

Abstract
Human α-defensin 5 (HD5, HD5ox to specify the oxidized and disulfide linked form) is a 32-residue cysteine-rich host-defense peptide, expressed and released by small intestinal Paneth cells, that exhibits antibacterial activity against a number of Gram-negative and –positive bacterial strains. To ascertain the contributions of its disulfide array to structure, antimicrobial activity, and proteolytic stability, a series of HD5 double mutant peptides where pairs of cysteine residues corresponding to native disulfide linkages (Cys3—Cys31, Cys5—Cys20, Cys10—Cys30) were mutated to Ser or Ala residues were overexpressed in E. coli, purified and characterized. A hexa mutant peptide, HD5[Serhexa], where all six native Cys residues are replaced by Ser residues was also evaluated. Removal of a single native S—S linkage influences oxidative folding and regioisomerization, antibacterial activity, Gram-negative bacterial membrane permeabilization, and proteolytic stability. Whereas the majority of the HD5 mutant peptides show low-micromolar activity against Gram-negative E. coli ATCC 25922 in colony counting assays, the wild-type disulfide array is essential for low-micromolar activity against Gram-positive S. aureus ATCC 25923. Removal of a single disulfide bond attenuates the activity observed for HD5ox against this Gram-positive bacterial strain. This observation supports the notion that the HD5ox mechanism of antibacterial action differs for Gram-negative and Gram-positive species (Wei, G.; de Leeuw, E., Pazgier, M., Yuan, W., Zou, G., Wang, J., Ericksen, B., Lu, W.-Y.; Lehrer, R. I.; Lu, W. (2009) J. Biol. Chem. 284, 29180-29192), and that the native disulfide array is a requirement for its activity against S. aureus.
Defensins comprise a family of ribosomally derived, low-molecular-weight (∼3-4 kDa), cysteine-rich host defense peptides that are biosynthesized by lower and higher eukaryotes.1-4 In humans, defensins are produced by epithelial cells of the skin and other organs (human β-defensins, HBD1-4), neutrophils (human neutrophil peptides, HNP1-4), and small intestinal Paneth cells (human α-defensins, HD5-6).3,5,6 In the oxidized forms, human defensins, which are comprised of ∼30 to 40-residues, exhibit regiospecific disulfide linkages with the pairings Cys1—Cys6, Cys2—Cys4, Cys3—Cys5 or Cys1—Cys5, Cys2—Cys4, Cys3—Cys6 for α- (HNPs fall into this category) or β-defensins, respectively.3 This conventional Cys numbering scheme indicates the ordering of Cys residues starting from the peptide N-terminus. These disulfide arrays stabilize the peptide fold, provide triple stranded β-sheet secondary structure, and confer protease resistance. Although the primary sequences of α-defensins vary significantly, the distribution of Cys residues is conserved, in addition to a single Gly residue and an Arg-Glu salt bridge.1
Human defensin 5 (HD5, Figure 1), the focus of this work, is a 32-residue α-defensin expressed by Paneth cells of the human small intestine.7 It is localized to subcellular granules.8 Paneth cells are secretory cells located at the base of the crypts of Lieberkü hn that contribute to mucosal immunity and protect nearby stem cells from bacterial invasion by releasing an antimicrobial cocktail, which contains lysozyme and defensins, in response to bacterial invasion.9 Paneth cells also provide essential niche signals for nearby Lgr5 stem cells.10 Studies of the murine α-defensin cryptdin-4, which is localized to the granules of murine Paneth cells and exhibits broad-range antibacterial activity in vitro,11 revealed that Paneth cells release defensins into the intestinal lumen in response to Gram-negative and –positive bacteria and various bacterial antigens, including lipopolysaccharide and muramyl dipeptide.12 Moreover, the HD5 transgenic mouse is resistant to oral Salmonella typhimurium challenge (1 × 109 CFU, lethal dose for wild-type mouse),13 which indicates that HD5 is a key contributor to host defense in humans. Recent HD5 transgenic mouse model studies showed that expression of HD5 results in an inversion of the Firmicutes/Bacteroidetes ratio, two predominant commensal bacterial phyla found in the gut.14 This observation suggests that HD5 also plays a homeostatic role in regulating the composition of the commensal intestinal microbiota.14,15 Furthermore, alterations in HD5 expression levels16,17 and impaired proteolytic processing of the HD5 precursor peptide18 have been associated with gastrointestinal disorders, which include Crohn's disease and other inflammatory conditions of the bowel.19,20 In addition to host defense and mucosal immunity, other physiological roles for defensin peptides exist, including chemotatic and immunomodulatory functions.21 Changes in defensin expression levels and/or defensin polymorphisms are associated with cancer22 and cystic fibrosis,23 and some defensins exhibit anti-HIV or anti-parasitic activities.24
Figure 1.
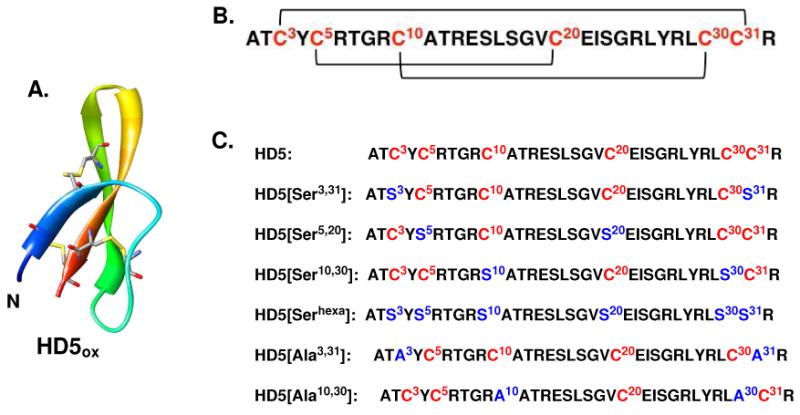
(A) X-ray crystal structure of HD5ox (PDB 1ZMP, ref 38). The N-terminus is labeled in blue and the C-terminus is labeled in red. (B) Illustration of the Cys3—Cys31, Cys5—Cys20, Cys10—Cys30 disulfide linkages in native HD5ox. (C) Nomenclature employed and amino acid sequences for the peptides studied in this work. Cys residues are depicted in red and the Ser/Ala mutations in blue.
Many unresolved questions regarding the physiological roles of HD5 and other defensin peptide exist, including those pertaining to antimicrobial mechanisms of action and structure. Along these lines, defensins are often described as non-specific membrane-disrupting peptides that exert an antibacterial effect by damaging the bacterial cell membrane. This classification stems, at least in part, from the overall cationic charge of these peptides and their propensity to exhibit antibacterial activity against a wide range of bacterial strains. Indeed, certain plant and mammalian defensins interact with synthetic membranes and membrane mimics.25,26 Assays, including those employing nucleotide-binding dyes,27 membrane potential-sensitive probes,28 and colorimetric indicator strains,29 demonstrate that membrane permeabilization occurs following incubation of bacteria and fungi with select defensin peptides. Various spectroscopic techniques, including solid-state NMR30 and small-angle X-ray scattering31 have provided recent structural insights into this phenomenon for select human defensins.
When considering mechanisms of action, it is appropriate to consider defensins on a case-by-case basis because the overall positive charge, an important factor for electrostatic interactions with the bacterial cell membrane, and primary amino acid sequences of the peptides vary dramatically. These structural differences may confer diverse mechanisms of action particular to a given peptide, and membrane disruption may not be the sole factor contributing to antibacterial action.1,4,32 Along these lines, some peptides classified as defensin do not cause membrane damage against tested strains. The fungal defensin plectasin does not cause potassium leakage from Bacillus subtilis, or alter the membrane potential.33 Rather, plectasin inhibits cell wall biosynthesis by forming a 1:1 complex with lipid II (Kd = 180 nM).33 Oyster defensin34 and HBD-335 were subsequently shown to inhibit peptidoglycan biosynthesis in S. aureus and bind lipid II. Surface plasmon resonance measurements indicated that HNP-1 binds to lipid II, and an attenuation of antibacterial activity was found in antibacterial activity assays conducted by using S. aureus cultures treated with various inhibitors of cell wall biosynthesis prior to HNP-1 addition.36 Lipid II is a validated cellular target for antibiotics in clinical use.37 Vancomycin, the nonribosomal peptide and antibiotic of last-resort for multi-drug resistant Gram-positive infections, binds to the N-acyl-D-Ala-D-Ala termini of lipid II. The lantibiotic nisin, employed in the food industry, interacts with lipid II and subsequently forms pores in the bacterial cell membrane.37 Like nisin, HBD-3 may cause gross defects in the bacterial cell membrane after binding to lipid II.35
The precise mechanism of antibacterial action of HD5ox is unclear. Structural studies of HD5ox, where “ox” designates the oxidized and disulfide-linked form, reveal that it exhibits triple stranded β-sheet secondary structure stabilized by three disulfide bonds. It has the α-defensin connectivity of Cys3—Cys31, Cys5—Cys20, Cys10—Cys30, where the superscripts correspond to the amino acid position in the HD5 sequence, and dimerizes in the solid state.38 A series of structure/activity relationship studies have provided insights into the structural requirements for HD5 antibacterial action.39 The Arg6-Glu14 salt bridge of HD5ox is essential for proteolytic stability and proper folding in the absence of the propeptide and mutation of Glu14 to Gln has some strain-specific influence on antimicrobial activity.40 This mutation had no effect on the activity against S. aureus and resulted in slightly enhanced killing of E. coli. Enantiomeric HD5ox (D-form) exhibited attenuated activity against S. aureus relative to native HD5ox; no loss of activity against E. coli was observed.41 A similar trend occurred for a disulfide-null mutant where each Cys residue of HD5 was replaced by an α-aminobutyric acid moiety.42 This linearized peptide exhibited comparable activity against E. coli and a 4-to-5-fold reduction in activity against S. aureus relative to native HD5ox.42 When taken together, these studies suggest that the mechanisms of action of HD5ox against E. coli and S. aureus are fundamentally different.41 No systematic evaluation of the HD5ox disulfide array, which may be a critical determinant for antibacterial activity against S. aureus and/or other physiological functions, has been reported to date. A number of mutagenesis studies have addressed the disulfide array of other human defensin peptides43,44 and cryptdin-4,11 but it is inappropriate to generalize the results from any one study because of the differences in primary amino acid sequence and overall cationic charge exhibited by these peptides.
In this work, we describe the preparation and characterization of a HD5 mutant peptide family where native S—S bonds have been systematically deleted by mutating pairs of Cys residues to Ser or Ala (Figure 1). We show that deletion of a native disulfide bond has profound consequences for oxidative folding and regioisomerization, proteolytic stability, and antibacterial activity. In particular, loss of any native disulfide bond attenuates the activity of HD5ox against S. aureus ATCC 25923. These observations indicate that HD5 exerts different mechanisms of action against Gram-negative and Gram-positive bacteria.
Experimental Procedures
Materials and General Methods
All solvents, reagents and chemicals were purchased from commercial suppliers in the highest available purity and used as received. Milli-Q water (18.2 ΩOhms, 0.22 μm filter) was employed to prepare all buffers, aqueous solutions, and peptide/oligonucleotide stock solutions. Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA) and used as received (standard desalting protocol). A Biorad MyCycler thermocycler was employed for all polymerase chain reactions (PCR). Chemically-competent E. coli TOP10 and BL21(DE3) cells were prepared in-house via standard protocols. A Qiagen miniprep kit was employed for plasmid isolations. Pfu Turbo DNA polymerase was purchased from Stratagene and T4 DNA ligase was obtained from New England Biolabs. All restriction enzymes were purchased from New England Biolabs. DNA sequencing and MALDI-TOF mass spectrometry were performed by staff in the Biopolymers Facility at the Massachusetts Institute of Technology. Details for the Fmoc-solid phase peptide synthesis of HD5[Ser5,20] are included as Supporting Information.
Instrumentation
Analytical and semi-preparative high-performance liquid chromatography (HPLC) were performed on an Agilent 1200 instrument equipped with a thermostated autosampler set at 4 °C and thermostated column compartment set at 20 °C unless noted otherwise. A 5-μm pore Clipeus C18 column (4.6 × 250 mm, Higgins, Inc.) was employed for all analytical HPLC experiments using a flow rate of 1 mL/min. A 5-μm pore ZORBAX C18 column (9.6 × 250 mm, Agilent Technologies) was employed for semi-preparative HPLC. A flow-rate of 5 mL/min was typically employed for semi-preparative HPLC. For all separations, solvent A was 0.2% TFA/H2O and solvent B was 0.2% TFA/MeCN, and absorption was monitored at 220 and 280 nm. An Agilent 8450 UV-visible spectrophotometer was employed for all routine optical absorption measurements. Peptide stock solution concentrations were routinely quantified by using calculated extinction coefficients, the values of which are listed in Table S1 (Supporting Information). Purified peptide samples were stored as lyophilized powders or as aqueous stock solutions at -20 °C. A BioTek Synergy HT plate reader was employed for the E. coli ML35 and turbidity assays described below. An Aviv Model 202 circular dichroism spectrometer operated at room temperature was utilized to collect CD spectra.
Cloning, Overexpression and Purification of His6-HD5
A synthetic gene containing the E. coli optimized nucleotide sequence for mature HD5 was obtained from DNA 2.0 in pJ201 (Supporting Information). The synthetic gene contained KpnI, SpeI and NdeI restriction sites on the 5′ end and a XhoI restriction site on the 3′ end. A TEV protease cleavage site (ENLYFQG) was incorporated into the 5′ end between the NdeI site and the 5′ end of the HD5 gene. The pJ201-TEV-HD5 plasmid was dissolved in 10 μL of water and transformed into chemically-competent E. coli TOP10 cells, and the plasmid was isolated from cultures (5 mL, containing 50 μg/mL kanamycin) by using a miniprep kit. The plasmid was digested with NdeI and XhoI (2.5 h, 37 °C) and gel purified, which afforded the fragment containing the HD5 gene. This fragment was ligated into NdeI and XhoI-digested pET28b with T4 DNA ligase (2 h, room temperature). The ligation reaction was used to transform chemically-competent E. coli TOP10 cells. Several of the resulting colonies were selected and used to inoculate 5 mL of LB media containing 50 μg/mL kanamycin. The cultures were grown to saturation and a miniprep kit was used to isolate the plasmid pET28b-TEV-HD5. Its identity was verified by DNA sequencing.
Quick-change site-directed mutagenesis (described below) was employed to obtain a plasmid encoding His6-HD5 with a CNBr cleavage site, hereafter pET28b-Met-HD5, using pET28b-TEV-HD5 as a template. The Gly residue of “ENLYFQG” was mutated to a Met residue, providing a Met moiety immediately adjacent to the N-terminal residue of mature HD5. The mutagenesis primers (M-1 and M-2) are listed in Table S2.
The pET28b-Met-HD5 expression plasmid was transformed into chemically-competent E. coli BL21(DE3) cells. Cultures from single colonies were grown to saturation in LB media containing 50 μg/mL kanamycin (37 °C, 175 rpm, t ∼ 16 h). The saturated cultures were diluted 1:100 into fresh LB containing 50 μg/mL kanamycin, incubated at 37 °C with shaking at 175 rpm, and induced with 500 or 100 μM IPTG at OD600 ∼ 0.6. Varying the IPTG concentration had no effect on the yield of His6-HD5. The cultures were incubated at 37 °C for an additional 3-4 h (OD600 ∼ 1.0 – 1.5) and subsequently pelleted by centrifugation (4,200 rpm × 30 min, 4 °C). The cell pellets were transferred to pre-weighed 50-mL polypropylene centrifuge tubes, flash frozen in liquid N2, and stored at -80 °C. The overexpression cultures were routinely grown in multiple 2-L portions contained in 4-L baffled flasks and combined to provide pellets from four or six liters of culture. Approximately 2 g/L of E. coli cells were obtained from each preparation.
The purification of His6-HD5 was established by modifying literature protocols for the purification of defensin peptides.45-47 In a typical peptide purification, cells from a 6-L pellet were thawed on ice and resuspended in ice-cold lysis buffer (6 M GuHCl, 100 mM Tris-HCl, pH 8) to provide a final volume of ∼40 mL. An aliquot (1 mL, 100 mM) of PMSF was added and the resuspended cells were transferred to an ice-cold stainless steel beaker and lysed by two rounds of sonication (1 sec on followed by 4 sec off for 1.5 min; 10% amplitude; Branson sonicator). Another 1-mL aliquot of 100 mM PMSF was added to the crude lysate, which was immediately clarified by centrifugation (13,000 rpm × 30 min, 4 °C). The supernatant was incubated with 3 mL of pre-washed (3 × 20 mL water) Ni-NTA resin (Qiagen) for 1.5 – 2 h at 4 °C with gentle mixing. The suspension was poured into a fritted glass column and the unbound fraction was eluted. The resin was subsequently washed with 45 mL of wash buffer (6 M GuHCl, 20 mM Tris-HCl, 300 mM NaCl, pH 8) and His6-HD5 was eluted by using 30 mL of elution buffer (6 M GuHCl, 10 mM Tris-HCl, 300 mM NaH2PO4, 200 mM NaCl, 1 M imidazole, pH 6.5). This fraction was transferred to a 3,500 MWCO Spectropor3 dialysis bag and dialyzed against 5% acetic acid (4 L, 24 h, 4 °C) followed by 0.1% acetic acid (4 L, 24 h, 4 °C). The lengthy dialysis was necessary to remove the GuHCl completely, and the step-wise decrease in acetic acid concentration was employed to dilute the acid percentage prior to lyophilization. The dialyzed sample was lyophilized to dryness, which afforded His6-HD5 as a white fluffy powder (15 – 20 mg/L). The purity of His6-HD5 was routinely evaluated by analytical HPLC. For a typical analytical HPLC run, a sample of the peptide was dissolved in 75 mM HEPES buffer pH 7.5 (200 μL) and incubated at room temperature for up to 3 h. The solution was divided into two 100-μL aliquots and TCEP (10 μL of a 100 mM stock solution in water) was added to one aliquot. Following an additional 15 min incubation at room temperature to reduce all disulfide linkages, 10 μL of 6% TFA(aq) was added to each sample. The samples were vortexed, centrifuged (13,000 rpm × 10 min, 4°C) and analyzed by analytical HPLC using a gradient of 10-60% B over 30 min. A comparison of the samples with and without TCEP provided insight into the degree of peptide oxidation that occurred during the protein purification (-TCEP) and purity of the sample (+TCEP). The retention time and results from MALDI-TOF mass spectrometry for His6-HD5 are included in Table 1.
Table 1. Characterization of Wild-Type HD5 and HD5 Mutants.
| Peptidea | Retention Time (min)b | Free Thiolc | [M+H]+ calc m/z | [M+H]+ obs m/z | Cys-Cys connectivityd |
|---|---|---|---|---|---|
| His6-HD5 | 23.4 | n.d.e | 6678.6 | 6676.3 | n.d.e |
| HD5red | 21.2 | 6.40 ± 0.69 | 3589.2 | 3588.1 | n.a.e |
| HD5ox | 16.0 | 0.81 ± 0.14 | 3583.1 | 3583.3 | (3–31)(5–20)(10–30) |
| HD5[Ser3,31]red | 20.6 | 4.02 ± 0.02 | 3557.1 | 3556.8 | n.a. |
| HD5[Ser3,31]ox (5-20)(10-30) | 16.3 | 0.31 ± 0.08 | 3553.0 | 3552.7 | (5–20)(10–30) |
| HD5[Ser3,31]ox (5-30)(10-20) | 17.2 | 0.36 ± 0.04 | 3553.0 | 3551.8 | (5–30)(10–20) |
| HD5[Ser3,31]ox (5-10)(20-30) | 17.4 | −0.29 ± 0.05 | 3553.0 | 3552.7 | (5–10)(20–30) |
| HD5[Ser5,20]red | 20.7 | 2.76 ± 0.42 | 3557.1 | 3555.9 | n.a.e |
| HD5[Ser5,20]ox (3-31)(10-30) | 16.5 | 0.0 ± 0.48 | 3553.0 | 3551.8 | (3–31)(10–30) |
| HD5[Ser5,20]ox (3-30)(10-31) | 17.0 | −0.17 ± 0.24 | 3553.0 | 3551.2 | (3–30)(10–31) |
| HD5[Ser10,30]red | 19.6 | 4.25 ± 0.11 | 3557.1 | 3556.6 | n.a.e |
| HD5[Ser10,30]ox (3-20)(5-31) | 16.0 | 0.19 ± 0.67 | 3553.0 | 3552.5 | (3–20)(5–31) |
| HD5[Ser10,30]ox (3-31)(5-20) | 16.9 | −0.28 ± 0.57 | 3553.0 | 3552.6 | (3–31)(5–20) |
| HD5[Ala3,31]red | 20.4 | 3.25 ± 0.04 | 3525.1 | 3523.5 | n.a.e |
| HD5[Ala3,31]ox (5-20)(10-30) | 16.3 | 0.79 ± 0.23 | 3521.0 | 3520.6 | (5–20)(10–30) |
| HD5[Ala3,31]ox (5-30)(10-20) | 16.7 | 0.69 ± 0.03 | 3521.0 | 3522.1 | (5–30)(10–20) |
| HD5[Ala3,31]ox (5-10)(20-30) | 17.2 | 0.35 ± 0.14 | 3521.0 | 3523.0 | (5–10)(20–30) |
| HD5[Ala10,30]red | 20.8 | 3.85 ± 0.09 | 3525.1 | 3523.1 | n.a.e |
| HD5[Ala10,30]ox (3-20)(5-31) | 16.2 | −0.46 ± 0.01 | 3521.0 | 3521.9 | (3–20)(5–31) |
| HD5[Ala10,30]ox (3-31)(5-20) | 17.1 | 0.62 ± 0.42 | 3521.0 | 3521.8 | (3–31)(5–20) |
| HD5[Ala10,30]ox (3-5)(20-31) | 17.4 | 0.59 ± 0.28 | 3521.0 | 3523.5 | (3–5)(20–31) |
| HD5[Serhexa] | 17.7 | n.a. e | 3492.8 | 3492.9 | n.a.e |
See Figure 1 for amino acid sequences. The numbers in parentheses indicate the S—S bonding pattern.
Retention times determined by using analytical RP-HPLC on a C18 column and a gradient of 10-60% B in 30 min.
Free thiol content determined by using the DTDP assay. The reported errors are the standard deviation of the mean.
See ref. 38 for HD5ox.
n.d. = not determined; n.a. = not applicable.
Tag Cleavage and Purification of HD5red
Cyanogen bromide in 80% formic acid was employed to cleave His6-HD5 and afford unmodified HD5red (32-aa; “red” specifies the reduced form with six free –SH from the Cys residues). In a typical cleavage reaction, a solution of 20 mg/mL CNBr in 80% formic acid was prepared and added to an equal volume of 2 mg/mL His6-HD5 in 80% formic acid. The reaction was covered in tin foil and incubated at room temperature for 5 h with gentle mixing. The reaction was diluted 1:1 or 1:2 with water, mixed for an additional 10 min, and transferred to a Spectropor3 3,500 MWCO dialysis membrane. The cleavage reaction was dialyzed against water (4 L per ∼50 mg of peptide cleaved) for 24 h at 4 °C. At this point, the samples were placed into fresh water and dialyzed again (4 L per ∼50 mg of peptide, 24 h, 4 °C). The samples were transferred to pre-weighed 50-mL polypropylene centrifuge tubes and lyophilized to dryness, which afforded a white powder. The product was analyzed by HPLC as described above for His6-HD5. The retention times for HD5red and HD5ox and MALDI-TOF data are listed in Table 1.
For large-scale purification of HD5red from the cleavage reaction, the lyophilized powder was suspended in 75 mM HEPES buffer adjusted to pH 8.2 (∼2 mg/mL), which yielded a cloudy solution, and incubated at room temperature for ≥3 h. An aliquot of 100 mM TCEP (1:30 dilution) was added and the mixture was incubated for an additional 15 min at room temperature. The solution was acidified by addition of 6% aqueous TFA (1:10 dilution) and aliquoted into 1.5-mL centrifuge tubes, centrifuged (13,000 rpm × 10 min, 4 °C), and purified by semi-preparative HPLC using a solvent gradient of 10-40% B in 15 min. The HD5red-containing fractions from multiple HPLC injections were combined and lyophilized to dryness, which afforded pure HD5red as a white powder in yields of 1-2 mg/L of culture.
Site-Directed Mutagenesis
A modified Quick-Change site-directed mutagenesis protocol (Stratagene) was employed to generate the HD5 mutants (Table 1). For the first round of mutagenesis, pET-28b-Met-HD5 was used as the template. The templates employed in subsequent rounds, primers and primer pairings are listed in Tables S2 and S3. The PCR protocol used for all mutagenesis reactions was: 95.0 °C for 30 min (1×); 55.0 °C for 1 min; 68 °C for 17 min (25×); 4.0 °C hold (1×).
Following PCR amplification using Pfu Turbo DNA polymerase, the residual template plasmid was digested with DpnI (2 μL added to a 50 μL PCR reaction in two 1-μL aliquots at t = 0 and 1.5 h) for 3 h at 37 °C. The DpnI digests were transformed into chemically-competent E. coli TOP10 cells. Overnight cultures (5 mL, 50 μg/mL kanamycin) were grown from single colonies and the purified plasmids were obtained by using a miniprep kit. The DNA sequences and presence of the desired mutation(s) were verified by DNA sequencing. Overexpression and purification of the His6-tagged and corresponding reduced mutant peptides were conducted as described above for His6-HD5. Yields for the mutant peptides ranged from ∼2.5 mg/L for His6-HD5[Ser5,20] to ∼15 mg/L for His6-HD5[Ser10,30].
Oxidative Folding
Purified and fully reduced peptides were folded by using a modification of an established protocol.48 In a typical folding experiment, the reduced peptide (1 mg) was dissolved in 0.5 mL of a freshly-prepared solution containing 8 M GuHCl, 3 mM glutathione and 0.3 mM glutathione disulfide. A 1.5-mL aliquot of 250 mM NaHCO3(aq) was added to the mixture, which caused the pH value to increase to 8.3. This mixture was incubated at room temperature for 4 h, centrifuged (13,000 rpm × 10 min), and the folded peptide(s) were purified by semi-preparative HPLC using a gradient of 10-30% B in 12 min. For large-scale folding reactions (i.e. ∼20 mg peptide), the same ratios were employed and the reaction was concentrated by using a 3,000 MWCO Amicon spin-filter prior to HPLC purification.
Thiol Quantification Assays
Quantification of free thiol residues in the reduced and oxidized defensin peptides was performed by using 2,2′-dithiodipyridine (DTDP, 4 mM in 12 mM HCl) and GSH (3 mM in 10 mM HCl) as a standard. To obtain a calibration curve, 10, 20, 30 or 40 μL of the GSH standard was added to the assay buffer (100 mM NaH2PO4, 200 μM EDTA, pH 7; degassed for 1 h by bubbling Ar) containing 125 μL of 4 mM DTDP (3 mL final volume). The solutions were incubated for 15 min at room temperature and the absorbance at 341 nm was measured. The absorbance at 341 nm for the reagent blank (no GSH present) was also measured. The 341 nm absorbance from the reagent blank (Ao) was subtracted from the absorbance of the solutions containing GSH, and the calibration curve was obtained by plotting corrected absorbance vs. GSH concentration.
To determine the number of free thiol residues in each peptide sample, peptide stock solutions were prepared in water (oxidized peptides) or 0.01 M HCl (reduced peptides) and the concentrations determined by UV-visible absorption spectroscopy. Aliquots of the peptide stock solutions were diluted with Ar-purged assay buffer containing DTDP as described above to provide final peptide concentrations of 2-4 or 6-7 μM for the reduced and oxidized peptides, respectively. The peptide solutions were incubated at room temperature for 15 min and absorbance at 341 nm was recorded and corrected by subtracting A0. The number of free thiols in the peptide was determined by using the glutathione standard curve. These assays were conducted at least twice for each peptide sample and with samples from independent purifications. The 4 mM DTDP stock solution can be stored in aliquots at -20 °C for several months.
Protease Digests and Regioisomer Identification
To identify the regioisomers of HD5[Ser3,31]ox and HD5[Ala3,31]ox, a 200-μL solution of each peptide (80 μM, 0.28 mg/mL) was prepared in an aqueous solution containing 20 mM CaCl2 and 0.001% TritonX-100 (pH 5.5). A 50-μL aliquot was removed to use as the no enzyme control. A 15-μL aliquot of trypsin (0.025 mg/mL in 20 mM CaCl2, 0.001% TritonX-100) was added to the remaining 150-μL solution of peptide. The mixture was incubated at room temperature for 14-16 h and quenched by addition of 6% TFA(aq) (15 μL). The quenched reactions were vortexed, centrifuged (13,000 rpm × 10 min, 4 °C) and stored on ice. The peptide fragments were separated and isolated by analytical HPLC (10-60% B in 30 min). The isolated fractions were lyophilized to dryness, and the resulting products were dissolved in 0.2% TFA and analyzed by MALDI-TOF mass spectrometry (reflector mode). The m/z values of the peptide fragments were assigned and used to identify the disulfide linkages as detailed in the Supporting Information section.
To identify the regioisomers of HD5[Ser10,30]ox and HD5[Ala10,30]ox, the peptides were first digested with trypsin as described above and the proteolytic products purified by HPLC. The fractions containing S—S linked peptide fragments (identified by MALDI-TOF) were subsequently digested with chymotrypsin. The samples from the trypsin digest were dissolved in 150 μL of 20 mM CaCl2, 0.001% TritonX-100 and a 15-μL aliquot of chymotrypsin (16 μg/mL) was added. The digests were incubated overnight at room temperature and the proteolysis products were separated and isolated by analytical HPLC (10-60% B in 30 min) and analyzed by MALDI-TOF mass spectrometry (Supporting Information).
Circular Dichroism Spectroscopy
Peptide solutions (20 μM, 280 μL) were prepared in 5 mM sodium phosphate buffer adjusted to pH 7.0. A 1-mm path-length quartz CD cell (Hellma) was employed for all measurements. The CD spectra were collected from 260 – 190 nm at 1 nm intervals (10 sec averaging time, three independent scans per wavelength). The data obtained from the three scans were averaged by using Excel (Microsoft Office 97) and plotted. To determine the effect of a membrane mimic on the peptide fold, the CD spectrum of each peptide was obtained in the presence of 10 mM SDS. For these studies, a 2.8-μL aliquot of 1 M SDS was added to 280 μL of the peptide solution. The samples were incubated at room temperature for ∼2 h and the CD data subsequently collected as described above.
Protease Susceptibility Assays
To determine the relative susceptibility of HD5ox, HD5[Ala3,31]ox, HD5[Ser3,31]ox and HD5[Serhexa] to trypsin digestion, 150-μL solutions of 80 μM peptide were prepared in buffer containing 100 mM Tris-HCl, 20 mM CaCl2, 0.001% Triton-X 100, pH 8.2 and a 25-μL aliquot was removed for the no enzyme control. A 12.5-μL aliquot of trypsin (25 μg/mL) was added and the reactions were mixed with a pipette and incubated at room temperature. A 25-μL aliquot of each reaction was removed at t = 2, 5, 10, 30, 60 min and quenched via addition of 3% TFA (6 μL). The samples were vortexed immediately, stored on ice, centrifuged (13,000 rpm × 10 min, 4 °C) to remove any precipitate, and analyzed by analytical HPLC using a solvent gradient of 10-60% B over 30 min.
Inner-Membrane Permeabilization Studies
The E. coli ML35 strain49 and standard protocols29,50 were employed to study the effect of HD5ox and the mutant peptides on E. coli inner membrane integrity. E. coli ML35 was grown overnight (37 °C, 16 h) with shaking in 5 mL of TSB. The overnight culture was diluted 1:100 into 6 mL of fresh TSB and grown for ∼2 h at 37 °C with shaking until the OD600 value reached ∼0.6, at which time the culture was diluted to obtain OD620 of 0.35 (1 × 108 CFU/mL). This culture was subsequently diluted 1:100 in two steps of 1:10 into fresh 12.5 mM sodium phosphate buffer adjusted to pH 7.4 and containing 1.25% TSB to provide the bacterial culture for the permeabilization assay.
A 96-well plate format was employed for the inner-membrane permeabilization assay. To each well was added 10 μL of peptide (10× sterile-filtered aqueous solution, variable concentrations) and 10 μL of 25 mM ONPG. An 80-μL aliquot of the bacterial culture was subsequently added and the plate was immediately transferred to a plate reader preheated to 37 °C. The plate was incubated at 37 °C for 2 h with shaking in the plate reader and the absorbance at 405 nm was measured in 5 min intervals. A no-peptide control, obtained by adding 10 μL of sterile water instead of the 10× peptide solution, was run with each experiment. Two or more independent assays were conducted for each peptide on different days.
Antibacterial Activity Assays – Colony Forming Unit Method
Bacteria (E. coli ATCC 25922 or S. aureus ATCC 25923) were grown overnight with shaking (37 °C, 16 h) in 5 mL of TSB. The overnight culture was diluted 1:100 into 6 mL of fresh TSB and grown for ∼2 h at 37 °C with shaking until the OD650 reached ∼0.5 (E. coli) or ∼0.6 (S. aureus). A 5-mL portion of the culture was transferred to a sterile culture tube and centrifuged (3500 rpm × 10 min, 4 °C) to pellet the bacterial cells. The supernatant was discarded and the cell pellet was resuspended in 5 mL of AMA buffer (10 mM sodium phosphate buffer supplemented with 1% TSB, pH 7.4). The cell suspension wa centrifuged (3500 rpm × 10 min, 4 °C) and the supernatant discarded. The resulting ce pellet was resuspended in 5 mL of AMA buffer and diluted with AMA buffer to obtain a OD650 value of 0.5 (2.5 × 108 CFU/mL) for E. coli or 0.6 (1 × 108 CFU/mL) for S. aureu For assays employing E. coli ATCC 25922, the bacterial suspension was further dilute 1:250 in three steps (1:10 × 1:10 × 1:2.5) into 2 mL of AMA buffer. For assay employing S. aureus ATCC 25923, the bacterial suspension was further diluted 1:100 i two steps (1:10 × 1:10) into 2 mL of AMA buffer. The diluted cultures were use immediately.
Antibacterial activity assays were performed in 96-well plates. Each we contained 10 μL of a 10× aqueous sterile-filtered peptide stock solution or a no-peptide control. A 90-μL aliquot of the diluted bacterial culture was added to each well and the plate was incubated for 1 h (37 °C, 150 rpm). Immediately after the 1 h incubation, a 50-μL aliquot from each well was removed and diluted with 450 μL of AMA buffer (10-1 dilution). This solution was vortexed gently and further diluted serially from 10-2 to 10-4 in 10-fold increments by adding a 100-μL volume from each dilution to 900-μL of fresh buffer. A 100-μL aliquot from each dilution was manually plated on trypticase soy agar plates by using a spinning inoculating table and incubated for 14-16 h at 37 °C. The number of colony forming units obtained for each peptide treatment was determined b colony counting. Only plates with 30 – 200 colonies were considered in the analysis With the exception of the experiments where S. aureus was treated with the two HD5[Ser5,20]ox regioisomers, all assays were conducted with at least two independently prepared and purified samples of each peptide and in three independent trials. The resulting averages are reported. Assays with HD5[Ser3,31]ox (5-20)(10-30) were not conducted because of insufficient sample.
Antibacterial Activity Assays – Turbidity Method
This method is based on a literature protocol51 and follows the one described above for determining colony-forming units. After the bacteria cultures were treated with peptide (or water control) for 1 h at 37 °C as described above, a 100-μL aliquot of 2× Mueller Hinton Broth (MHB) was added to each well and the plate was incubated at 37 °C with shaking at 150 rpm. Using a plate reader, the OD600 was measured in 1 h intervals over 5 h and a final OD600 reading was performed at 14-20 h. The OD600 values were plotted against time to provide a growth curve. These assays were repeated at least in duplicate. Assays with HD5[Ser3,31] (5-20)(10-30) were not conducted because of insufficient sample.
Results
Preparation of HD5ox and Mutants
A number of defensin peptides, including HD5, have been overexpressed in heterologous hosts as fusion proteins. Guided by literature procedures,45-47 we overexpressed HD5 and its mutants as His6 fusion proteins in E. coli BL21(DE3), and a Met residue was incorporated adjacent to the N-terminal Ala residue of HD5 to provide a CNBr cleavage site for tag removal. This design strategy provides the native HD5 following tag cleavage and is important because small modifications to the peptide N- or C-termini may alter the antibacterial activity of the peptide. Following IPTG induction and growth at 37 °C, the His-tagged peptides were isolated from the insoluble fraction by using a denaturing protocol and purified by Ni-NTA chromatography. Following extensive dialysis to remove GuHCl and lyophilization, His6-HD5 was isolated as a white fluffy solid. Because SDS-PAGE does not reveal small-molecule impurities and impurities arising from small peptide fragments, we chose to evaluate purified His6-HD5 by analytical reverse-phase HPLC. In the absence of reducing agent, the analytical HPLC trace for His6-HD5 varies dramatically depending on the preparation and one example is provided in Figure 2. One major peak, multiple small peaks (Figure 2), or a broad hump may be observed. These variations reflect peptide oxidation and formation of various disulfide linkages (uncharacterized) during purification. Addition of excess reducing agent (i.e. TCEP, DTT) results in formation of one major peak of longer retention time (Figure 2). This single peak (∼23 min) corresponds to the fully reduced His6-HD5 fusion peptide (Table 1). Analogous behavior was also observed in the HPLC analyses of the His-tagged mutant peptides (data not shown). His6-HD5 was routinely isolated in yields of ∼15-20 mg/L of bacterial culture and the yields of the mutant peptides reproducibly varied from ∼2.5 mg/mL for His6-HD5[Ser5,20] to ∼15 mg/mL for His6-HD5[Ser10,30].
Figure 2.
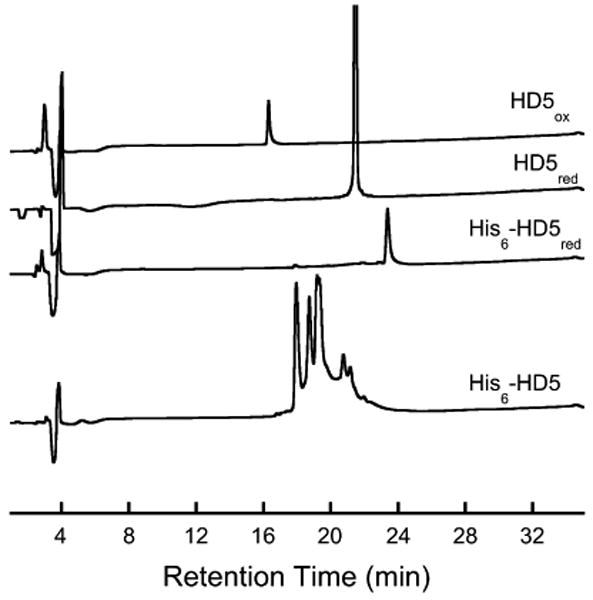
Representative analytical HPLC traces (220 nm absorption) of His6HD5, purified HD5red and purified HD5ox (variable sample concentrations). TCEP was added to Ni-NTA purified His6-HD5 to provide the fully reduced sample His6-HD5red (75 mM HEPES buffer, pH 7.4).
The His6 tag was removed by using 10 equiv (w/w) of CNBr in 80% formic acid. Literature protocols for CNBr cleavages and reaction work-up conditions vary. We found that a 1:1 dilution of the cleavage reaction with water followed by extensive dialysis against water was optimal for large-scale (>20 mg) reactions. When shorter dialysis times were employed, the yield of HD5red diminished by ∼20% and analytical HPLC of the crude reactions revealed multiple and more abundant additional peaks of lower retention time (unidentified). Following dialysis and lyophilization, the crude reaction was dissolved in aqueous buffer (75 mM HEPES, pH 8.2) and incubated for 3 h at room temperature. Subsequent addition of TCEP converted of any oxidized peptide to the reduced form. A typical yield of pure HD5red (Figure 2) following tag cleavage, reduction and semi-preparative HPLC purification is 1-2 mg/L. This optimized cleavage and purification protocol was employed for obtaining the HD5 double mutants. The final incubation step and TCEP addition were omitted for HD5[Serhexa]. Representative analytical HPLC traces of the purified mutant peptides are exhibited in Figure 3 (Ser double mutants) and in Figures S1 and S2 (Ala double mutants and hexa mutant).
Figure 3.
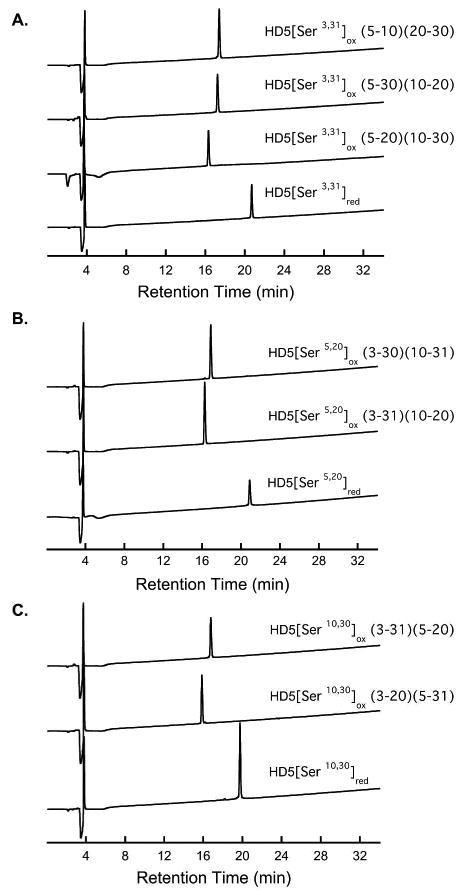
Representative analytical HPLC traces (220 nm absorption) of purified HD5 double mutants in the reduced and oxidized forms (10-60% B in 30 min). (A) HD5[Ser3,31]. (B) HD5[Ser5,20]. (C) HD5[Ser10,30]. The disulfide linkages are indicated in parentheses. HPLC traces of the purified Ala mutants are available as Supporting Information.
Peptide Folding, S—S Connectivity Determination
Small-scale (∼1 mg of peptide) oxidative folding experiments performed at room temperature revealed that the HPLC peak of HD5red disappears and a new peak of shorter retention time forms at t < 4 h and persists at longer time points. The reaction product was isolated and characterization by MALDI-TOF, which revealed a m/z value corresponding to the oxidized form (Table 1), and thiol quantification provided a free thiol count of < 1, which further supported complete oxidation of the peptide scaffold.
Incubation of the double mutant peptides under the same folding conditions resulted in formation of multiple new peaks in the analytical HPLC traces (Figures 3 and S1-S5). Folding of HD5[Ser5,20]red and HD5[Ser10,30]red each resulted in formation of two major peaks with approximately equal integrated area whereas three new peaks of varying intensity were observed following the folding of HD5[Ser3,31]red (Figures S3-S5). Likewise, folding of HD5[Ala3,31]red and HD5[Ala10,30]red resulted in the formation of three new peaks. In all cases, the product peaks were separated and purified by HPLC, and characterized by MALDI-TOF and thiol quantification assays (Table 1). The results from mass spectrometry and thiol quantification established that the each new HPLC peak corresponded to a fully oxidized double mutant with two S—S bonds, confirming that multiple regioisomers form upon oxidative folding of the HD5 double mutants.
Several methods were therefore employed to assign the S—S linkages in each regioisomer. For the HD5[Ser3,31] and HD5[Ala3,31], MALDI-TOF analysis of the peptide fragments resulting from trypsin digest was sufficient to unambiguously assign the S—S linkages (Table 1 and Figures S6-S11). Sequential digestion with trypsin and chymotrypsin was necessary and sufficient to characterize the oxidized HD5[Ser10,30] and HD5[Ala10,30] regioisomers Figures S12-S16). Because trypsin/chymotrypsin double digest could not provide unambiguous S—S determination for the HD5[Ser5,20]ox regioisomers, we synthesized authentic standards for the (3—31)(10—30) and (3— 30)(10—31) regioisomers by using Fmoc solid-phase peptide synthesis and orthogonal Cys protecting groups as detailed in the Supporting Information section (Scheme S1, Figure S17). Analytical HPLC analysis employing these synthetic standards provided the S—S connectivity assignments provided in Table 1 (Figures S18). These data reveal trends between HPLC retention times, which reflect relative hydrophilicity, and S—S connectivities. For instance, a comparison of the HPLC retention times for the 3-31 deletion mutants HD5[Ser3,31] and HD5[Ala3,31] illustrates that the native S—S connections afford the most hydrophilic regioisomers. Likewise, HD5[Ser5,20] (3-31)(10-30) has a shorter retention time than HD5[Ser5,20] (3-30)(10-31); however, this trend is not observed for the 10-30 deletion mutants. For both HD5[Ser10,30] and HD5[Ala10,30], the regioisomers with non-native (3-20)(5-31) linkages are most hydrophilic judging by HPLC retention time. These comparisons indicate that the relative HPLC retention times are not a consequence of the choice of Ser/Ala mutation. Rather, the variations stem from differences in the overall peptide fold resulting from variable S—S linkages.
Circular Dichroism Spectroscopy
Circular dichroism spectroscopy was therefore employed to investigate the ramifications of disulfide deletion and regioisomerization on peptide secondary structure (Figures 4 and S19-S20). In aqueous buffer (10 mM sodium phosphate buffer, pH 7.0), the CD spectrum of HD5ox exhibits a negative band at 198 nm and a positive band at 230 nm. Upon addition of 10 mM SDS, a membrane mimic, the negative band shifts slightly to 202 nm and the θ value at 190 nm becomes positive. These changes indicate that the secondary structure of HD5ox undergoes some reorganization in the presence of SDS. The changes in CD signature upon addition of SDS to the mutant peptides are, in general, more pronounced. The spectra all exhibit a negative band of varying intensity at ca. 200 nm that undergoes a slight red-shift following SDS addition. In aqueous buffer, the CD spectra of the mutants indicate a lack of overall secondary structure and the changes observed following addition of SDS suggest an increase in β-sheet character. In total, these data confirm that the loss of a native S—S bond increases the conformational flexibility of the peptide. As a result, the secondary structure largely exists as a random coil-like conformation in aqueous solution and the peptides can adopt increased β-sheet character in the presence of the membrane mimic SDS.
Figure 4.
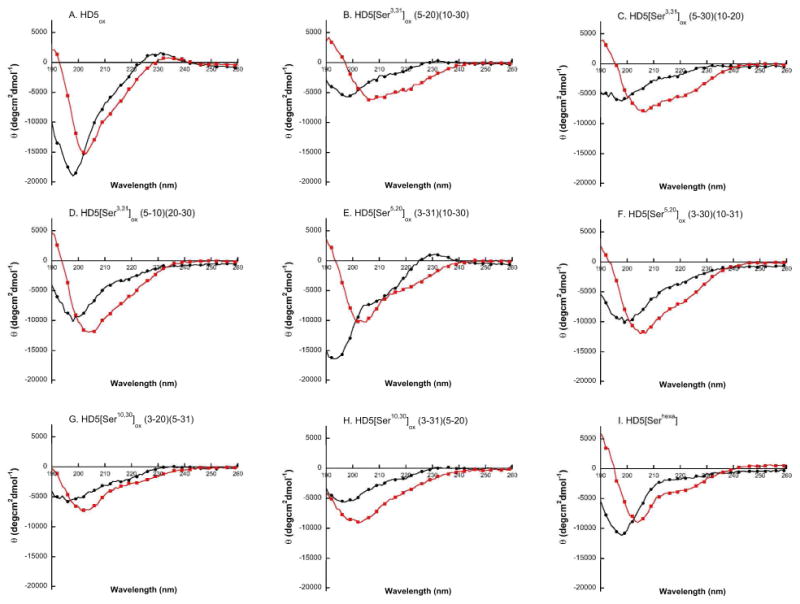
CD spectra of HD5ox and the Ser mutant peptides in the absence (black ●) and presence (red ■) of 10 mM SDS (5 mM sodium phosphate buffer, pH 7.0). (A) HD5ox. (B) HD5[Ser3,31]ox (5-20)(10-30). (C) HD5[Ser3,31]ox (5-30)(10-20). (D) HD5[Ser3,31]ox (5-10)(20-30). (E) HD5[Ser5,20]ox (3-31)(10-30). (F) HD5[Ser5,20]ox (3-30)(10-31). (G) HD5[Ser10,30]ox (3-20)(5-31). (H) HD5[Ser10,30]ox (3-31)(5-20). (I) HD5[Serhexa]. CD spectra for the Ala mutants are available as Supporting Information.
Antibacterial Activity Assays
Defensins are often described as non-specific membrane-penetrating peptides. To ascertain the effect of S—S deletion on antibacterial activity, E. coli ATCC 25922 and S. aureus ATCC 25923 were chosen as representative Gram-negative and −positive bacterial strains, respectively, the cell membranes of which have different compositions. The results from colony counting assays conducted by using HD5ox and the Ser mutant family (Figure 5) reveal that HD5ox exhibits low micromolar activity against both E. coli and S. aureus, in good agreement with literature reports,48,51 and that the antimicrobial behavior of the mutant peptides exhibit clear strain dependence. Deletion of any disulfide bond abrogates the antibacterial activity of HD5ox against S. aureus whereas low micromolar activity against E. coli is observed for most Ser mutants in the colony counting assay, including HD5[Serhexa] (Figure 5). Attenuation of activity against E. coli is observed only for the HD5[Ser5,20] (3-30)(10-31) regioisomer.
Figure 5.
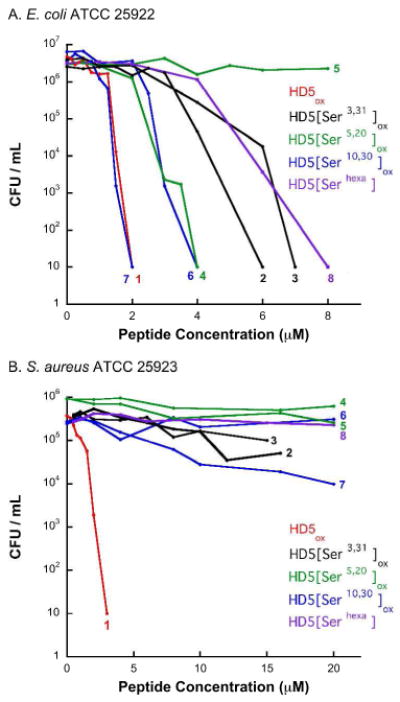
Antibacterial activity assays with the HD5ox mutant family (CFU method). 1, HD5ox; 2, HD5[Ser3,31]ox (5-30)(10-20); 3, HD5[Ser3,31]ox (5-10)(20-30); 4, HD5[Ser5,20]ox (3-31)(10-30); 5, HD5[Ser5,20]ox (3-30)(10-31); 6, HD5[Ser10,30]ox (3-31)(5-20); 7, HD5[Ser10,30]ox (3-20)(5-31). (AActivity of HD5ox and the Ser mutants against E. coli ATCC 25922. (B) Activity of HD5ox and the Ser mutants against S. aureus ATCC 25923. In both instances a CFU of 10 indicates no colony formation. The data represent the averages of three independent trials and the error bars are omitted for clarity. Plots including error bars are provided as Supporting Information.
A high-throughput liquid culture assay for quantifying defensin antibacterial activity by turbidity measurements has been described in the literature.51 In this work, a modified version of this turbidity assay was also performed (Figures S21-S28). Like the CFU assays, the turbidity assays indicate that HD5ox is bactericidal against E. coli ATCC 25922 in the low micromolar range (2 μM CFU assay vs. 4 μM turbidity assay). In contrast, bactericidal behavior was not observed in the turbidity assay for the mutant peptides in the concentration ranges evaluated. The variability in liquid culture vs. plate assay suggests that the colony counting may overestimate or the turbidity assay may underestimate the activity of the mutant peptides to some degree. For turbidity assays employing S. aureus ATCC 25923, no activity is observed for the Ser and Ala mutant peptides, as expected. Higher concentrations of HD5ox are required for killing of S. aureus in the turbidity assay, and this trend is in agreement with data reported previously.51
Inner-Membrane Permeabilization Assays
Membrane permeabilization assays were conducted by using E. coli ML35 to ascertain the ability of HD5ox and the mutants to permeate the inner membrane of this bacterial strain. E. coli ML35 is lactose permease deficient and contains a cytoplasmic β-galactosidase, which accepts the lactose mimic ONPG as a substrate. ONPG is unable to cross the intact E. coli ML35 cell membrane, but can enter the periplasm and cytoplasm if the outer and inner cell membranes are damaged. β-Galactosidase-catalyzed hydrolysis of ONPG results in formation of galactose and ONP, the latter of which has an optical absorption band centered at 420 nm and provides a colorimetric readout of membrane damage. Figure 6 exhibits the result of membrane permeabilization assays where E. coli ML35 was treated with 2 μM of each peptide. A rapid increased in optical absorption at 405 nm was observed following treatment with 2 μM HD5ox, indicating relatively efficient membrane disruption. In contrast, incubation of E. coli ML35 with 2 μM of mutant peptides resulted in little to no membrane disruption. This observation is in agreement with the antimicrobial activity assay results, where slightly elevated concentrations of the double mutant peptides were required for bacterial cell death relative to HD5ox. A series of concentration-dependent studies were therefore undertaken to ascertain the effect of increasing the mutant peptide concentration on E. coli ML35 membrane damage (Figures S29-S35). In most instances, increasing the peptide concentration causes an enhancement in ONP formation, which suggests that the native disulfide array and peptide topology of HD5ox are not essential for membrane disruption. Nevertheless, the native peptide confers the most rapid formation of ONP, which indicates that it is most efficient at causing inner membrane damage. It is possible that the mechanism of membrane disruption for HD5ox and the mutant peptides vary. The attenuation of activity observed for HD5[Ser5,20] (3-30)(10-31) against E. coli ATCC 25922 is manifest in the E. coli ML35 assays because no ONP formation is observed for this isomer.
Figure 6.
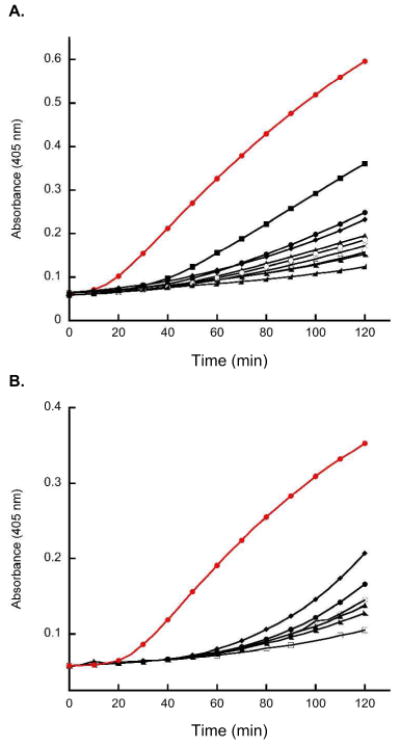
Representative plots from inner-membrane permeabilization assays. ONP formation monitored at 405 nm following incubation of E. coli ML35 with 2 μM of peptide and 2.5 mM ONPG at 37 °C (10 mM sodium phosphate buffer, 1% TSB, pH 7.4). (A) Comparison of HD5ox with the Ser mutant peptides. (red ●) HD5ox, (●) HD5[Ser3,31]ox (5-20)(10-30), (◣) HD5[Ser3,31]ox (5-30)(10-20), (◆) HD5[Ser3,31]ox (5-10)(20-30), (▲) HD5[Ser10,30]ox (3-20)(5-31), (■) HD5[Ser5,20]ox (3-31)(10-30), (◢) HD5[Ser5,20]ox (3-30)(10-31), (○) HD5[Ser10,30]ox (3-31)(5-20), (×) HD5[Serhexa]. (B) Comparison of HD5ox with the Ala mutant peptides. (red ●) HD5ox, (●) HD5[Ala3,31]ox (5-20)(10-30), (◣) HD5[Ala3,31]ox (5-30)(10-20), (◆) HD5[Ala3,31]ox (5-10)(20-30), (▲) HD5[Ala10,30]ox (3-20)(5-31), (○) HD5[Ala10,30]ox (3-31)(5-20). Results from inner-membrane permeabilization assays where E. coli ML35 were treated with varying concentrations of each peptide are available as Supporting Information.
Protease Degradation Assays
It is accepted that the defensin disulfide array confers protease resistance,11 and HD5ox is a very poor substrate for trypsin.40 In contrast, treatment of HD5red with proteases such as trypsin results in rapid proteolytic cleavage (data not shown). To ascertain the consequence of deletion of a single S—S bond to proteolytic stability, enzymatic activity assays were conducted by using trypsin and HD5[Ala3,31]ox (5-10)(20-30), HD5[Ser3,31]ox (5-10)(20-30), HD5[Ser3,31]ox (5-20)(10-30), HD5[Serhexa] and HD5ox as potential substrates and analyzed by analytical HPLC. The data in Figure 7 reveal that all mutant peptides are substrates for trypsin whereas HD5ox is not accepted as a substrate by the enzyme. With the exception of HD5[Ser3,31]ox (5-20)(10-30), the full-length mutant peptides are consumed within 2 min of trypsin digestion. Whereas HD5[Serhexa] is fully degraded within 2 min, longer incubation times are required for complete proteolytic breakdown of the initial products of HD5[Ala3,31]ox and HD5[Ser3,31]ox digestion. Although the presence of two disulfide bonds decreases the accessibility of certain trypsin cleavage sites, loss of a single S—S bond results in rapid proteolytic degradation by the protease regardless of native or non-native regioisomerization of the two intact disulfide bonds. The complete disulfide array of native HD5ox is therefore essential for protease resistance.
Figure 7.
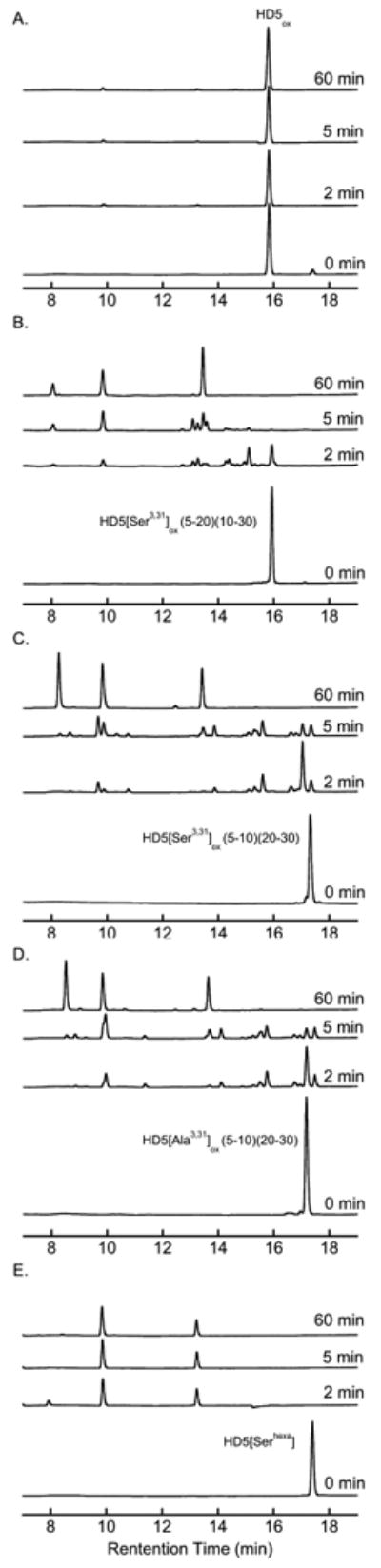
Analytical HPLC traces (220 nm) for trypsin susceptibility assays. (A) HD5ox. (B) HD5[Ser3,31] (5-20)(10-30). (C) HD5[Ser3,31]ox (5-10)(20-30). (D) HD5[Ala3,31]ox (5-10)(20-30). (E) HD5[Serhexa].
Discussion
Defensins are cysteine-rich host defense peptides and, as a family, are celebrated for their broad-range antimicrobial activities and other physiological roles. Despite similar topologies resulting from regiospecific disulfide bond formation, the amino acid sequences and compositions of individual defensin peptides differ substantially. As a result, it is important to evaluate defensins from the perspectives of structure and physiological function on a case-by-case basis. A number of structural/activity relationship studies on human, murine and invertebrate defensins that addressed the necessity of the disulfide array illustrate this point. Shuffling of the native disulfide array of HBD-3 or removal of all Cys residues by substitution with α-aminobutyric acid had no effect on the antibacterial activity of HBD-3 against E. coli ATCC 25922, but influenced the chemotactic activity of this peptide.43 Later work indicated that the activity of linear HBD-3 analogs varies with choice of mutation.52 In a similar study of HNP-1 mutants, variants with only one or two disulfide bonds were slightly more active than the wild-type peptide against E. coli, S. aureus and P. aeruginosa, and disulfide shuffling did not compromise antibacterial activity against the tested strains.44 Deletion of the Cys residues all together resulted in a ca. 8- to 14-fold decrease in activity against these three strains.44 Linear bovine β-defensin 2 analogs exhibited similar activity to the wild-type peptide.53 A thorough study of the disulfide array of murine cryptdin-4 revealed that mutations of S—S bonding pairs of Cys residues to Ala resulted in no loss of function and increased protease susceptibility.11 Although first hypothesized that the interactions between cryptdin-4 analogs and the bacterial membrane are independent of the disulfide arrays,11 further investigations suggested that the mechanisms of membrane damage for wild-type versus the disulfide null mutant peptides vary.50 Similar conclusions have been drawn by others in studies of designer defensin peptides.54 Lastly, the disulfide arrays exhibited by other types of cysteine-rich antimicrobial peptides, including protegrin-155 and gomesin,56 have also been subject to mutagenesis and evaluation. For instance, linear analogs of gomesin, an antimicrobial peptide produced by the spider, afforded 8- to 64-lower antibacterial activity compared to the wild-type strain.56
Recently, reduction of the disulfide bonds in select defensin peptides by addition of a reducing agent such at DTT has been employed to explore the consequences of disulfide bond deletion. Antibacterial activities conducted with reduced cryptdin-457 and HBD-158 in the presence of DTT revealed that the reduced peptides exhibit potent activity against the anaerobic Gram-positive commensal bacterial strains Bifidobacterium and Lactobacillus. This behavior is not observed for the oxidized forms. These studies bring into question the physiological distribution of defensins as reduced and oxidized peptides, in addition to the physiological significance of the reduced peptides and the disulfide array.
In this work, we present a systematic investigation of HD5 mutant peptides where either one native or all three native disulfide bonds are deleted by replacing pairs of Cys with pairs of Ser or Ala residues. HPLC analysis and characterization of the products of oxidative folding assays by enzymatic digest and mass spectrometry, or by the use of authentic standards prepared by chemical synthesis, allowed for unambiguous regioisomer assignments for all oxidized peptides in this study (Table 1). CD spectroscopy validated that, in all instances, loss of a single S—S bond results in a loss of secondary structure relative to HD5ox (Figure 4). This behavior is reminiscent of that observed for other antimicrobial Cys-rich peptides upon S—S deletion including HNP-1(44) and HBD-1[Ser35],59 and confirms that the native disulfide array is essential for maintaining the native peptide fold. In agreement with expectations and with studies of cryptdin-4 disulfide mutants,11 removal of a single S—S bond confers marked susceptibility to proteolytic cleavage by trypsin (Figure 7). Trypsin is a serine protease that preferentially cuts after cationic residues including Arg and Lys and it is the processing enzyme for the HD5 propeptide.60
The results from antibacterial activity assays conducted by using the Cys^Ser mutant peptides reveal very different trends for Gram-negative E. coli and Gram-positive S. aureus. Only wild-type HD5ox exhibited low micromolar antibacterial activity against S. aureus in colony counting assays; loss of a single disulfide bond resulted in attenuation of activity against this strain. Most mutants, in contrast, displayed activity against E. coli. The attenuation of activity against E. coli for HD5[Ser5,20] (3-30)(10-31) is striking and the origins of this behavior are currently unclear. The behavior of HD5[Serhexa] is in agreement with that of an α-aminobutyric acid-substituted HD5 analog reported by others.42 The loss of activity against S. aureus is therefore not the results of the choice of Cys substitution.
Cryptdin-4 variants lacking one S—S bond remained active against S. aureus,11 in contrast to our observations with the HD5 mutant family. We hypothesize that this difference stems from the variations in overall positive charge exhibited by these peptides. Cryptdin-4 contains nine Arg residues and, at neutral pH, has an overall charge of +8 whereas HD5 has six Arg residues and an overall charge of ca. +4. Cryptdin-4 variants may therefore have an enhanced ability to interact with bacterial cell membranes via electrostatic interactions and thereby retain activity with S—S loss.50
Precisely how HD5ox kills various Gram-negative and –positive bacterial strains is not yet clear. Elucidating the detailed mechanism of action will require further investigations of structure and putative physiological targets. Along these lines, it is important to determine if the attenuation of antibacterial activity observed for S. aureus upon S—S deletion is specific for this strain or occurs with other Gram-positive bacteria, including strains that inhabit the gut. Ascertaining the origins of the disulfide-dependent attenuation of activity against S. aureus, which may result from proteolytic degradation, a failure to assemble into a necessary oligomeric state, or the inability to interact with a specific cellular target, will also provide insight into how HD5ox exerts its antibacterial function. Efforts to address such possibilities are in progress.
Supplementary Material
Acknowledgments
We thank Ms. Debbie Pheasant for assistance with the CD spectrophotometer, Mr. Timothy Caradonna for assistance with the antimicrobial activity assays and for proofreading, Professor Barbara Imperiali for use of her centrifuge, and Professor JoAnne Stubbe for use of her Milli-Q water purification system.
This work was supported by Grant DP2OD007045 from the Office of the Director, National Institutes of Health (EMN) and the Department of Chemistry at the Massachusetts Institute of Technology (EMN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Office of the Director, National Institutes of Health or the National Institutes of Health. Instrumentation for circular dichroism spectroscopy is maintained in the Department of Chemistry Biophysical Instrumentation Facility.
Abbreviations
- CFU
Colony forming unit
- CNBr
Cyanogen bromide
- GSH
Glutathione (reduced)
- GSSG
Glutathione (oxidized)
- GuHCl
Guanidinium hydrochloride
- HBD
Human β-defensin
- HD5
Human α-defensin 5
- HD5red
Reduced human defensin 5
- HD5ox
Oxidized human defensin 5
- HNP
Human neutrophil peptide (an α-defensin)
- ONP
O-nitrophenol
- ONPG
O-Nitrophenyl-β-galactopyranoside
- PMSF
Phenylmethylsulfonyl fluoride
- SDS
Sodium dodecylsulfate
- TCEP
Tris(2-carboxyethyl)phosphine
- TFA
Trifluoroacetic acid
- TSB
Trypticase soy broth
- OD
Optical density
Footnotes
Supporting Information Available: Peptide molecular weights and extinction coefficients (Table S1), primers and primer pairings used for PCR (Tables S2 and S3), nucleotide sequence for the HD5 synthetic gene, details for the Fmoc solid-phase peptide synthesis of the HD5[Ser5,20] regioisomers, Scheme S1, Figure S1-S36. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ouellette AJ. Paneth cell α-defensins in enteric innate immunity. Cell Mol Life Sci. 2011;68:2215–2229. doi: 10.1007/s00018-011-0714-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ouellette AJ. Defensin-mediated innate immunity in the small intestine. Best Practice & Research Clinical Gastroenterology. 2004;18:405–419. doi: 10.1016/j.bpg.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 4.Aerts AM, Francois IEJA, Cammue BPA, Thevissen K. The mode of antifungal action of plant, insect and human defensins. Cell Mol Life Sci. 2008;65:2069–2079. doi: 10.1007/s00018-008-8035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouellette AJ. Paneth cell α-defensin synthesis and function. CTMI. 2006;306:1–25. doi: 10.1007/3-540-29916-5_1. [DOI] [PubMed] [Google Scholar]
- 6.Schneider JJ, Unholzer A, Schaller M, Schäfer-Korting M, Korting HC. Human defensins. J Mol Med. 2005;83:587–595. doi: 10.1007/s00109-005-0657-1. [DOI] [PubMed] [Google Scholar]
- 7.Jones DE, Bevins CL, Ghosh D, Ganz T. Paneth cells of the human small intestine express an antimicrobial peptide gene. J Biol Chem. 1992;267:23216–23225. [PubMed] [Google Scholar]
- 8.Porter EM, Liu L, Oren A, Anton PA, Ganz T. Localization of human intestinal defensin 5 in Paneth cell granules. Infect Immun. 1997;65:2389–2395. doi: 10.1128/iai.65.6.2389-2395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter EM, Bevins CL. The multifaceted paneth cell. Cell Mol Life Sci. 2002;59:156–170. doi: 10.1007/s00018-002-8412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van der Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–419. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maemoto A, Qu X, Rosengren KJ, Tanabe H, Henschen-Edman A, Craik DJ, Ouellette AJ. Functional analysis of the α-defensin disulfide array in mouse cryptdin-4. J Biol Chem. 2004;279:44188–44196. doi: 10.1074/jbc.M406154200. [DOI] [PubMed] [Google Scholar]
- 12.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 13.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422:522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 14.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salzman NH, Underwood MA, Bevins CL. Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol. 2007;19:70–83. doi: 10.1016/j.smim.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H, Fellermann K, Ganz T, Stange EF, Bevins CL. Reduced Paneth cell α-defensins in ileal Crohn's disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi J, Aono S, Lu W, Ouellette AJ, Hu X, Ji Y, Wang L, Lenz S, van Ginkel FW, Liles M, Dykstra C, Morrison EE, Elson CO. A novel role for defensins in intestinal homeostasis: regulation of IL-1β secretion. J Immunol. 2007;179:1245–1253. doi: 10.4049/jimmunol.179.2.1245. [DOI] [PubMed] [Google Scholar]
- 18.Elphick D, Liddell S, Mahida YR. Impaired luminal processing of human defensin-5 in Crohn's disease. Am J Pathol. 2008;172:702–713. doi: 10.2353/ajpath.2008.070755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J. Defensins and Paneth cells in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1284–1292. doi: 10.1002/ibd.20197. [DOI] [PubMed] [Google Scholar]
- 20.Ouellette AJ, Bevins CL. Paneth cell defensins and innate immunity of the small bowel. Inflamm Bowel Dis. 2001;7:43–50. doi: 10.1097/00054725-200102000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Menendez A, Finlay BB. Defensins in the immunology of bacterial infections. Curr Opin Immunol. 2007;19:385–391. doi: 10.1016/j.coi.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Droin N, Hendra JB, Ducoroy P, Solary E. Human defensins as cancer biomarkers and antitumor molecules. J Proteomics. 2009;72:918–927. doi: 10.1016/j.jprot.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Tesse R, Cardinale F, Santostasi T, Polizzi A, Manca A, Mappa L, Iacoviello G, De Robertis F, Logrillo VP, Armenio L. Association of β-defensin-1 gene polymorphisms with Pseudomonas aeruginosa airway colonization in cystic fibrosis. Genes Immun. 2008;9:57–60. doi: 10.1038/sj.gene.6364440. [DOI] [PubMed] [Google Scholar]
- 24.Verma C, Seebah S, Low SM, Zhou L, Liu SP, Li J, Beuerman RW. Defensins: antimicrobial peptides for therapeutic development. Biotechnol J. 2007;2:1353–1359. doi: 10.1002/biot.200700148. [DOI] [PubMed] [Google Scholar]
- 25.de Paula VS, Razzera G, Barreto-Bergter E, Almeida FCL, Valente AP. Portrayal of complex dynamic properties of sugarcane defensin 5 by NMR: multiple motions associated with membrane interaction. Structure. 2011;19:26–36. doi: 10.1016/j.str.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Satchell DP, Sheynis T, Shirafuji Y, Kolusheva S, Ouellette AJ, Jelinek R. Interactions of mouse paneth cell α-defensins and α-defensin prescursors with membranes. Prosegment inhibition of peptide association with biomimetic membranes. J Biol Chem. 2003;278:13838–13846. doi: 10.1074/jbc.M212115200. [DOI] [PubMed] [Google Scholar]
- 27.van der Weerden NL, Hancock REW, Anderson MA. Permeabilization of fungal hyphae by the plant defensin NaD1 occurs through a cell wall-dependent process. J Biol Chem. 2010;285:37513–37520. doi: 10.1074/jbc.M110.134882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thevissen K, Ghazi A, De Samblanx GW, Brownlee C, Osborn RW, Broekaert WF. Fungal membrane responses induced by plant defensins and thionins. J Biol Chem. 1996;271:15018–15025. doi: 10.1074/jbc.271.25.15018. [DOI] [PubMed] [Google Scholar]
- 29.Lehrer RI, Barton A, Daher KA, Harwig SSL, Ganz T, Selsted ME. Interaction of human defensins with Escherichia coli. J Clin Invest. 1989;84:553–561. doi: 10.1172/JCI114198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Lu W, Hong M. The membrane-bound structure and topology of a human α-defensin indicate a dimer pore mechanism for membrane disruption. Biochemistry. 2010;49:9770–9782. doi: 10.1021/bi101512j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt NW, Mishra A, Lai GH, Davis M, Sanders LK, Tran D, Garcia A, Tai KP, McCray PB, Jr, Ouellette AJ, Selsted ME, Wong GCL. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J Am Chem Soc. 2011;133:6720–6727. doi: 10.1021/ja200079a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilmes M, Cammue BPA, Sahl HG, Thevissen K. Antibiotic activities of host defense peptides: more to it than lipid bilayer perturbation. Nat Prod Rep. 2011;28:1350–1358. doi: 10.1039/c1np00022e. [DOI] [PubMed] [Google Scholar]
- 33.Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, Jansen A, Nielsen AK, Mygind PH, Raventós DS, Neve S, Ravn B, Bonvin AMJJ, De Maria L, Andersen AS, Gammelgaard LK, Sahl HG, Kristensen HH. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328:1168–1172. doi: 10.1126/science.1185723. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt P, Wilmes M, Pugnière M, Aumelas A, Bachere E, Sahl HG, Schneider T, Destoumieux-Garzón D. Insight into invertebrate defensin mechanism of action: oyster defensins inhibit peptidoglycan biosynthesis by binding to Lipid II. J Biol Chem. 2010;285:29208–29216. doi: 10.1074/jbc.M110.143388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sass V, Schneider T, Wilmes M, Körner C, Tossi A, Novikova N, Shamova O, Sahl HG. Human β-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun. 2010;78:2793–2800. doi: 10.1128/IAI.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Leeuw E, Li C, Zeng P, Li C, Diepeveen-de Buin M, Lu WY, Breukink E, Lu W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584:1543–1548. doi: 10.1016/j.febslet.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discovery. 2006;5:321–332. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 38.Szyk A, Wu Z, Tucker K, Yang D, Lu W, Lubkowski J. Crystal structures of human α-defensins HNP4, HD5, and HD6. Protein Sci. 2006;15:2749–2760. doi: 10.1110/ps.062336606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pazgier M, Li X, Lu W, Lubkowski J. Human defensins: synthesis and structural properties. Curr Pharm Des. 2007;13:3096–3118. doi: 10.2174/138161207782110381. [DOI] [PubMed] [Google Scholar]
- 40.Rajabi M, de Leeuw E, Pazgier M, Li J, Lubkowski J, Lu W. The conserved salt bridge in human α-defensin 5 is required for its precursor processing and proteolytic stability. J Biol Chem. 2008;283:21509–21518. doi: 10.1074/jbc.M801851200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei G, de Leeuw E, Pazgier M, Yuan W, Zou G, Wang J, Ericksen B, Lu WY, Lehrer RI, Lu W. Through the looking glass, mechanistic insights from enantiomeric human defensins. J Biol Chem. 2009;284:29180–29192. doi: 10.1074/jbc.M109.018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Leeuw E, Burks SR, Li X, Kao JPY, Lu W. Structure-dependent functional properties of Human Defensin 5. FEBS Lett. 2007;581:515–520. doi: 10.1016/j.febslet.2006.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Z, Hoover DM, Yang D, Boulègue C, Santamaria F, Oppenheim JJ, Lubkowski J, Lu W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human β-defensin 3. Proc Natl Acad Sci USA. 2003;100:8880–8885. doi: 10.1073/pnas.1533186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandal M, Nagaraj R. Antibacterial activities and conformations of synthetic α-defensin HNP-1 and analogs with one, two and three disulfide bridges. J Peptide Res. 2002;59:95–104. doi: 10.1034/j.1399-3011.2002.01945.x. [DOI] [PubMed] [Google Scholar]
- 45.Figueredo S, Mastroianni JR, Tai KP, Ouellette AJ. Expression and purification of recombinant α-defensins and α-defensin precursors in Escherichia coli. Methods Mol Biol. 2010;618:47–60. doi: 10.1007/978-1-60761-594-1_4. [DOI] [PubMed] [Google Scholar]
- 46.Pazgier M, Lubkowski J. Expression and purification of recombinant human α-defensins in Escherichia coli. Protein Expression Purif. 2006;49:1–8. doi: 10.1016/j.pep.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Marquès L, Oomen RJFJ, Aumelas A, Le Jean M, Berthomieu P. Production of an (Arabidopsis halleri) foliar defensin in Escherichia coli. J Appl Microbiol. 2009;106:1640–1648. doi: 10.1111/j.1365-2672.2008.04131.x. [DOI] [PubMed] [Google Scholar]
- 48.Wu Z, Ericksen B, Tucker K, Lubkowski J, Lu W. Synthesis and characterization of human α-defensins 4-6. J Peptide Res. 2004;64:118–125. doi: 10.1111/j.1399-3011.2004.00179.x. [DOI] [PubMed] [Google Scholar]
- 49.Lehrer RI, Barton A, Ganz T. Concurrent assessment of inner and outer membrane permeabilization and bacteriolysis in E. coli by multiple-wavelength spectrophotometry. J Immunol Methods. 1988;108:153–158. doi: 10.1016/0022-1759(88)90414-0. [DOI] [PubMed] [Google Scholar]
- 50.Hadjicharalambous C, Sheynis T, Jelinek R, Shanahan MT, Ouellette AJ, Gizeli E. Mechanisms of α-defensin bactericidal action: comparative membrane disruption by cryptdin-4 and its disulfide-null analogue. Biochemistry. 2008;47:12626–12634. doi: 10.1021/bi800335e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ericksen B, Wu Z, Lu W, Lehrer RI. Antibacterial activity and specificity of the six human α-defensins. Antimicrob Agents Chemother. 2005;49:269–275. doi: 10.1128/AAC.49.1.269-275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chandrababu KB, Ho B, Yang D. Structure, dynamics, and activity of an all-cysteine mutated human β-defensin 3 peptide analogue. Biochemistry. 2009;48:6052–6061. doi: 10.1021/bi900154f. [DOI] [PubMed] [Google Scholar]
- 53.Krishnakumari V, Sharadadevi A, Singh S, Nagaraj R. Single disulfide and linear analogues corresponding to the carboxy-terminal segment of bovine β-defensin-2: effects of introducing the β-hairpin nucleating sequence D-Pro-Gly on antibacterial activity and biophysical properties. Biochemistry. 2003;42:9307–9315. doi: 10.1021/bi034403y. [DOI] [PubMed] [Google Scholar]
- 54.Antcheva N, Morgera F, Creatti L, Vaccari L, Pag U, Pacor S, Shai Y, Sahl HG, Tossi A. Artificial β-defensin based on a minimal defensin template. Biochem J. 2009;421:435–447. doi: 10.1042/BJ20082242. [DOI] [PubMed] [Google Scholar]
- 55.Lai JR, Huck BR, Weisblum B, Gellman SH. Design of non-cysteine-containing antimicrobial β-hairpins: structure-activity relationship studies with linear protegrin-1 analogues. Biochemistry. 2002;41:12835–12842. doi: 10.1021/bi026127d. [DOI] [PubMed] [Google Scholar]
- 56.Fázio MA, Oliveira VX, Jr, Bulet P, Miranda MTM, Daffre S, Miranda A. Structure-activity relationship studies of gomesin: Importance of the disulfide bridges for conformation, bioactivities, and serum stability. Biopolymers. 2006;84:205–218. doi: 10.1002/bip.20396. [DOI] [PubMed] [Google Scholar]
- 57.Masuda K, Sakai N, Nakamura K, Yoshioka S, Ayabe T. Bactericidal activity of mouse α-defensin cryptdin-4 predominantly affects noncommensal bacteria. J Innate Immun. 2010;3:315–326. doi: 10.1159/000322037. [DOI] [PubMed] [Google Scholar]
- 58.Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, Buchner J, Schaller M, Stange EF, Wehkamp J. Reduction of disulfide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nature. 2011;469:419–423. doi: 10.1038/nature09674. [DOI] [PubMed] [Google Scholar]
- 59.Circo R, Skerlavaj B, Gennaro R, Amoroso A, Zanetti M. Structural and functional characterization of hBD-1(Ser35), a peptide deduced from a DEFB1 polymorphism. Biochem Biophys Res Commun. 2002;293:586–592. doi: 10.1016/S0006-291X(02)00267-X. [DOI] [PubMed] [Google Scholar]
- 60.Ghosh D, Porter E, Shen B, Lee SK, Wilk D, Drazba J, Yadav SP, Crabb JW, Ganz T, Bevins CL. Paneth cell trypsin is the processing enzyme for human defensin-5. Nat Immunol. 2002;3:583–590. doi: 10.1038/ni797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.