

Abstract
This Letter describes a chemical lead optimization campaign directed at a weak mGlu5 NAM discovered while developing SAR for the mGlu5 PAM, ADX-47273. An iterative parallel synthesis effort discovered multiple, subtle molecular switches that afford potent mGlu5 NAMs, mGlu5 PAMs as well as mGlu5 partial antagonists.
The metabotropic glutamate receptor subtype 5 (mGlu5) has become a prominent molecular target for a number of CNS pathologies.1,2 mGlu5 negative allosteric modulators (NAMs) are being actively pursued for anxiety, pain, Parkinson’s disease, cocaine addiction and Fragile × Syndrome, while mGlu5 positive allosteric modulators (PAMs) are under development for the treatment of schizophrenia.3–9 The prototypical mGlu5 allosteric ligand is MPEP (1),10 a NAM, and many allosteric ligands, both PAM and NAM, bind at the MPEP-site.1–10 Recently, we reported on the discovery of molecular switches in a series of MPEP-site phenylethynyl pyrimidines in which incorporation of a single methyl group in either the 3- or 4-position converted an mGlu5 partial antagonist lead 2 (IC50 = 486 nM, 71% partial) into either a NAM 3 (IC50 = 7.5 nM) or PAM 4 (EC50 = 3.3 μM, 4.2-fold shift), respectively (Fig. 1).11 Further SAR identified additional, subtle molecular switches that afforded centrally penetrant and in vivo active mGlu5 NAMs and PAMs.12 After these key findings, we began to take note of pharmacology switches, and identified these in multiple mGlu5 allosteric modulator scaffolds.13,14 Interestingly, our initial SAR work in the mGlu5 PAM ADX-47273 5 series in 2009 produced potent PAMs, such as 6 (EC50 = 240 nM, 14-fold shift), and ago-PAMs such as 7 (EC50 = 170 nM, 20-fold shift), but only one weak NAM 8 (IC50 = 8.7 μM).15 This was the first indication that pharmacology switching is possible in the ADX-47273 series by replacing an aryl amide, as in 6, with a cyclobutyl amide in 8.15 While we were exploring this finding, a manuscript appeared in 2010 describing the identification of racemic mGlu5 NAM 9, closely related to our NAM 8, from an HTS screen, and the parallel synthesis of over 1,300 analogs.16 However, within this manuscript, there is little discussion of the impact of stereochemistry and no mention of pharmacology switching. Here, we present our SAR study, developed though an iterative parallel synthesis approach, that afforded potent mGlu5 PAMs, NAMs and partial antagonists from subtle modifications to the ADX-47273 scaffold.
Figure 1.

Structures of selected MPEP-site allosteric ligands that display a range of mGlu5 pharmacology with subtle modifications.
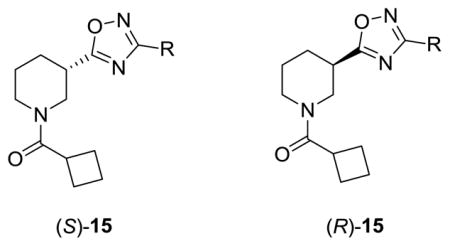
Our initial library evaluated two dimensions: stereochemistry at the 3-postion and replacement for the 2-pyridyl moiety while holding the cyclobutyl amide constant. In our earlier work in the ADX-47273 series,15 the (S)-stereochemistry at the 3-position was essential for mGlu5 PAM activity, and it was important to ascertain the stereochemical bias, if any, to produce NAMs. In the event, (S)-10 was converted to the methyl ester 11, followed by acylation to yield 12. Saponification provides 13, which is then coupled to various (Z)-N′-hydroxylimidamides 14 and refluxed to deliver analogs (S)-15 (Scheme 1). The analogous (R)-15 congeners were made via the same scheme except (R)-10 was used.
Scheme 1.

Reagents and conditions: (a) SOCl2, MeOH (99%); cylcobutane carbonyl chloride, DIEA, DCM (96%); (c) LiOH, THF, H2O (95%); (d) EDCI, HOBt, DIEA, dioxane, reflux, 24 h (45–59%).
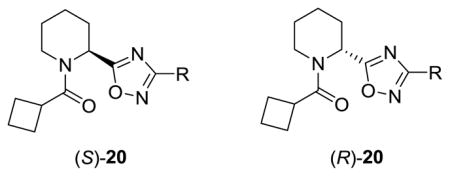
As shown in Table 1, the stereochemical preference we identified in our earlier PAM work in this series carried over into the NAM pharmacology with the (S)-enantiomer preferred, ie., (S)-15e (IC50= 0.2 μM) versus (R)-15e (IC50= 3.1 μM). Significantly, 3-substituted aryl congeners (S)-15e–f, proved most enlightening, affording submicromolar mGlu5 NAMs, with in the case of (S)-15e, an ~41-fold increase in potency over 8.15 These data led us to consider if there is stereochemical bias for pharmacological mode of action within the 9 scaffold. Thus we prepared small, enantiopure libraries of analogs (S)-20 and (R)-20, from either (S)-16 and (R)-16, respectively, and evaluated them in our mGlu5 assays (Scheme 2). As shown in Table 2, this effort found that both enantiomers afford comparable activity and mode of pharmacology. This library provided an efficacious submicromolar PAM (S)-20c (EC50 = 730 nM, 71% Glu Max) as well as several submicromolar NAMs ((S)- and (R)-20e–f) which also afforded a full blockade of the EC80, and in the case of (S)-20f, an 77 nM NAM. Based on these data, our next round of library synthesis employed both the 20e NAM scaffold and the 20c PAM scaffold, and focused on evaluating other amide moieties beyond the cyclobutyl amide. These analogs 21 and 22 were readily prepared following a variation of Scheme 2.
Table 1.
Structures and activities of analogs (S)-15 and (R)-15.
 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Pharmacology | IC50 (μM)a | EC50 (μMa) | Glu Max (%)a |
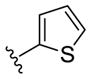
| (S)-15a |
|
NAM | 9.3 | NA | 67 |
| (R)-15a | Inactive | - | - | - | |
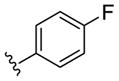
| (S)-15b |
|
Inactive | - | - | - |
| (R)-15b | Inactive | - | - | - | |
| (S)-15c |
|
NAM | >10 | NA | 33 |
| (R)-15c | NAM | 9.9 | NA | 19 | |
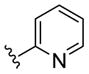
| (S)-15d |
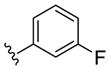
|
NAM | 2.4 | NA | 31 |
| (R)-15d | NAM | >10 | NA | 60 | |
| (S)-15e |
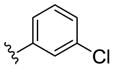
|
NAM | 0.2 | NA | 2.4 |
| (R)-15e | NAM | 3.1 | NA | 18 | |
| (S)-15f |
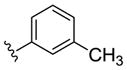
|
NAM | 0.7 | NA | 2.5 |
| (R)-15f | NAM | 4.7 | NA | 14 | |
| (S)-15g |
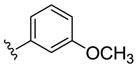
|
NAM | 1.8 | NA | 2.1 |
| (R)-15g | NAM | >10 | NA | 54 | |
Average of at least three independent determinations. NA, not applicable.
Scheme 2.

Reagents and conditions: (a) SOCl2, MeOH (99%); cylcobutane carbonyl chloride, DIEA, DCM (95%); (c) LiOH, THF, H2O (95%); (d) EDCI, HOBt, DIEA, dioxane, reflux, 24 h (40–55%).
Table 2.
Structures and activities of analogs (S)-20 and (R)-20.
 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Pharmacology | IC50 (μM)a | EC50 (μMa) | Glu Max (%)a |
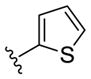
| (S)-20a |
|
NAM | 2.6 | NA | 11 |
| (R)-20a | NAM | 3.5 | NA | 7 | |
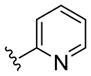
| (S)-20b |
|
NAM | 10 | NA | 33 |
| (R)-20b | Inactive | - | - | - | |
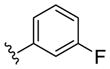
| (S)-20c |
|
PAM | NA | 0.7 | 71 |
| (R)-20c | PAM | NA | 0.6 | 37 | |
| (S)-20d |
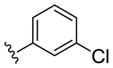
|
NAM | 0.9 | NA | 6 |
| (R)-20d | NAM | 10 | NA | 38 | |
| (S)-20e |
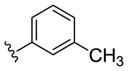
|
NAM | 0.5 | NA | 3 |
| (R)-20e | NAM | 0.3 | NA | 6 | |
| (S)-20f |
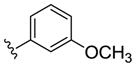
|
NAM | 0.08 | NA | 1.8 |
| (R)-20f | NAM | 0.3 | NA | 0.5 | |
Average of at least three independent determinations. NA, not applicable.
The library of 20e analogs, 21a–i, afforded both NAMs and partial antagonists,17 with no evidence of PAM activity (Table 3). Interestingly, the 3- and 5-membered saturated ring amides 21a and 21c, afforded partial antagonists, while the 4- and 6-membered saturated ring amides 21b and 21d afforded full non-competitive antagonists (NAMs). In contrast, the library of 20c analogs, 22a–i, afforded predominantly PAMs and ago-PAMs. For example, 22a proved to be a potent (EC50 = 78 nM, 70% Glu Max) mGlu5 PAM, more potent than the previous PAMs 6 and 7 we developed in the ADX-47273 series.15 In addition, we observed an interesting trend here with the 3- and 5-membered saturated ring amides 22a and 22c affording ago-PAMs, while the 4- and 6-membered saturated ring amides 22b and 22d displaying pure PAM activity. Again, very subtle perturbations to the core scaffold engender opposing modes of mGlu5 pharmacology.
Table 3.
Structures and activities of analogs 21 and 22.
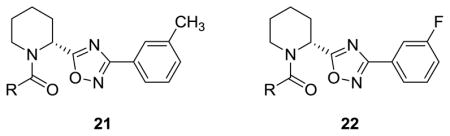 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Pharmacology | IC50 (μM)a | EC50 (μMa) | Glu Max (%)a |
| 21a |
|
Part. Antag. | 0.28 | NA | 16 |
| 22a | Ago-PAM | NA | 0.08 | 70 | |
| 21b/20c |
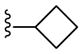
|
NAM | 0.37 | NA | 5.8 |
| 22b/20e | PAM | NA | 0.65 | 38 | |
| 21c |
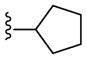
|
Part. Antag. | 0.6 | NA | 24 |
| 22c | Ago-PAM | NA | 0.31 | 80 | |
| 21d |
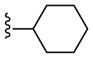
|
NAM | 2.5 | NA | 9.2 |
| 22d | PAM | NA | 3.5 | 58 | |
| 21e |
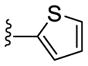
|
NAM | 0.73 | NA | 0.9 |
| 22e | NAM | 10 | NA | 34 | |
| 21f |
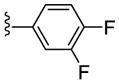
|
NAM | 10 | NA | 46 |
| 22f | PAM | NA | 1.4 | 93 | |
| 21g |
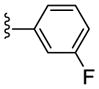
|
NAM | 4.7 | NA | 16 |
| 22g | PAM | NA | 0.46 | 59 | |
| 21h |
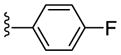
|
NAM | 10 | NA | 47 |
| 22h | PAM | NA | 2.3 | 91 | |
| 21i |
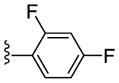
|
Inactive | - | - | - |
| 22i | PAM | NA | 2.9 | 95 | |
Average of at least three independent determinations. NA, not applicable.
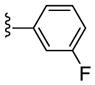
At this point, we elected to examine the impact of contracting the piperidine ring to a pyrrolidine ring while maintaining the original cyclobutyl amide and surveying a diverse group of subsitutents on the oxadiazole ring. This initial library employed racemic proline to afford racemic analogs 23, following a variation of scheme 2. As shown in Table 4, this modification afforded inactive compounds, weak NAMs (IC50s ~ 10 μM) and two low micromolar PAMs (23a and 23b). Based on these data, we made a second generation library holding constant the 3-fluorobenzene moiety of 23a, and surveyed a diverse collection of amide moieties to replace the cyclobutyl group. As shown in Table 5, this effort afforded predominantly pure PAMs 24 with a range of potencies and efficacies. To address the role of sterochemical preference, we separated racemic 23a into pure enantiomers (S)-23a and (R)-23a by chiral SFC. In this case, (R)-23a is a potent mGlu5 PAM (EC50 = 530 nM) while (S)-23a is a very weak PAM (EC50 = 7,000 nM) (Fig. 2). Note, this is the opposite stereochemical preference observed within the 3-piperidinyl-based mGlu5 PAMs 5–7.15
Table 4.
Structures and activities of analogs 23.
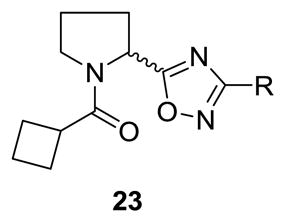 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Pharmacology | IC50 (μM)a | EC50 (μMa) | Glu Max (%)a |
| 23a |
|
PAM | NA | 1.5 | 64 |
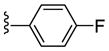
| 23b |
|
PAM | NA | 2.7 | 72 |
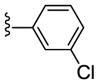
| 23c |
|
NAM | 10 | NA | 25 |
| 23d |
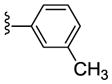
|
NAM | 2.4 | NA | 34 |
| 23e |
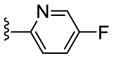
|
Inactive | NA | NA | NA |
| 23f |
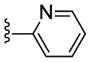
|
Inactive | NA | NA | NA |
| 23g |
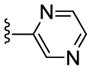
|
Inactive | NA | NA | NA |
Average of at least three independent determinations. NA, not applicable.
Table 5.
Structures and activities of analogs 24.
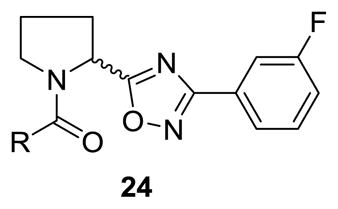 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Pharmacology | IC50 (μM)a | EC50 (μMa) | Glu Max (%)a |
| 24a/23a |
|
PAM | NA | 2.9 | 68 |
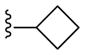
| 24b |
|
PAM | NA | 1.5 | 63 |
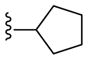
| 24c |
|
PAM | NA | 1.0 | 75 |
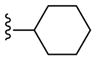
| 24d |
|
PAM | NA | 10 | 8 |
| 24e |
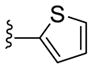
|
PAM | NA | 0.43 | 60 |
| 24f |
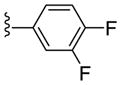
|
PAM | NA | 3.7 | 62 |
| 24g |
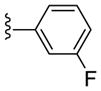
|
PAM | NA | 2.6 | 47 |
| 24h |
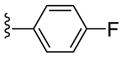
|
PAM | NA | 3.6 | 59 |
| 24i |
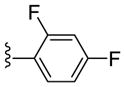
|
PAM | NA | 10 | 61 |
Average of at least three independent determinations. NA, not applicable.
Figure 2.

Resolved enantiomers of 23a. All the mGlu5 PAM activity resides in the (R)-enantiomers, (R)-23a.
In summary, an iterative parallel synthesis optimization approach for our weak mGlu5 NAM 8, identified multiple regioisomeric and stereochemical ‘molecular switches’ that modulated modes (NAM, partial antagonist, PAM, ago-PAM) of mGlu5 pharmacology. From 8 (IC50 = 8.7 μM), potent PAMs (EC50 = 78–200 nM) and NAMs (IC50 = 77–400 nM), were developed. In many cases, the perturbations in structure were subtle which led to opposing modes of pharmacology and suggests subtle conformational changes within the GPCR either facilitate or prohibit coupling to the G-protein. These data, coupled with our earlier work in a structurally distinct MPEP-site scaffold 2, suggests that metabolites of MPEP-site allosteric ligands must be characterized, as metabolites may engender opposing modes of mGlu5 modulation. Additional studies with these ligands, as well as their metabolites, are in progress and will be reported in due course.
Acknowledgments
The authors thank NIDA (DA023947-01) for support of our programs in developing mGlu5 NAMs and partial antagonists for the treatment of addiction. The authors would like to thank Nathan Kett, Chris Denicola and Sichen Chang for the purification of compounds utilizing the mass-directed HPLC system.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.a) Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]; b) Conn PJ, Pin JP. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.a) Lea PM, IV, Faden AI. CNS Drug Reviews. 2006;12(2):149–166. doi: 10.1111/j.1527-3458.2006.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kew JNC. Pharmacol Ther. 2004;104:233–244. doi: 10.1016/j.pharmthera.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Lindsley CW, Emmitte KA. Curr Opin, Drug Disc Dev. 2009;12:446–457. [PubMed] [Google Scholar]
- 4.Conn PJ, Lindsley CW, Jones C. Trends in Pharm Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn PJ, Christopolous A, Lindsley CW. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindsley CW, Wisnoski DD, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. J Med Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 7.Hammond AS, Rodriguez AL, Niswender CM, Lindsley CW, Conn PJ. ACS Chem Neuro. 2010;1:702–716. doi: 10.1021/cn100051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Z, Wisnoski DD, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg Med Chem Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 9.Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Conn PJ, Lindsley CW, Emmitte KA. Bioorg Med Chem Lett. 2010;20:4390–4394. doi: 10.1016/j.bmcl.2010.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Veliocelebi G, Kuhn R. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:4098–5101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma S, Kedrowski J, Rook JM, Smith JM, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. J Med Chem. 2009;52:4103–4106. doi: 10.1021/jm900654c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez AL, Williams R, Zhou S, Lindsley SR, Le U, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2009;19:3209–3212. doi: 10.1016/j.bmcl.2009.04.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Rodriguez AL, Williams R, Weaver CD, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2009;19:6502–6506. doi: 10.1016/j.bmcl.2009.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engers DW, Rodriguez AL, Oluwatola O, Hammond AS, Venable DF, Williams R, Sulikowski GA, Conn PJ, Lindsley CW. Chem Med Chem. 2009;4:505–511. doi: 10.1002/cmdc.200800357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wágner G, Wéber C, Nyéki O, Nógrádi K, Bielik A, Molnár L, Bobok A, Horváth A, Kiss B, Kolok S, Nagy J, Kurkó D, Gál K, Greiner I, Szombathelyi Z, Keser GM, Dómany G. Bioorg Med Chem Lett. 2010;20:3737–3741. doi: 10.1016/j.bmcl.2010.04.075. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. Mol Pharmacol. 2005;68:1793–1802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]