

Abstract
The mechanisms subserving the ability of glucocorticoid signaling within the medial prefrontal cortex (mPFC) to terminate stress-induced activation of the hypothalamic–pituitary–adrenal (HPA) axis are not well understood. We report that antagonism of the cannabinoid CB1 receptor locally within the mPFC prolonged corticosterone secretion following cessation of stress in rats. Mice lacking the CB1 receptor exhibited a similar prolonged response to stress. Exposure of rats to stress produced an elevation in the endocannabinoid 2-arachidonoylglycerol within the mPFC that was reversed by pretreatment with the glucocorticoid receptor antagonist RU-486 (20 mg/kg). Electron microscopic and electrophysiological data demonstrated the presence of CB1 receptors in inhibitory-type terminals impinging upon principal neurons within layer V of the prelimbic region of the mPFC. Bath application of corticosterone (100 nm) to prefrontal cortical slices suppressed GABA release onto principal neurons in layer V of the prelimbic region, when examined 1 h later, which was prevented by application of a CB1 receptor antagonist. Collectively, these data demonstrate that the ability of stress-induced glucocorticoid signaling within mPFC to terminate HPA axis activity is mediated by a local recruitment of endocannabinoid signaling. Endocannabinoid activation of CB1 receptors decreases GABA release within the mPFC, likely increasing the outflow of the principal neurons of the prelimbic region to contribute to termination of the stress response. These data support a model in which endocannabinoid signaling links glucocorticoid receptor engagement to activation of corticolimbic relays that inhibit corticosterone secretion.
Introduction
Exposure to stressful stimuli evokes a well characterized activation of the hypothalamic–pituitary–adrenal (HPA) axis that results in the secretion of glucocorticoids into the circulation (Pecoraro et al., 2006). In the short term, glucocorticoids optimize physiological and metabolic conditions such that an organism can appropriately respond to the threat at hand by mobilizing glucose stores, trafficking leukocytes, and enhancing vigilance and attention (McEwen et al., 1997; Pecoraro et al., 2006). However, persistent glucocorticoid secretion can produce detrimental effects on cardiovascular, metabolic, and neural systems, and is associated with many disease states such as hypertension, type II diabetes, and mood disorders (McEwen, 2008; Chrousos, 2009). Accordingly, secretion of glucocorticoids is tightly regulated by neural and hormonally mediated negative feedback processes that limit the magnitude and duration of HPA axis activity through both rapid and delayed processes. Rapid feedback inhibition of the HPA axis is accomplished by local actions of glucocorticoids at the pituitary and the paraventricular nucleus of the hypothalamus (PVN), but the long-loop feedback inhibition of HPA axis activity is driven by upstream corticolimbic structures that communicate with the hypothalamus (Herman et al., 2003; Pecoraro et al., 2006).
Neuroendocrine and neuroanatomical studies have identified the medial prefrontal cortex (mPFC) as a critical site of action for glucocorticoid-mediated termination of HPA axis activity following exposure to stress. Glucocorticoid receptors (GRs) are present within the mPFC, and corticosterone implants within this region dampen stress-induced activation of the HPA axis and accelerate the return of circulating glucocorticoid concentrations to baseline (Diorio et al., 1993). Lesions of the mPFC, in particular the prelimbic region of the mPFC, impair termination of HPA axis activity following cessation of stress (Diorio et al., 1993; Figueiredo et al., 2003; Spencer et al., 2005; Radley et al., 2006, 2009). Elegant anatomical studies have delineated the circuit subserving prefrontal cortical regulation of the HPA axis. This circuit involves activation of glutamatergic afferents from the prelimbic region of the mPFC, which then activate inhibitory relays to the PVN in the bed nucleus of the stria terminalis, and possibly the peri-PVN region (Spencer et al., 2005; Radley et al., 2006, 2009; Ulrich-Lai and Herman, 2009). Despite the mapping of this circuit, surprisingly little is known about the mechanisms by which glucocorticoids modulate mPFC neuronal activity to promote activation of output projections that contribute to termination of HPA axis activity.
Several lines of evidence suggest that the endocannabinoid (eCB) system could be involved in coordinating the effects of glucocorticoids on mPFC neuronal activation. First, both in vitro and in vivo studies have demonstrated that glucocorticoids increase eCB signaling (Di et al., 2005; Malcher-Lopes et al., 2006; Hill et al., 2010b). Second, the eCBs N-arachidonylethanolamine [anandamide (AEA)] and 2-arachidonoylglycerol (2-AG), modulate the balance of excitation and inhibition within a given neural circuit through their ability to inhibit synaptic release of neurotransmitters via activation of presynaptic cannabinoid CB1 receptors (CB1Rs) (Freund et al., 2003). Third, disruption of eCB/CB1R signaling promotes activation of the HPA axis, indicating that this system negatively regulates activation of the HPA axis (Patel et al., 2004; Steiner and Wotjak, 2008; Hill and McEwen, 2010). Together, these data suggest that glucocorticoids could recruit eCB signaling to increase the outflow of the mPFC and terminate corticosterone secretion. The current study sought to examine this hypothesis.
Materials and Methods
Neuroendocrine studies.
Seventy-day-old male Sprague Dawley rats (300 g; Charles River) were used to determine the role of CB1R signaling within the mPFC. The rats were pair housed (except following surgical procedures, when they were individually housed) in standard maternity bins lined with contact bedding. Colony rooms were maintained at 21°C, and on a 12 h light/dark cycle, with lights on at 9:00 A.M. All rats were given ad libitum access to Purina Rat Chow and tap water. All protocols were approved by the Canadian Council for Animal Care and the Animal Care Committee of the University of British Columbia. All studies occurred during the first third of the light cycle, during the daily nadir of HPA axis activity.
For microinjection studies, animals were subjected to stereotaxic surgery. Rats were anesthetized with 100 mg/kg ketamine hydrochloride and 7 mg/kg xylazine, and implanted with bilateral 23 gauge stainless steel guide cannulae into the mPFC (coordinate: anterior–posterior +3.0; medial-lateral ±0.7; dorsal-ventral −3.4) (Paxinos and Watson, 1998). Four steel screws and dental acrylic were used to permanently affix the guide cannulae to the skull. Stainless steel stylets (30 gauge) were inserted into the guide cannulae until the time of infusion. Immediately following surgery, antibiotic ointment was applied to the skull and surrounding incision. All rats were allowed 1 week of recovery before testing and were individually housed during this period.
For stress testing, subjects were put into a polystyrene tube (diameter 6 cm, length 20 cm) with breathing holes. Tubes were long enough to completely encase the rat and too narrow for turning or other large movements. Rats were left in the tubes for 30 min, then removed and returned to their home cage. Subjects were randomly divided to receive either AM251 (0.28 ng/side) or vehicle (1 part dimethyl sulfoxide, 9 parts 0.9% sterile saline), and animals received bilateral infusions of either solution 10 min before the initiation of restraint stress. A 30 gauge injection cannula extending 0.8 mm below the tips of the guide cannulae was used for infusions. Drug solutions or vehicle were delivered at a rate of 0.2 μl/28 s using a microsyringe pump (Model 341, Sage Instruments). Injection cannulae were left in place for an additional 1 min to allow for diffusion. Following infusions, animals were returned to their home cages for 10 min before stress induction. Blood samples (∼100 μl) were taken immediately at stress offset, and then at 30 and 60 min following the cessation of stress. A separate cohort of animals was prepared in an identical fashion and received infusions of AM251 or vehicle under identical conditions, except that they were not exposed to restraint stress. These animals were bled at the same time points following vehicle or AM251 administration to determine whether AM251 administration alone activated the HPA axis. All rats were killed in a carbon dioxide chamber 24 h following testing. Brains were removed and fixed in a 4% formalin solution. The brains were frozen, sliced in 50 μm sections, and mounted. Placements were verified with reference to the atlas of Paxinos and Watson (1998), and histological analysis demonstrated that ∼85% of cannula placements were in boundaries of the nuclei of interest (see Fig. 1A, representative histology). Subjects with cannulae outside of the desired structure were excluded from subsequent analysis.
Figure 1.
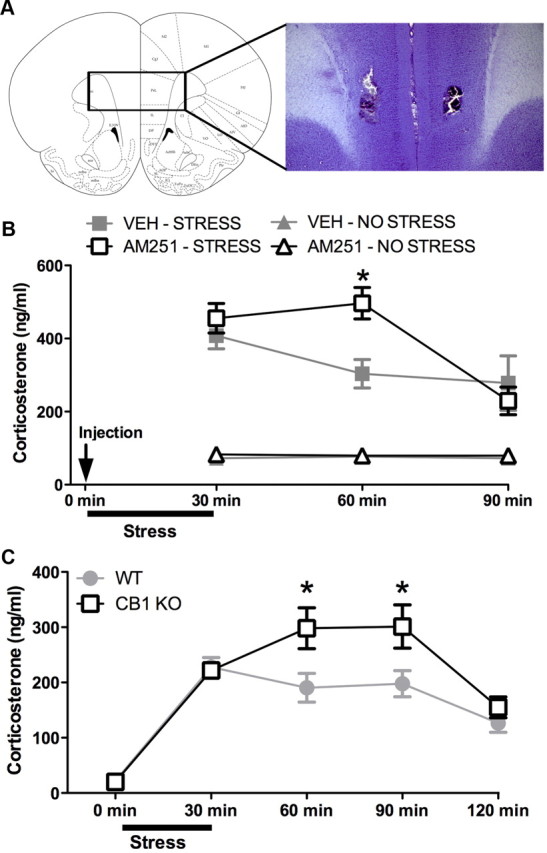
CB1R signaling within the medial prefrontal cortex is required for the decline of corticosterone levels following cessation of stress exposure. A, Representative photomicrograph of a cannula track terminating in the medial prefrontal cortex. B, Local administration of the CB1R antagonist AM251 (0.28 ng/side) into the medial prefrontal cortex of male Sprague Dawley rats prolonged corticosterone secretion following exposure to 30 min restraint stress (n = 6/condition; *p < 0.05 indicates a significant difference between AM251- and vehicle (VEH)-treated animals that have been exposed to stress). Animals exposed to stress are identified by square symbols, and animals not exposed to stress are identified by triangle symbols. Intracortical injections of VEH or AM251 occurred 10 min before stress induction. C, ICR mice lacking the CB1R (CB1KO) exhibited a prolongation in corticosterone secretion following exposure to 30 min of restraint stress compared to control (WT) (n = 8–9/condition; *p < 0.05 indicates a difference between CB1R-deficient mice (CB1KO) and wild-type mice).
Male ICR mice, aged 9–12 weeks (housed in groups of 5 per cage), were used to determine the course of HPA axis recovery following stress in CB1R-deficient mice. Mice were maintained on a 12 h light/dark cycle with lights on at 6:00 A.M., and food and water were available ad libitum. CB1 receptor-null mice were bred in-house from a founder line generously provided by Roche Laboratories and backcrossed for nine generations onto the ICR strain (Pan et al., 2008). Wild-type mice derived from the same backcrossing were used as controls in those studies. Genotypes were determined by PCR using DNA isolated from ear tissue obtained at weaning. All procedures performed with mice were approved by the Institutional Animal Use and Care Committee of the Medical College of Wisconsin. Mice were restrained for 30 min by anchoring the proximal portion of the tail to a laboratory bench top with strips of cloth tape. Blood (∼10 μl) was collected from a tail nick into a hematocrit tube immediately before restraint, immediately after the restraint, and at various time points during recovery.
For all neuroendocrine studies, blood samples were centrifuged (for rat, 3000 × g for 10 min; for mouse, 1000 × g for 1 min), after which plasma was removed and stored at −80°C. Corticosterone (in 5 μl of plasma for rat and 2 μl for mouse) was measured in duplicate using commercial RIA kits (MP Biomedicals), as previously described (Hill et al., 2010a).
Biochemical studies.
Male rats were used for biochemical studies, and housing conditions were identical to those described above. Subjects were randomly assigned to one of the following four conditions: vehicle (1:1 ratio of 0.9% saline/propylene glycol)/no stress; RU486 (20 mg/kg; Sigma)/no stress; vehicle/stress (30 min restraint stress); and RU-486/stress. Stress procedures were identical to those described above for the rat studies. Since the effects of intra-mPFC administration of the CB1R antagonist AM251 on stress-induced corticosterone secretion emerged at 30 min after stress cessation, we used this time point for biochemical analysis of eCB content. Animals in the stress condition were returned to their home cage for 30 min following the conclusion of the 30 min restraint session before being terminated. RU-486 injections occurred 30 min before stress onset, and time points for termination in vehicle and RU-486 conditions with no stress were performed at times following injection comparable to those in the stress condition. All subjects were rapidly decapitated. The mPFC was dissected as previously described (Hill et al., 2010b) (also see Fig. 2A, diagrammatic representation of region, which was dissected for analysis), frozen in liquid nitrogen within 5 min of decapitation, and stored at −80°C until analysis. Once we had established the effect of stress on eCBs in the mPFC, we sought to determine whether this response occurred throughout the entire PFC or was restricted to regions that were known to be involved in HPA axis regulation (Radley et al., 2006). A separate cohort of rats was exposed to 30 min of restraint stress and then returned to their home cage for 30 min, or acted as cage controls, after which they were rapidly decapitated and a dorsal region of the frontal cortex (composed of motor and anterior cingulate cortices) (see Fig. 2A, dissected region) was collected for analysis.
Figure 2.

Stress-induced mobilization of endocannabinoid content within the medial prefrontal cortex depends on the glucocorticoid receptor. A, Diagrammatic representation of the regions of the frontal cortex, which were dissected out for AEA and 2-AG analysis: left, mPFC; right, dorsal frontal cortex. B, The tissue content of 2-AG, but not AEA, was elevated within the medial prefrontal cortex of male Sprague Dawley rats 30 min following a 30 min exposure to restraint stress. This effect was blocked by systemic preadministration of RU-486 (20 mg/kg), a glucocorticoid receptor antagonist (n = 7–8/condition; *p < 0.05). C, Restraint stress had no effect on 2-AG or AEA content within the dorsal region of the frontal cortex, primarily composed of motor cortex (n = 8/condition).
Brain regions were subjected to a lipid extraction process as described previously (Patel et al., 2003). Tissue samples were weighed and placed into borosilicate glass culture tubes containing 2 ml of acetonitrile with 84 pmol of [2H8]anandamide and 186 pmol of [2H8]2-AG. Tissue was homogenized with a glass rod and sonicated for 30 min. Samples were incubated overnight at −20°C to precipitate proteins, then centrifuged at 1500 × g to remove particulates. The supernatants were removed to a new glass tube and evaporated to dryness under N2 gas. The samples were resuspended in 300 μl of methanol to recapture any lipids adhering to the glass tube, and dried again under N2 gas. Final lipid extracts were suspended in 20 μl of methanol and stored at −80°C until analysis. The contents of the two primary eCBs (AEA and 2-AG) were determined in the lipid extracts using isotope-dilution, liquid chromatography-mass spectrometry as described previously (Patel et al., 2005).
Immunofluorescence.
For immunofluorescence studies of CB1R expression in the mPFC, we used both rats and mice. Male Sprague Dawley rats were obtained and housed as described for the neuroendocrine and biochemical studies above. Male C57BL/6J mice (10 weeks of age) were obtained from Charles River and housed in groups of 5 per cage under a 12 h light/dark cycle with lights off at 7:00 A.M. and food and water available ad libitum. All procedures involving these mice were approved by the Institutional Animal Use and Care Committee of the Rockefeller University.
To obtain tissue for immunofluorescence staining, both rats and mice were transcardially perfused with 4% paraformaldehyde, and whole brains were removed and fixed in 4% v/v formaldehyde overnight and then in 30% w/v sucrose in PBS at 4°C for 72 h. Brains were cut into 40 μm slices on a freezing microtome, and free-floating slices of the prefrontal cortex (from 3.2 to 2.7 mm rostral to bregma) were processed for immunofluorescence. Free-floating slices were washed in PBS and then blocked in PBS containing 5% normal horse serum and 0.1% TritonX100 (PBS-T) for 1 h. Tissue slices were then incubated for 24 h at room temperature in a mixture of PBS with 5% normal donkey serum, 0.1% Triton X100, and a guinea pig anti-CB1R C-terminus antibody (1:1000 dilution) (Berghuis et al., 2007). Following incubation, tissue slices were washed repeatedly in PBS and then incubated in PBS with 5% normal horse serum, 0.1% Triton X100, and Alexa Fluor 680-conjugated donkey anti-guinea pig IgG (H+L; 1:200 dilution) for 2 h at room temperature. Tissue slices were then washed in PBS repeatedly, mounted on gel-coated slides, and coverslipped. Immunofluorescence was examined within the PFC using a fluorescence microscope.
Electron microscopy.
The animal protocol followed in this study strictly adhered to NIH Guidelines for the Care and Use of Laboratory Animals in Research and was approved by the Animal Care Committee at Weill Medical College of Cornell University. Adult male Sprague Dawley rats (housed 2 per cage) and adult male C57BL/6J mice (housed 5 mice per cage) were deeply anesthetized with sodium pentobarbital (150 mg/kg, intraperitoneal injection) before perfusion. Rats underwent rapid vascular perfusion via the ascending aortic arch with sequential delivery of 10 ml of heparin (1000 U/ml), 60 ml of 3.75% acrolein/2% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB; pH 7.4), and 250 ml of 2% PFA in 0.1 m PB. In mice, brain tissue was fixed via rapid vascular perfusion through the left ventricle of the heart with 5 ml of heparin-saline, 30 ml of 3.75% acrolein/2% PFA in PB, and 100 ml of 2% PFA. Brains were removed from the cranium and fixed in 2% PFA in PB at 4°C for 30 min, then mounted in chilled 0.1 m PB for sectioning. Sections were cut coronally at 40 μm thickness on a Leica Vibratome. Sections were collected through the mPFC from 3.20 to 2.70 mm rostral to bregma (Paxinos and Watson, 1998). Sections were placed in 1% sodium borohydride in 0.1 m for 30 min to reduce excess aldehyde linkages and then were rinsed thoroughly in successive washes of 0.1 m PB. To increase antibody and immunoreagent penetration, tissue was subjected to one round of rapid freeze-thaw. Sections were incubated in a cryoprotectant solution (25% sucrose and 3.5% glycerol in 0.05 m PB) for 15 min before sequential immersion in liquid borohydride (Freon, Refron), liquid nitrogen, and room temperature 0.1 m PB. The sections were then rinsed in 0.1 m Tris-buffered saline (TBS). A guinea pig polyclonal antiserum directed against the C terminus of the rat CB1R was used (Berghuis et al., 2007). Mouse monoclonal antibody to CaMKII was commercially obtained from ThermoScientific, which recognizes both the phosphorylated and nonphosphorylated α subunit of CaMKII (Fong et al., 2002). Free-floating sections were incubated in 0.5% bovine serum albumin (BSA) in 0.1 m TBS for 30 min to block nonspecific binding before incubation in the primary antibody solution for 12 h at room temperature followed by 36 h at 4°C. Anti-CB1R was used at a 1:3000 dilution, while mouse anti-CaMKII was diluted to 1:300. For immunoperoxidase labeling, sections were rinsed multiple times in 0.1 m TBS following primary antibody incubation. They were then incubated in a 1:400 dilution of biotinylated donkey anti-guinea pig IgG (Jackson ImmunoResearch Laboratories) at room temperature for 30 min. After successive 0.1 m TBS rinses, sections were incubated in an avidin–biotin peroxidase complex (Vectastain Elite ABC Kit, Vector Laboratories) for 30 min, rinsed again with 0.1 m TBS, and placed in 0.022% 3,3′-diaminobenzidine with 0.0033% hydrogen peroxide in 0.1 m TBS for 6 min. Dual labeling with immunogold-silver was performed on the same sections and directly followed immunoperoxidase labeling. The sections of tissue were first rinsed in 0.01 m PBS and then blocked in a solution of 0.8% BSA with 0.1% gelatin for 10 min before 2 h secondary antibody incubation in a 1:50 dilution of donkey anti-mouse IgG conjugated with ultrasmall colloidal gold (Electron Microscopy Sciences). Sections were washed with 0.01 m PBS and placed in 2% glutaraldehyde in 0.01 m PBS for 10 min to fix gold-conjugated IgG to the tissue. Following 0.01 m PBS and 0.2 m citrate buffer rinses, the gold particles were silver enhanced using the IntenSE M kit (GE Healthcare) for 7 min at room temperature and immediately placed back into citrate buffer. The tissue was rinsed with 0.1 m PB and laid flat for 60 min of fixation in 2% osmium tetroxide in 0.1 m PB. After thorough rinsing with 0.1 m PB, sections were subjected to a dehydration series consisting of increasing ethanol concentrations followed by propylene oxide, and then incubated overnight in a 1:1 mixture of propylene oxide and EPON (Electron Microscopy Sciences). Sections of tissue were then transferred to 100% EPON for 2 h and finally flat embedded between two sheets of Aclar plastic and placed into a drying oven for 72 h. Flat-embedded tissue sections containing the mPFC were cut into trapezoids, glued to EPON blocks, trimmed with a glass knife, and cut into ultrathin (60 nm) sections using an Ultra45° diamond knife (Diatome) on a Leica EM UC6 Ultramicrotome. These sections were taken from the EPON–tissue interface where there is optimal penetration of the antisera. After collection on 400 mesh copper grids (Electron Microscopy Sciences), the ultrathin sections were counterstained by being placed for 20 min in 5% uranyl acetate and 6 min in Reynolds lead citrate (Reynolds, 1963). Philips CM10 transmission electron microscope (FEI) interfaced with an AMT Advantage HR/HR-B CCT Camera System (Advanced Microscopy Techniques) was used to examine the ultrathin sections and capture images. Adobe Photoshop (version 11.0) was used to adjust brightness and contrast of the images, and PowerPoint 2008 for Mac was used to assemble plates and add lettering. Immunolabeled profiles were classified according to the detailed criteria set forth by Peters et al. (1991). Soma were considered labeled for CaMKII immunogold if they contained five or more silver-enhanced gold particles (excluding particles observed in the nucleus), large dendrites were considered labeled if they contained two or more particles, while small dendrites (minimum axis diameter <500 μm) and spines were considered labeled if they contained one or more particles.
Electrophysiological studies.
Male ICR mice (∼2 months of age, housed 5 mice per cage; Harlan Laboratories) were killed, and coronal slices containing the mPFC were prepared and placed into artificial CSF (ACSF) as described previously (Pan et al., 2008). All procedures performed with mice were approved by the Institutional Animal Use and Care Committee of the Medical College of Wisconsin. Pyramidal neurons (which represent principal neurons) in layer V were identified visually by infrared–differential contrast interference videomicroscopy based upon pyramidal shaped soma with a prominent apical dendrite that projects to the pial surface. Whole-cell patch-clamp recordings were made from pyramidal neurons, whose identity was confirmed by examination of firing characteristics in response to the injection of depolarizing and hyperpolarizing currents; in particular, they exhibit spike frequency adaptation (Satake et al., 2008). All recordings were performed at 32 ± 1°C by using an automatic temperature controller (Warner Instrument). The pipette solution contained the following (in mm): K-gluconate 100, KCl 50, HEPES 10, EGTA 0.2, MgCl2 2, MgATP 4, Na2GTP 0.3, and Na2-phosphocreatine 10 at pH 7.2 (with KOH). The ionotropic glutamate receptor antagonists CNQX (10–20 μm) and AP-5 (50 μm) were added to inhibit excitatory responses. For recording of evoked IPSCs, pyramidal neurons were voltage-clamped at −60 mV, and IPSCs were evoked at 0.1 Hz by a tungsten stimulation electrode placed near the apical dendrites. In some studies, depolarization-induced suppression of inhibition (DSI) was induced by depolarization from −60 to 0 mV for 5 s. For the studies of the effects of corticosterone, slices were allowed to recover from harvest for 1 h, then were incubated with 100 nm corticosterone in ethanol (0.001% final concentration) or ethanol alone at 32°C following the methodology established by Verkuyl et al. (2005). Twenty minutes after the addition of corticosterone, the slices were washed and stored in normal ACSF (without corticosterone or ethanol) for at least 1 h (and up to 4 h) at room temperature before being transferred into the recording chamber. For studies examining interactions between corticosterone application and the eCB system, AM251 (2 μm in 0.05% DMSO) was applied at the same time as corticosterone and was also present in the incubation media during patch-clamp recordings. Spontaneous miniature IPSCs (mIPSCs) were recorded from the pyramidal neurons at a holding potential of −70 mV. Action potential generation was blocked with tetrodotoxin (TTX; 0.5 μm). In paired-pulse ratio (PPR) paradigm, IPSCs were evoked at 0.05 Hz using a bipolar tungsten electrode placed adjacent to the recorded neuron. A paired-pulse stimulation with a 100 ms interstimulus interval was applied. TTX was not present in the incubation media.
Statistics.
Data for the effects of intra-mPFC administration of AM251- or CB1R-deficient mice on stress-induced corticosterone secretion was analyzed with a repeated-measures ANOVA with time being the within factor and either drug treatment or genotype being the between factor. For eCB ligand analysis, a univariate ANOVA was used with both stress and drug treatment as fixed factors. For electrophysiological data examining the effects of CORT and AM251 on mIPSCs, a one-way ANOVA was used with post hoc analysis performed using a Student–Newman–Keuls test. For all analysis, p < 0.05 was used as an indication of significance.
Results
CB1R signaling contributes to the return of glucocorticoid hormone secretion to control following cessation of stress exposure
We examined the effects of local antagonism of CB1R within the mPFC to determine whether CB1R signaling in this region regulates the time course of changes in circulating glucocorticoid concentrations following exposure to stress. Bilateral administration of the CB1R antagonist AM251 (0.28 ng/side) (Fig. 1A, representative histological cannula placement) into the mPFC resulted in a significant interaction between AM251 administration and time following stress exposure on plasma corticosterone concentration (F(2,20) = 3.49, p < 0.05) (Fig. 1B). Post hoc analysis revealed that AM251 administration did not alter the concentration of plasma corticosterone measured immediately at stress offset (30 min following stress induction; p > 0.05). However, at 30 min following cessation of stress (60 min following stress induction), antagonism of CB1R signaling within the mPFC resulted in significantly elevated concentrations of corticosterone relative to vehicle-infused animals (p < 0.05). There was no interaction between the effects of intra-mPFC AM251 administration and time on corticosterone concentrations in animals that were not exposed to stress (F(2,20) = 0.07, p > 0.05) (Fig. 1B). In nonstressed animals, there were no main effects of either AM251 administration (F(1,20) = 0.14, p > 0.05) or time (F(1,20) = 0.03, p > 0.05) on circulating concentrations of corticosterone.
We also examined circulating concentrations of corticosterone over time following stress exposure in mice lacking CB1R protein expression (CB1R−/−). Statistical analysis revealed a significant interaction between genotype and time on circulating concentrations of corticosterone (F(4,140) = 4.41, p < 0.01) (Fig. 1C). Post hoc analysis showed that neither basal concentrations of corticosterone (p > 0.05) nor corticosterone concentrations measured immediately after stress offset (30 min following stress induction; p > 0.05) differed between wild-type and CB1R−/− mice. However, the CB1R−/− mice exhibited significantly elevated corticosterone responses to stress compared with the wild-type mice at 60 min (p < 0.01) and 90 min (p < 0.01) after stress initiation (30 and 60 min following stress offset, respectively). These findings mirror what was seen following intra-mPFC administration of AM251 to rats and support the hypothesis that CB1R activation is required for appropriate termination of glucocorticoid secretion following the cessation of stress.
Exposure to stress mobilizes eCBs within the mPFC
The requirement for CB1R activation in the mPFC for termination of the stress response suggests that stress recruits eCB signaling in this brain region. To test this hypothesis, we determined the effects of stress exposure on eCB tissue content within the mPFC (composed of prelimbic and infralimbic cortices). Since intra-mPFC infusion of AM251 significantly affected circulating corticosterone concentrations 30 min after stress cessation, we measured eCB content in mPFC harvested 30 min following stress cessation. The role of corticosterone activation of GRs was determined in rats pretreated with the GR antagonist RU486 (20 mg/kg). Analysis of lipid extracts of mPFC sections for eCB concentrations (Fig. 2A, region of tissue analyzed) revealed a significant interaction between stress exposure and treatment with RU486 on 2-AG content (F(1,23) = 5.98, p < 0.03) (Fig. 2B). Post hoc analysis demonstrated that tissue content of 2-AG within the mPFC was elevated 30 min following the cessation of stress (p < 0.01); systemic pretreatment with RU486 prevented the increase in 2-AG produced by stress. There was no significant main effect of either stress (F(1,23) = 0.10, p > 0.05) or RU486 treatment (F(1,23) = 0.01, p > 0.05), and no interaction (F(1,23) = 2.25, p > 0.05) (Fig. 2B) between stress exposure and RU486 treatment on the tissue content of AEA.
To determine the regional specificity of the stress effect on eCBs within frontal cortical subregions, we also assessed the effect of stress on eCB tissue contents within the dorsal PFC, a region that is composed of the motor and cingulate cortices (Fig. 2A, region of tissue analyzed). Stress exposure did not significantly affect 2-AG (t(12) = 0.86, p > 0.05) (Fig. 2C) or AEA (t(12) = 1.78, p > 0.05) (Fig. 2C) tissue contents within the dorsal frontal cortex.
CB1Rs are present on GABAergic terminals and inhibit GABA release at synapses with principal neurons within layer V of the prelimbic region of the mPFC
Increased activity of projection neurons from the mPFC is required for termination of the HPA axis (Radley et al., 2006, 2009), and CB1R activation is known to inhibit GABA release in brain regions, including the frontal cortex (Chiu et al., 2010). Thus, we explored the hypothesis that stress-induced eCB signaling disinhibits pyramidal neurons in the mPFC via inhibition of GABA release. Our first objective was to examine CB1 receptor expression in the mPFC of both rats and mice since both species respond similarly to loss of CB1R signaling. Immunohistofluorescence studies using a CB1R polyclonal antibody revealed dense CB1R expression throughout layers II/III and V of the prelimbic region of the mPFC in both mice (Fig. 3A,C) and rats (Fig. 3B,D), a region of the mPFC particularly important for glucocorticoid-mediated regulation of the HPA axis and termination of stress-induced corticosterone secretion (Diorio et al., 1993; Radley et al., 2006, 2009).
Figure 3.
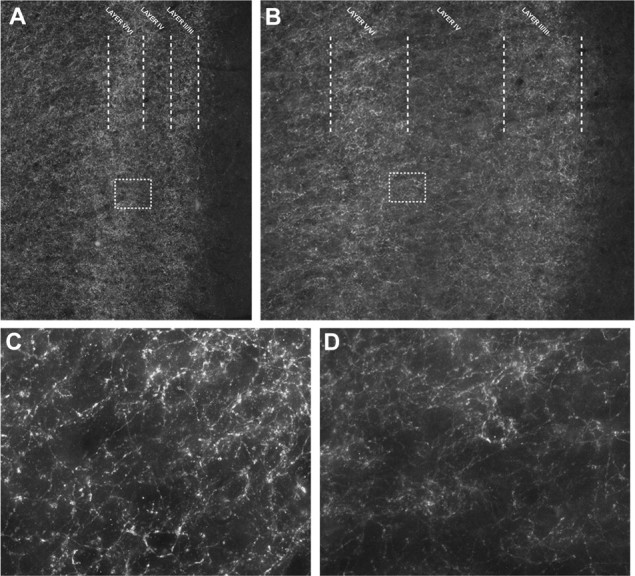
Distribution of CB1Rs within the prelimbic region of the medial prefrontal cortex. A, B, Immunofluorescence of CB1Rs within the prelimbic region of the medial prefrontal cortex (at 10× magnification) demonstrates prominent expression of the CB1R within both layers II/III and layers V/VI of the prefrontal cortex of male C57BL/6J (A) and male Sprague Dawley rats (B). C, D, High magnification (60× magnification) of CB1R expression within layer V of the prelimbic region of the prefrontal cortex reveals a punctate network of CB1R expression around cell bodies in both mouse (C) and rat (D).
Immunoelectron microscopic examination of ultrathin sections of layers III–VI of the prelimbic mPFC of rats and mice was performed to determine the cellular localization of the CB1R. Sections were immunolabeled for both the CB1R and calcium/calmodulin-dependent kinase II (CaMKII), a serine/threonine-specific kinase expressed by cortical pyramidal neurons (Fong et al., 2002; McDonald et al., 2002; Muller et al., 2006). Twenty-four grid squares within layer V of the prelimbic mPFC from two rats and two mice over a total area of 69,984 μm2 were examined. Micrographs were sampled randomly from fields where both CaMKII (immunogold) and CB1R (immunoperoxidase) labels were observed. Of 150 CB1R-immunolabeled axon and axon terminal profiles counted, 70 axon terminals formed visible synaptic contacts, while 41 were apposed to dendrites without visible postsynaptic membrane specializations. Sixty-eight of the 70 CB1R-labeled axon terminals formed symmetric synapses, 79.4% (54/68) of which were on CaMKII containing somatodendritic profiles. Of these 54 CB1R terminals, 85% contacted CaMKII-labeled somata (Fig. 4A,B), and the remainder contacted dendrites (Fig. 4C). Neither the total expression of CB1R per unit area nor the percentage of CaMKII-positive targets significantly differed between the rat and mouse. Of 150 total profiles examined, only 5 showed dual immunolabeling for CB1R and CaMKII. These data support the hypothesis that CB1R in the mPFC are predominantly located on GABAergic terminals.
Figure 4.
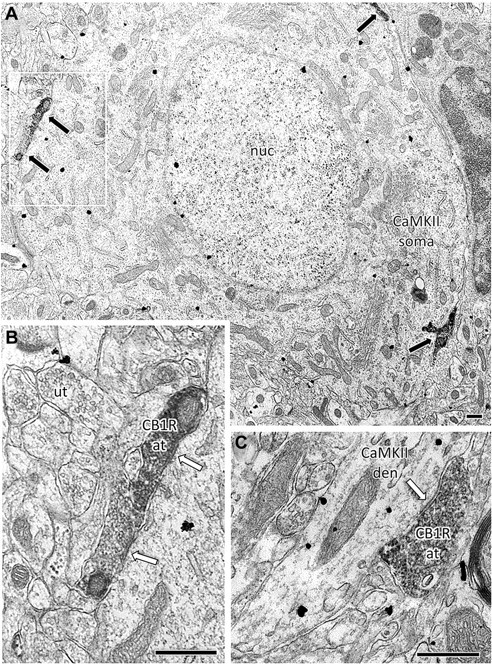
CB1Rs are predominantly localized on GABAergic terminals impinging upon pyramidal neurons in the prelimbic region of the medial prefrontal cortex. Electron micrographs of contacts between CB1R- and CaMKII-immunolabeled profiles in layers V–VI of the rodent prelimbic PFC. A, A composite image of nine high-magnification micrographs shows multiple CB1R-expressing terminals (immunoperoxidase, black arrows) targeting a large pyramidal-shaped CaMKII-positive (immunogold, arrowheads) soma in the male Sprague Dawley rat prelimbic PFC. The area outlined in white is enlarged in B. B, Two CB1R-labeled axon terminals form symmetric inhibitory-type somatic contacts (white arrows). C, A CB1R-labeled terminal forms a symmetric synapse with a CaMKII-labeled dendrite in the male C57BL/6J mouse prelimbic PFC. at, Axon terminal; den, dendrite; nuc, nucleus; ut, unlabeled terminal. Scale bars, 500 nm.
In light of the predominant expression of CB1R on GABAergic terminals, whole-cell patch-clamp electrophysiology in slices from adult male mice was used to examine CB1 receptor regulation of GABA-mediated IPSCs in layer V principal neurons in the mPFC. Ionotropic glutamate receptor antagonists CNQX (20 μm) and d-AP-5 (20 μm) were included in the ACSF. Visually identified principal neurons exhibited spike frequency adaptation (Fig. 5A), a common feature of pyramidal neurons (Satake et al., 2008). We recorded three types of eCB/CB1 receptor-mediated responses: DSI; long-term depression (LTD) of IPSCs (I-LTD); and CB1 agonist-induced depression of IPSCs. Depolarization of principal neurons to 0 mV for 5 s induced a transient suppression of evoked IPSCs (i.e., DSI), which was abolished by 2 μm AM251 (Fig. 5B). Following baseline recordings of evoked IPSCs at 0.1 Hz, a 10 Hz 5 min stimulation was applied to induce I-LTD. This stimulation protocol induced a long-lasting depression of IPSCs in mPFC slices, which was blocked by 2 μm AM251 (Fig. 5C). This is consistent with I-LTD described in other brain regions, as well as a recent report of a comparable phenomenon in the frontal cortex that is influenced by dopaminergic signaling (Edwards et al., 2006; Chiu et al., 2010). Bath application of the CB1R agonist WIN 55212–2 (2 μm) induced significant depression of evoked IPSCs, which was blocked by the CB1R antagonist AM 251 (4 μm) (Fig. 5D). Thus, GABAergic synapses onto principal neurons in layer V of the prelimbic region of the mPFC exhibit characteristics of eCB/CB1 receptor mediated regulation and synaptic plasticity.
Figure 5.

CB1 receptor regulation of GABA-mediated currents within layer V principal neurons of the mPFC. A, Current-clamp recordings from a pyramidal neuron within layer V of the prelimbic region of the medial PFC of ICR mice at a membrane potential of −60 mV during injection of depolarizing and hyperpolarizing current pulses of different intensities (pulses are shown in inset below the responses). A typical pyramidal neuron shows spike frequency adaptation. B, Depolarization from −60 to 0 mV for 5 s induced DSI (n = 11), which was blocked by the CB1R antagonist AM 251 (2 μm; n = 8; p < 0.05 vs control). C, Repetitive stimulation (10 Hz for 5 min; indicated by arrow) of synaptic afferents induced I-LTD (n = 7, p < 0.05 vs baseline), which was blocked by AM 251 (n = 7, p > 0.05 vs baseline). D, Bath application of the CB1R agonist WIN 55212–2 (2 μm) depressed evoked IPSP amplitude, and this depression was reversed after addition of AM 251 (4 μm, n = 7).
Glucocorticoids suppress GABA release in the mPFC via an endocannabinoid mechanism
We hypothesize that stress-induced increases in corticosterone levels activate principal neuron outflow via CB1 receptor-mediated disinhibition of principal neurons in layer V of the prelimbic region of the mPFC. To test this hypothesis, corticosterone (100 nm) was applied to slices of mPFC, and spontaneous mIPSCs and evoked IPSCs were analyzed. mIPSCs were recorded in the presence of the Na+ channel blocker TTX (0.5 μm). Since the neuroendocrine and biochemical studies reported above indicate that the effects of glucocorticoids on eCB signaling are measureable 1 h following the onset of stress, we performed electrophysiological recordings a minimum of 1 h following incubation with corticosterone (consistent with the methodology of Verkuyl et al., 2005). Incubation of the slices with corticosterone for 20 min followed by at least 1 h of rest significantly decreased the frequency of mIPSCs, an effect that was blocked by AM 251 (F(2,22) = 4.69, p < 0.05) (Fig. 6A–D). Corticosterone had no significant effect on the mean amplitude of mIPSCs (F(2,22) = 0.48, p > 0.05) (Fig. 6E) or the cumulative probability distribution of the amplitude (p > 0.05) (Fig. 6F).
Figure 6.

Corticosterone (CORT) depresses GABA-mediated currents in layer V principal neurons of the mPFC via recruitment of endocannabinoid signaling. A–C, mIPSCs recorded in layer V pyramidal neurons in medial prefrontal cortical slices from ICR mice encompassing the prelimbic cortex that were treated with vehicle (control), or CORT (100 nm, 20 min) alone or in combination with AM 251 (2 μm). D, CORT treatment decreased the mean frequency of mIPSCs, and this effect was blocked by AM 251 (n = 8–9; *p < 0.05). E, F, CORT treatment had no significant effect on the mean amplitude of mIPSCs (E) or the cumulative amplitude distributions of mIPSCs (F). G, CORT treatment (100 nm, 20 min) increased the paired-pulse ratio in layer V pyramidal neurons of the PFC, and this effect was blocked by AM 251 (2 μm; n = 8–10, *p < 0.05).
To further delineate the mechanism of corticosterone-mediated inhibition of IPSCs, we compared the PPR (100 ms) in control and corticosterone-treated slices. In control slices, the PPR, calculated as IPSC2/IPSC1, was significantly <1, indicative of a paired-pulse depression of IPSCs (Wilcox and Dichter, 1994). In corticosterone-treated slices, the PPR was significantly increased (F(2,23) = 4.03, p < 0.05) (Fig. 6G), which is consistent with an effect of corticosterone to inhibit presynaptic GABA release. The effect of corticosterone on PPR was abolished by AM 251.
Discussion
Termination of HPA axis activation following exposure to stress is essential to limit the duration of glucocorticoid secretion and prevent the deleterious effects of persistently elevated levels of glucocorticoid hormones on cardiovascular, immune, metabolic, and neural systems. Our data demonstrate that stress, via activation of GRs, increases 2-AG levels within the mPFC, and that eCB signaling contributes to the appropriate termination of glucocorticoid secretion following cessation of stress. Immunohistochemical and EM data indicate that CB1Rs are expressed almost entirely by GABAergic terminals in layer V of the prelimbic region of the mPFC, particularly on axons that synapse onto the soma of principal neurons. Functional studies support a role for CB1R to inhibit GABA release within this same neuronal population. Our data also demonstrate that incubation of slices from the mPFC with corticosterone induces eCB-mediated inhibition of GABA release onto principal neurons. Collectively, these data support the novel hypothesis that glucocorticoid hormones released following stress exposure activate eCB/CB1R signaling within the mPFC, inhibiting GABA release onto layer V pyramidal neurons in the prelimbic cortex, and promoting the termination of corticosterone secretion. Consistent with this model, pharmacological disruption of GABAA receptor signaling within the mPFC also decreases stress-induced activation of the HPA axis (Weinberg et al., 2010). Together, these data indicate that prefrontal cortical eCB signaling links glucocorticoids and neuronal activation within the mPFC and contributes to the long negative feedback loop to inhibit corticosterone secretion following cessation of stress.
The present data are consistent with the established role of the mPFC, and particularly the prelimbic region of the mPFC, in the regulation of the HPA axis and termination of the stress response. Lesion studies have demonstrated that selective ablation of the prelimbic region of the PFC does not alter the magnitude, but rather the duration, of corticosterone secretion following exposure to a psychogenic stressor (Diorio et al., 1993; Radley et al., 2006, 2009). In the present study, both genetic deletion of the CB1R, and local antagonism of CB1R signaling within the mPFC prolonged the elevation in stress-induced levels of circulating corticosterone. Our histological and electrophysiological experiments demonstrate the presence of CB1Rs on GABAergic terminals impinging upon pyramidal neurons within layer V of the prelimbic region of the mPFC. These findings support the hypothesis that activation of CB1R signaling in this brain region could result in disinhibition of excitatory projections from the prelimbic mPFC to other brain regions.
The circuit by which the efferent projection neurons from the prelimbic region of the PFC inhibit the HPA axis involves a secondary activation of inhibitory GABAergic neurons within subregions of the bed nucleus of the stria terminalis (BNST) (Spencer et al., 2005; Radley et al., 2009) or the peri-PVN region (Herman et al., 2005). Activation of either of these inhibitory circuits feeding into the PVN dampens neuronal activation of the corticotropin-releasing hormone (CRH)-secreting cells of the PVN (Herman et al., 2005). Layer V of the prelimbic cortex is the primary site for projection neurons that extend to subcortical limbic structures (Gabbott et al., 2005) such as the BNST; thus, the identification of eCB-mediated regulation of neuronal excitability within layer V neurons in the prelimbic region of the mPFC provides a neurochemical and functional mechanism that compliments the previously established neuroanatomical networks involved in prefrontocortical regulation of HPA axis activity. Recent data indicate that excitatory afferents arising from the ventral subiculum activate the same inhibitory relays within the BNST as are activated by excitatory afferents originating from the mPFC (Radley and Sawchenko, 2010). These data suggest that both hippocampal and mPFC projections are involved in the long glucocorticoid-mediated negative feedback loop via inputs into the BNST. In the current study, disruption of eCB signaling within the mPFC attenuated, but did not completely prevent, the return of circulating glucocorticoids to baseline concentrations. It is possible that the projections from the ventral subiculum to the BNST, which would not be affected by CB1 receptor blockade in the mPFC, are responsible for the ultimate return of circulating corticosterone concentrations to baseline.
It is possible that disruption of CB1R within the mPFC regulates corticosterone secretion through an HPA axis-independent pathway. For example, ventral regions of the mPFC can regulate autonomic outflow during conditions of stress (Neafsey, 1990), and it has recently been demonstrated that subregions of the BNST are capable of modulating corticosterone secretion independent of ACTH levels (Choi et al., 2007). As such, it is plausible that CB1Rs within the mPFC could contribute to higher-order regulation of autonomic outflow, and that the ability of prefrontal cortical CB1Rs to regulate corticosterone secretion involves the activity of the sympatho-medullary arm of the stress response (Bornstein et al., 2008). Further studies are required to differentiate between these possibilities.
Within the context of HPA axis regulation, however, the activation of efferent projections from the mPFC has been shown to function as a prominent pathway in glucocorticoid-mediated negative feedback. Local activation of glucocorticoid receptors within the mPFC accelerates the poststress decline in circulating levels of corticosterone following exposure to stress (Diorio et al., 1993), and the downregulation of glucocorticoid receptors within the mPFC following chronic stress or in aging is associated with impaired glucocorticoid negative feedback regulation (Mizoguchi et al., 2003, 2009). Unlike the local glucocorticoid effects in the PVN that rapidly decrease activation of CRH neurosecretory cells governing HPA axis output and promote fast-feedback inhibition on the HPA axis (Di et al., 2003; Evanson et al., 2010), disruption of the mPFC to PVN circuit prolongs the recovery to normal circulating glucocorticoids levels following stress exposure (Diorio et al., 1993; Radley et al., 2006, 2009). As such, these data demonstrate that glucocorticoid-mediated negative feedback possesses both short-loop (locally within the PVN) and long-loop (distally within the mPFC and ventral subiculum) components. Moreover, there is evidence that eCB signaling contributes to both of these phases of glucocorticoid feedback. The current data create an argument for a role of prefrontal cortical eCB signaling in the long-loop phase of glucocorticoid feedback. And, it has recently been reported that local antagonism of the CB1R within the PVN impairs fast-feedback inhibition of HPA axis activity by glucocorticoids (Evanson et al., 2010). In the present study, we report that mice globally deficient in CB1Rs exhibit a larger peak in corticosterone secretion following stress, which is consistent with these mice lacking fast-feedback inhibition due to the absence of CB1R signaling within the PVN.
Taking these data together, we propose the following model for the integration of eCB signaling into the temporal phases of glucocorticoid feedback. Glucocorticoid hormones are released into the circulation in response to stress. In the PVN, glucocorticoids evoke a rapid induction of eCB release through a nongenomic pathway, which results in a rapid suppression of glutamatergic inputs to CRH neurosecretory cells and decreases the excitatory drive to the HPA axis (Di et al., 2003; Evanson et al., 2010). In the mPFC, glucocorticoids produce a time-delayed increase in 2-AG, which, via CB1R activation, suppresses GABAergic inputs to principal neurons. This suppression of GABAergic inputs to principal neurons could act to increase the outflow of these projection neurons to inhibitory relays within the BNST, and thus contribute to the long loop of glucocorticoid negative feedback. In sum, our model proposes a temporally and structurally specific role of eCB signaling in distinct phases of glucocorticoid feedback.
The mechanism by which glucocorticoids increase eCB signaling within the mPFC was not elucidated in the current study, but appears to be distinct from the process that occurs within the PVN. In the PVN, glucocorticoid regulation of eCB signaling is not blocked by an antagonist of the nuclear GR and is driven by glucocorticoid-induced G-protein signaling (Di et al., 2003). Furthermore, administration of glucocorticoids in the absence of stress can rapidly (∼10 min) increase eCB content within the hypothalamus but not the mPFC (Hill et al., 2010b). Thus, we hypothesize that the actions of glucocorticoids on eCB content within the mPFC require coincident increases in neuronal activation, which occur following exposure to stress to produce a detectable increase in 2-AG content using bulk tissue measurements.
Termination of HPA axis activation following exposure to stress is an essential process for maintaining optimal health in the face of persistent stress. The data presented herein suggest an important role of the eCB system within the mPFC in the termination of glucocorticoid release following exposure to stress. Increased 2-AG within the mPFC following exposure to stress provides a mechanism of coincidence detection that can fine tune the excitability of pyramidal neurons within the prelimbic region of the mPFC and contribute to termination of corticosterone secretion following cessation of stress exposure. These data contribute to our general understanding of the mechanisms subserving glucocorticoid-mediated negative feedback and stress recovery. Furthermore, they provide a mechanism that could underlie modulation by glucocorticoids of neuronal sensitivity in extrahypothalamic structures that contribute to feedback and recovery.
Footnotes
This research was supported by National Institutes of Health Grants DA09155 (C.J.H.); DA024741 (Q.-s.L.); MH41256 (B.S.M.); MH40342 (V.M.P.); and DA021696 and DA011322 (K.M.); and operating grants from the Canadian Institutes of Health Research (CIHR) to B.B.G. and V.V. M.N.H. and I.N.K. are recipients of postdoctoral fellowships from CIHR; R.J.M. and T.T.L. are recipients of Canadian Graduate Student Scholarships Doctoral Award from CIHR; and M.L.F. is the recipient of Training Grant 5 T32 DA7274-19 from the National Institute of Drug Abuse.
References
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Bornstein SR, Engeland WC, Ehrhart-Bornstein M, Herman JP. Dissociation of ACTH and glucocorticoids. Trends Endocrinol Metab. 2008;19:175–180. doi: 10.1016/j.tem.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Chiu CQ, Puente N, Grandes P, Castillo PE. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci. 2010;30:7236–7248. doi: 10.1523/JNEUROSCI.0736-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DC, Furay AR, Evanson NK, Ostrander MM, Ulrich-Lai YM, Herman JP. Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic-pituitary-adrenal axis activity: implications for the integration of limbic inputs. J Neurosci. 2007;27:2025–2034. doi: 10.1523/JNEUROSCI.4301-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5:374–381. doi: 10.1038/nrendo.2009.106. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 2005;569:751–760. doi: 10.1113/jphysiol.2005.097477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13:3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE. Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol. 2006;95:67–75. doi: 10.1152/jn.00813.2005. [DOI] [PubMed] [Google Scholar]
- Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151:4811–4819. doi: 10.1210/en.2010-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo HF, Bruestle A, Bodie B, Dolgas CM, Herman JP. The medial prefrontal cortex differentially regulates stress-induced c-fos expression in the forebrain depending on type of stressor. Eur J Neurosci. 2003;18:2357–2364. doi: 10.1046/j.1460-9568.2003.02932.x. [DOI] [PubMed] [Google Scholar]
- Fong DK, Rao A, Crump FT, Craig AM. Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J Neurosci. 2002;22:2153–2164. doi: 10.1523/JNEUROSCI.22-06-02153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:791–797. doi: 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, Hillard CJ, Gorzalka BB, Viau V. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A. 2010a;107:9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010b;35:1333–1338. doi: 10.1016/j.psyneuen.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, Tasker JG. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–6650. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ, Muller JF, Mascagni F. GABAergic innervation of alpha type II calcium/calmodulin-dependent protein kinase immunoreactive pyramidal neurons in the rat basolateral amygdala. J Comp Neurol. 2002;446:199–218. doi: 10.1002/cne.10204. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Central effects of stress hormones in health and disease: Understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008;583:174–185. doi: 10.1016/j.ejphar.2007.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, Goldfarb RH, Kitson RP, Miller AH, Spencer RL, Weiss JM. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- Mizoguchi K, Ishige A, Aburada M, Tabira T. Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience. 2003;119:887–897. doi: 10.1016/s0306-4522(03)00105-2. [DOI] [PubMed] [Google Scholar]
- Mizoguchi K, Ikeda R, Shoji H, Tanaka Y, Maruyama W, Tabira T. Aging attenuates glucocorticoid negative feedback in rat brain. Neuroscience. 2009;159:259–270. doi: 10.1016/j.neuroscience.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ. Pyramidal cells of the rat basolateral amygdala: synaptology and innervation by parvalbumin-immunoreactive interneurons. J Comp Neurol. 2006;494:635–650. doi: 10.1002/cne.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey EJ. Prefrontal cortical control of the autonomic nervous system: anatomical and physiological observations. Prog Brain Res. 1990;85:147–165. doi: 10.1016/s0079-6123(08)62679-5. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Rademacher DJ, Hillard CJ. Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther. 2003;306:880–888. doi: 10.1124/jpet.103.054270. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- Patel S, Carrier EJ, Ho WS, Rademacher DJ, Cunningham S, Reddy DS, Falck JR, Cravatt BF, Hillard CJ. The postmortal accumulation of brain N-arachidonylethanolamine (anandamide) is dependent upon fatty acid amide hydrolase activity. J Lipid Res. 2005;46:342–349. doi: 10.1194/jlr.M400377-JLR200. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th Ed. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pecoraro N, Dallman MF, Warne JP, Ginsberg AB, Laugero KD, la Fleur SE, Houshyar H, Gomez F, Bhargava A, Akana SF. From Malthus to motive: how the HPA axis engineers the phenotype, yoking needs to wants. Prog Neurobiol. 2006;79:247–340. doi: 10.1016/j.pneurobio.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HdF. The fine structure of the nervous system. New York: Oxford UP; 1991. [Google Scholar]
- Radley JJ, Sawchenko PE. Evidence for a common relay subserving prefrontal cortical and hippocampal inhibition of the neuroendocrine stress response. Soc Neurosci Abstr. 2010;36:792–4. [Google Scholar]
- Radley JJ, Arias CM, Sawchenko PE. Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci. 2006;26:12967–12976. doi: 10.1523/JNEUROSCI.4297-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Gosselink KL, Sawchenko PE. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J Neurosci. 2009;29:7330–7340. doi: 10.1523/JNEUROSCI.5924-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake T, Mitani H, Nakagome K, Kaneko K. Individual and additive effects of neuromodulators on the slow components of afterhyperpolarization currents in layer V pyramidal cells of the rat medial prefrontal cortex. Brain Res. 2008;1229:47–60. doi: 10.1016/j.brainres.2008.06.098. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Buller KM, Day TA. Medial prefrontal cortex control of the paraventricular hypothalamic nucleus response to psychological stress: possible role of the bed nucleus of the stria terminalis. J Comp Neurol. 2005;481:363–376. doi: 10.1002/cne.20376. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Wotjak CT. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res. 2008;170:397–432. doi: 10.1016/S0079-6123(08)00433-0. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkuyl JM, Karst H, Joëls M. GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur J Neurosci. 2005;21:113–121. doi: 10.1111/j.1460-9568.2004.03846.x. [DOI] [PubMed] [Google Scholar]
- Weinberg MS, Johnson DC, Bhatt AP, Spencer RL. Medial prefrontal cortex activity can disrupt the expression of stress response habituation. Neuroscience. 2010;168:744–756. doi: 10.1016/j.neuroscience.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox KS, Dichter MA. Paired pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J Neurosci. 1994;14:1775–1788. doi: 10.1523/JNEUROSCI.14-03-01775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]