dávila M, Ortín J, Domingo E. Multiple genetic variants arise in the course of replication of foot-and-mouth disease virus in cell culture. Virology. 1983;128:310–318. doi: 10.1016/0042-6822(83)90258-1. [DOI] [PubMed] [Google Scholar]
- 38.García-Arriaza J, Manrubia SC, Toja M, Domingo E, Escarmís C. Evolutionary transition toward defective RNAs that are infectious by complementation. J Virol. 2004;78:11678–11685. doi: 10.1128/JVI.78.21.11678-11685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toja M, Escarmís C, Domingo E. Genomic nucleotide sequence of a foot-and-mouth disease virus clone and its persistent derivatives. Implications for the evolution of viral quasispecies during a persistent infection. Virus Res. 1999;64:161–171. doi: 10.1016/s0168-1702(99)00089-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.