

Abstract
Insulin-like growth factor-1 (IGF-1) plays a central role in cellular growth, differentiation, survival, and cell cycle progression. It is expressed early during development and its effects are mediated through binding to a tyrosine kinase receptor, the insulin-like growth factor-1 receptor (IGF-1R). In the circulation, the IGFs bind to IGF-binding proteins (IGFBPs), which determine their bioavailability and regulate the interaction between the IGFs and IGF-1R. Studies in animal models and in humans have established critical roles for IGFs in skeletal growth and development. In this review we present new and old findings from mouse models of the IGF system and discuss their clinical relevance to normal and pathological skeletal physiology. © 2010 American Society for Bone and Mineral Research.
Keywords: INSULIN-LIKE GROWTH FACTOR; GROWTH HORMONE, BONE ACCRUAL; SKELETAL GROWTH; CORTICAL-BONE
Introduction
From sulfation factor, discovered in the 1950s, to somatomedin in the 1970s, to insulin-like growth factor 1 (IGF-1) in the 1980s, we have learned that IGFs play a central role in growth, development, and metabolism. While originally IGFs were discovered in serum and found to be produced by the liver, numerous studies in the 1980s demonstrated that virtually all tissues express IGF-1. The recognition of IGF-1 as an endocrine (serum) and an autocrine/paracrine (tissue) hormone has helped the scientific community develop an understanding of how these two systems function coordinately. It has become clear that liver IGF-1 is regulated largely by the ambient levels of growth hormone (GH) secreted from the pituitary, but this is not necessarily the case for non–hepatic tissue production of IGF-1. Additionally, the intimate relationship between GH and IGF-1 has made it difficult to attribute discrete actions to each hormone, and in the past two decades, a great deal of effort has been made to unravel these functions. In this article we discuss the effects of GH/IGF-1 on the skeleton as derived from mouse models. The models are presented in the context of what they tell us about IGF-1's role as a global growth factor, an endocrine factor, and a tissue growth factor and are placed, where possible, in the context of human IGF-1 deficiencies.
IGF as a Global Growth Factor
Given that the bulk of circulating IGF-1 is produced through GH action on the liver, it was expected that inhibition of GH signaling in mice would retard growth significantly. Indeed, in Snell dwarf (dw/dw) mice, deletion of the transcription factor gene pit1 resulted in a loss of GH production(1) and reduced bone length owing to a reduction in cartilage hypertrophy and delayed epiphyseal ossification.(2–3) Such alterations are likely the mechanism behind marked growth retardation at 2 weeks of age and complete growth arrest at the onset of puberty (4 weeks of age).(4) Similar phenotypes were found in the Ames dwarf mouse (dt/dt), where df (prop-1, an upstream regulator of pit-1) is absent and GH is again not produced. Further, Ames dwarf male and female mice had decreases in body weight that were accompanied by reductions in lean mass, bone area, and bone mineral content (BMC).(5) It should be noted, however, that deficiencies in prolactin and thyroid-stimulating hormone (TSH) are also present in both Snell(6,7) and Ames(8) dwarf mice. Thus the aforementioned phenotypic changes may not be due entirely to GH effects. When Stat5b, a downstream effector of the GH receptor (GHR), was ablated in mice, body weights and bone lengths were reduced in a manner similar to Ames and Snell dwarf mice, although this effect was mainly apparent in male mice.(9) The defects of Snell and Ames dwarf as well as Stat5b−/− mice resulted in blunted GHR activity and are similar to the clinical characteristics of Laron syndrome in humans, where the GHR is mutated. The clinical details of Laron syndrome are extensive and beyond the scope of this review, but they have been published previously.(10) These patients have low spinal and femoral neck areal bone mineral density (BMD) but volumetric BMD is normal. Bone size is markedly decreased compared with controls.(11) It should be noted that the Stat5b−/− model has clinical significance of its own given that Stat5b mutations have been found in humans, and the phenotype of growth retardation is similar to that of Stat5b−/− mice. However, the Stat5b−/− mouse model has a sexually dimorphic phenotype (growth retardation is largely in males), whereas human cases of Stat5b deletions/mutations have demonstrated significant (and comparable) growth retardation in both females(12–14) and males.(15)
Total inactivation of the Igf1 gene in mice resulted in 80% perinatal lethality. The surviving pups were 50% smaller than controls, highlighting the importance of Igf-1 in early growth.(16) This has been confirmed in studies of embryonic growth showing that fetal mice had short-limb dwarfism delays in mineralization and increased chrondrocyte apoptosis.(17) In Igf1−/+ mice it was reported that body weights, femur lengths, and BMD values were reduced in females and males, but the differences in males appeared only after puberty.(18) In terms of bone morphology, Igf1−/+ mice were found to have significantly reduced cortical area and periosteal circumference by 2 months (after puberty) in both males and females. This finding is supported by another study of Igf1−/+ mice that indicated that, at least in terms of transverse size (periosteal circumference), the differences appear as early as 3 weeks of age.(19) Bikle and colleagues reported that Igf1−/− mice had a 24% reduction in cortical bone size and shortened femoral lengths, but trabecular bone density and connectivity were increased.(20) These investigators also demonstrated a defect in osteoclastogenesis and showed that osteoclasts were smaller with fewer nuclei in these knockouts. Additionally, expressions of RANKL and c-fms [the receptor for monocyte colony-stimulating factor (M-CSF)] were reduced significantly.(21) Interestingly, human patients with IGF1 gene deletions have been identified. Patients homozygous for IGF1 gene deletions exhibit growth retardation (short stature) and normal genitalia and proceed through puberty, but at a much slower rate.(22) Bone size is reduced at birth, and vertebral BMD was reduced, but this was due primarily to a decrease in bone volume.(23) IGF-1 replacement therapy had a significantly greater effect on bone volume than on bone density. Mice with deletion of the Igf1 receptor (Igf1r−/−) resembled Igf1 null mice in that pups were born smaller than controls (organ hypoplasia) and died shortly after birth.(24) In addition, the authors noted that primary ossification centers in the cranial and facial bones appeared later in Igf1r−/− mice compared with controls. Studies of Igf1r+/− mice found significant reductions in body weight by 4 weeks of age, although this was noticed only in males.(25) Although human patients with total IGF1R deletions have not been identified, patients with heterozygous mutations in the IGF1R receptor have been identified(26–29) and show various forms of intrauterine growth retardation and blunted postnatal growth,(30–34) as might be expected from studies of Igf1r−/− mice. When the downstream effector of Igf1r, the insulin-receptor substrate 1 (Irs-1) was mutated in mice, significant reductions in body weight were apparent from birth and through adulthood.(35) Interestingly, these Irs1−/− mice showed no delay in ossification of their long bones. In contrast, analysis of mice that had a spontaneous mutation in Irs1 resulting in failure to translate the protein showed growth retardation, low bone mineral density, reduced cortical and trabecular thickness, and low bone-formation rates.(36) The human phenotype is similar to the latter model in that weight and body length are reduced at birth.(37)
Reduced Igf-1 bioavailability was demonstrated in transgenic mice with ubiquitous expression of Igf-binding proteins (Igfbps) and the acid-labile subunit (ALS). Overexpression of ALS resulted in reduced body weight gains during the first 3 weeks of growth and significantly reduced body weights through puberty.(38) Overexpression of Igfbp-1 resulted in growth retardation and a delay in mineralization of several bones (ie, craniofacial, metacarpal, and vertebral).(39) Igfbp-2 overexpression also resulted in growth retardation and mineralization defects. Specifically, whole-body BMC, femoral BMC, tibial BMC, and femoral volume were reduced in Igfbp2 transgenics.(40) In Igfbp3 transgenics, overexpression demonstrated a similar phenotype with decreases in body weight, cortical bone density, cortical bone volume, and cortical thickness.(41) These deficiencies were likely a result of a surface-specific cellular deficiency because bone-formation rates were significantly reduced on the periosteal surface. Further, cancellous bone density and trabecular thickness were decreased in Igfbp3 transgenics. Similar changes in cortical and cancellous bone were found in Igfbp5 transgenics, although it was reported that the skeletal phenotype was more severe in males than in females.(42) Endosteal bone formation was not inhibited but rather was enhanced as a result of Igfbp-5 overexpression. It should be noted that these Igfbp transgenics often do not alter total serum Igf-1 levels. However, in Igfbp overexpression models, although serum Igf-1 levels may be normal, the amount of extracellular fluid Igf-1 that is available to bind to tissue receptors is reduced, and this likely contributes to the observed bone and growth phenotypes.
In 1988, Mathews and colleagues created a transgenic mouse line expressing human IGF-1 in nearly all tissues.(43) During puberty (4 to 6 weeks of age), both female and male transgenic mice exhibited significant increases in body weight that lasted throughout the study (52 weeks of age). Numerous organ weights were found to be larger in mice overexpressing human IGF-1 (owing to hyperplasia), indicating that IGF-1 action from birth and through puberty leads to proportional size increases in a variety of tissues. However, this global “scaling up” of body size did not hold true for longitudinal growth of the skeleton because the tibias and radii of transgenic mice were identical in length to those of control mice. Currently, there is no known human condition that directly mirrors the tissue IGF-1 overexpression model, although patients with trisomy 15q26, and thus ubiquitous upregulation of the IGF-1R, also exhibit overgrowth and tall stature.(44–45)
The abovementioned studies indicate a common theme of growth inhibition and, in many cases, impaired skeletal development in the presence of a global GH/IGF-1 deficiency (Table 1). However, specific changes in body composition, bone size/shape, and timing of ossification are highly variable and appear to depend on sex, genetic background, age, and where along the GH/IGF-1 signaling pathway the disruption exists. To more specifically control for these variables, mouse models were created to tease apart the endocrine (serum) role of Igf-1 from its autocrine/paracrine (tissue) role.
Table 1.
Mouse Models of the GH/Igf Axis and Their Human Counterparts
| Target | Mutant name | Genetic background | Skeletal phenotype | Ref | Human counterpart (ref) |
|---|---|---|---|---|---|
| Global GH action | Pit1−/− (dw/dw, Snell) | Mixed (from outbred) | GR, abnormal growth plate, decreased linear growth. | (2,72) | Mutation in Pit1 locus affect multiple targets. Patients exhibit short stature (MIM No. 173110). |
| Prop1−/− (dt/dt, Ames) | Mixed (from outbred) | GR, reduced bone area and BMC | (73,5) | Mutation in the Prop1 locus affect multiple targets. Patients exhibit short stature (MIM No. 601538). | |
| Ghrhr−/− (lit/lit) | C57BL6 | GR (60% of adult size), reduced cortical BMD, normal trabecular bone. | (74) | Isolated GHD type 1B (MIM No. 139191) | |
| Ghrbp−/− | C57BL6 | GR (60% of adult size) reduced tibial length and decreased hight of growth plate | (75–76) | Laron syndrome (MIM No. 262500) | |
| Stat5b−/− | 129 x BALB/c outcross | GR, impaired longitudinal growth, impaired endochondral ossification | (9,77) | GH insensitivity with immune deficiency (Stat5b gene deletion) (MIM No. 604260) | |
| GH antagonist triglyceride | C57BL/6J × SJL/J | GR (60% of adult size), fourfold increased body adiposity, reduced BMD | (78) and* | Not applicable | |
| GH triglyceride | C57BL/6J | GH overexpression increased body weight, tibial mass, and tibial density | (79) | Acromegaly (MIM No. 102200)/ gigantism, Sotos syndrome (MIM No. 117550) | |
| Global IGF-1 action | Igf1−/− | CD-1 | GR (30% of adult size), reduced cortical BMD, increased trabecular BMD | (16,80) | Igf1 mutation(26,81) |
| C57BL6 | Igf1 gene deletion(22) | ||||
| 75% NMRI genetic background | (MIM No. 608747) | ||||
| Igf1m/m (MIDI) | Not specified | GR, reduced femoral length and areal BMD | (82) | Not applicable | |
| Igf1+/− (haploinsufficiency) | CD1 | GR (70% of adult size), reduced femoral length and areal BMD | (18,19) | Not described in the literature | |
| MF1/DBA | |||||
| Igf1TG (ubiquitous expression) | Not specified | Increased body weight and organ growth, normal skeletal size and morphology | (43) | Not applicable | |
| Igf1r−/− | 129/Sv | Intrauterine GR, lethal (neonates at 45% of WT) delayed bone ossification | (24) | Not described in the literature | |
| Igf1r + /− (haploinsufficiency) | 129/Sv | GR (90% of adult size) | (25) | IGF-1 resistance (MIM No. 147370) | |
| Inbred mouse lines | C3H | Serum and skeletal Igf-1 levels were greater in C3H mice which have a significantly larger femoral total area and cortical area as compared to C57BL/6 mice | (83) | Not applicable | |
| C57BL/6 | |||||
| Global IGF-2 action | IGF2−/− | MF1 C57BL/6 Chimera | GR (60% of neonate size) | (84) | Not described in the literature |
| Igf2TG | 75% NMRI genetic background | Increased body weight, no effect on femoral architecture or BMD. | (80) | Not applicable | |
| Global IGF axis | Igf1−/− Igf2+/− (haploinsufficiency) | MF1 C57BL/6 Chimera | GR (30% of adult size) | (24) | Not described in the literature |
| Igf1−/− Igf1r−/− | Lethal (neonates at 45% of WT) | (24) | Not described in the literature | ||
| Igf2+/− Igf1r+/− (haploinsufficiency) | GR (30% of adult size) | (4) | Not described in the literature | ||
| Igf1−/− GHR−/− | Mixed Breeding (C57BL/6, 129Sv, MF!/DBA) | GR (17% of adult size) | (85) | Not described in the literature | |
| Global IGF-1R mediators | Irs1−/− | CD-1 C57BL/6 Chimera | Growth retardation (50% of adult size); no delay in long bone ossification | (35) | Not described in the literature. Heterozygous mutation is associated with metabolic disorder.(86) |
| Irs1 sml/sml | C3.SW-H2b/SnJ | Growth retardation (50% of adult size), low bone mineral density, reduced cortical and trabecular thickness, and low bone-formation rates | (36) | ||
| Akt1/2 | MF1 C57BL/6 Chimera | Intrauterine GR, lethal (neonates at 45% of WT), delayed bone ossification | (87) | Not described in the literature | |
| Foxo1/3/4 (conditional global deletion) | FVB/N C57BL6 mixed | Reduced BMD, significant reductions in trabecular bone | (88) | Not described in the literature | |
| Global IGF bioavailability | Igfbp1TG | C57BL/6/CBA | GR, reduced skeletal mineralization | (39) | Not applicable |
| Igfbp2−/− | C57BL6 | Sex-related decrease in BMD (male) | (89) | Not described in the literature | |
| Igfbp2 TG | Not specified | Decreased (10%) carcass weight, reduced bone length, bone cross-sectional area, and BMC | (40,90) | Not applicable | |
| Igfbp3−/− | C57BL6 | No effects on body weight or linear growth were noted | (91) | Not described in the literature | |
| Igfbp3 TG | CD-1 | Reduced volumetric and cortical BMD, increased resorption | (41) | Not applicable | |
| Igfbp4−/− | C57BL/6 | GR (85% of adult size) | (91) | Not described in the literature | |
| Igfbp4 TG | FVB/N | GR | (92) | Not applicable | |
| Igfbp5−/− | C57BL/6 | No effects on body weight or linear growth were noted | (91) | ||
| Igfbp5 TG | C57BL/6JxCBA/CA | Sex-related decrease in BMD, impaired mineralization, decreased BFR | (42) | Not applicable | |
| ALS−/− | CD-1 | GR (80% of adult size), reduced volumetric and cortical BMD, 10% reduction in femoral length | (93) | IGFALS deficiency (ALS gene deletion) | |
| C57BL/6 | (MIM No. 601489) | ||||
| ALS TG | Balb C | Modest GR | |||
| CD-1 | (38) | Not applicable | |||
| PappA−/− (IGFBP-4 protease) | C57BL/6 129Sv | GR (60% of adult size) | (94) | Not described in the literature | |
| PappATG | C57BL/6J XCBA/CA | Increased body weight | (95) | Not applicable | |
| Igfbp3−/– -4−/− -5−/− | C57BL/6 | GR (80% of adult size) | (91) | ||
| Igfbp4−/−; PappA−/− | 129/C57BL/6 | GR (90% of adult size) | (94) | ||
| Endocrine IGF-1 | Liver-specific Igf-1 TG (Hit) | C57BL/6 | Liver-specific Igf-1 TG | (52–53,96–97) | |
| FVB/N | |||||
| Liver-specific IGF1−/− (LID) | FVB/N | Normal growth, reduced volumetric and cortical BMD, 5% reduction in femoral length | (93) | ||
| C57BL/6 | |||||
| LID ALS−/− | FVB/N C57BL6 mixed | GR (70% of adult size), reduced cortical and trabecular BMD | |||
| Autocrine/paracrine IGF-1 activity or bioavailbility | Osteoblast-specific Igf1 TG | FVB/N | Increased volumetric and cortical BMD | (64) | |
| Osteoblast-specific Igfbp4 TG | FVB/N | Decreased in bone volume and cortical BMD | (61) | ||
| Osteoblast-specific IGF1R−/− | C57BL/6 x FVB/N | Normal growth, impaired mineralization | (58) | ||
| Osteoblast-specific PappA TG | C57BL/6J XCBA/CA | Increased calvarial BMD and tibial/femoral bone area and periosteal circumference | (98) | ||
| Chondrocyte-specific IGF1−/− | C57BL/6 X SJL | Body length, areal BMD, and BMC were reduced between 4 and 12 weeks | (56) | ||
| Skeletal muscle/bone-specific IGF1−/− (collagen 1 and 2 expressing cells) | C567BL/6 X FVB/N | Reduced body weight, femoral BMD, femoral bone size, mineral apposition rate, and bone-formation rate. | (57) | ||
| IGF1−/− liver-specific Igf2 TG | 75% NMRI genetic background | GR, adults show similar body weight and length to the Igf1−/− mice. | (80) | ||
| Endocrine and autocrine/paracrine IGF-1 interplay | Igf1−/− liver-specific Igf1 TG (KO-HIT) | FVB/N | Normalized skeletal growth and development due to threefold increase in endocrine (serum) Igf-1 levels. | (52–53) | |
| Igf1−/− liver-specific Igf1 KI | C57BL/6 CBA | Physiologic levels of liver-derived IGF-1 restored body size of the Igf1 null mice to ∼70% of WT size | (65) |
S Yakar personal note.
Mendelian Inheritance in Man = MIM; http://www.ncbi.nlm.nih.gov/omim/).
IGF-1 as an Endocrine Factor
Studies from inbred mouse strains with distant genetic backgrounds that have markedly different serum Igf-1 concentrations have reinforced the importance of serum Igf-1 for bone development.(46) The mouse strains with low Igf-1 (C57BL/6J) have reduced total BMD and cortical thickness, whereas mice with higher serum Igf-1 levels (C3H/HeJ) show increased total BMD and femoral cortical thickness. In addition, congenic mice (B6.C3H.6T) with a 40% reduction in serum Igf-1 also had reduced BMD and delayed development.(47) Given that the liver is responsible for nearly 75% of serum Igf-1 levels, ablation of Igf1 gene expression in the liver was a promising approach to quantify the effects of serum Igf-1 on skeletal growth and development. Liver-specific Igf-1-deficient (LID) mice were created using the Cre-LoxP system. Preliminary data indicated an 80% decrease in serum Igf-1 levels, no change in body weight, and a small but significant decrease in body length from 3 to 8 weeks of age.(48) A subsequent study examined skeletal growth of LID mice in detail from 4 to 52 weeks of age and confirmed these initial findings while presenting new data on the role of Igf-1 in skeletal development after puberty.(49) Results of this study indicated that a constitutive loss (from birth) of serum Igf-1 resulted in significant reductions in body weight after puberty. Although no alterations in trabecular bone were found, significant decreases in femoral total area (Tt.Ar), cortical area (Ct.Ar), and polar moment of inertia (Jo) were found beginning at 8 weeks of age. As a result, femurs were more slender (less robust) with reduced stiffness and reduced strength in bending. Thus reductions in serum Igf-1 tended to target cortical bone by preventing periosteal apposition during growth. Interestingly, marrow area (Ma.Ar) was not altered during early growth and actually decreased relative to controls from 16 to 32 weeks of age (endosteal infilling), suggesting that when serum Igf-1 levels are lowered early during growth, bones become more slender, but skeletal elements are able to activate a compensatory adaptive response by adding more bone endosteally to support increases in body weight. This was evident by an increase in relative cortical area (RCA) during growth (Fig. 1) such that the total amount of bone tissue present per total area of bone increased owing to endosteal infilling.
Fig. 1.
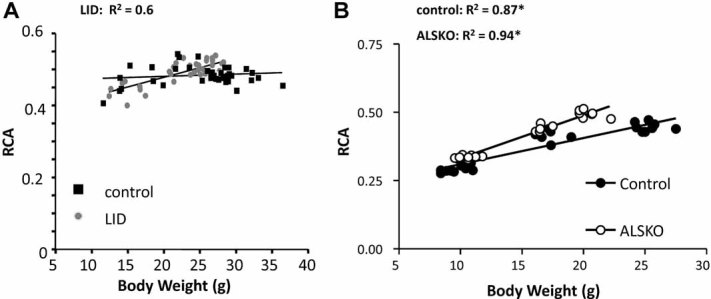
(A) Relative cortical area (RCA = Ct.Ar./Tt.Ar.) versus body weight for male LID and control mice during growth, as published previously.(49) (B) RCA versus body weight for male ALS KO and control mice during growth, as published previously.(50) For both LID and ALS KO mice, RCA is increased as body weight increases during growth compared with control mice, illustrating a small compensatory response of bone to decreased periosteal apposition.
Serum Igf-1 levels also have been reduced in mouse models by altering the expression of one or more Igf-1-binding proteins. In Igfbp3 knockout mice, serum Igf-1 levels were reduced by 40%, but body weight and length were increased by 8 weeks compared with controls.(48) Interestingly, these mice had decreased femoral trabecular bone volume fraction (BV/TV) and trabecular number (Tb.N) with no apparent changes in cortical bone size or tissue amount, suggesting that Igfbp3 may have an Igf-1-dependent or -independent effects on the skeleton. The ALS is an important binding protein of Igf-1 and Igfbp-3 in serum, and when it is ablated in mice (ALS KO), it results in 65% reductions in serum Igf-1 levels, similar to the LID model. A detailed skeletal analysis of ALS KO mice from 4 to 16 weeks indicated reductions in body weight and body length throughout growth in both female and male mice.(50) In addition, by adulthood (16 weeks), ALS KO mice, both females and males, had reduced Tt.Ar., Ct.Ar., Ma.Ar, Jo, and robustness (more slender). Similar to LID mice, ALS KO mice are also able to compensate for smaller, more slender bones through marrow infilling [reducing Ma.Ar and increasing their relative cortical area (RCA)]. Interestingly, the increased slenderness and compensatory increased marrow infilling (increased RCA) were more prominent in female than male ALS KO mice (Fig. 1). The data from male LID and ALS KO mice indicate a common role for serum Igf-1 in maintaining periosteal apposition during growth. The ALS KO mouse model is now of particular interest because human patients with ALS deficiency have been and continue to be reported in the literature. Although detailed quantification of skeletal structures in ALS-deficient patients has not been performed, a review of case reports has been published that indicates short stature and reduced BMD in a number of patients.(51)
Increased Igf-1 levels in serum were demonstrated in mice expressing hepatocyte-specific rat Igf1 transgene (HIT). HIT mice exhibit two- to threefold increases in serum Igf-1 levels, which were accompanied by increases in body weight, body length, femoral length, and femoral Tt.Ar, Ct.Ar, Ct.Th, Jo, and robustness.(52–53) Trabecular architecture also was examined in the HIT model, but only a few changes were observed at 16 weeks of age (Tb.Th increased in HIT mice). Thus the HIT phenotype is a scaling up of body size and cortical skeletal size with proportional changes in lean and fat mass. It should be noted that the HIT model is one of the few examples where Igf-1 alteration changed the composition of bone; HIT mice at 16 weeks were found to have a higher tissue mineral density (TMD) in both cortical and cancellous bone, indicating that more mineral was present in a given volume of bone than in control mice.
Together, studies of mice with reductions in serum Igf-1 revealed minor changes in body weight and length but significant decreases in transverse bone growth (Table 1). Reductions in serum Igf-1 during postnatal growth are extremely important in establishing bone robustness and suggest a possible role in determining increased fracture risk during adulthood and aging. On the other hand, increases in serum Igf-1 levels during growth lead to enhancement of all bone traits and may play a protective role later on during aging. They also emphasize the importance of serum Igf-1 in contributing to cortical size and bone density. Indeed, several papers have reported skeletal abnormalities in humans with low serum IGF-1.(54,55) In general, these have been patients with severe GH deficiency. These patients usually have normal volumetric BMD values and smaller bones. Increased fractures have been reported in some studies.(55) These studies are difficult to relate to the mouse models because, except for the case of Igf1 gene deletion,(23) GH deficiency also has been present.
IGF-1 as a Tissue Growth Factor
Tissue-specific regulation of IGF-1 is an important feature of many developmental processes. In the past 10 years, we have advanced our understanding of IGF-1 action on bone using cell-type specific IGF-1/IGF-1R inactivation. In a model where chondrocyte Igf-1 synthesis was disrupted, significant reductions in body weight, body length, total-body BMD, and femoral length were observed beginning at 4 weeks of age in both female and male mice.(56) Further, femoral width, as measured by periosteal circumference, was reduced in both sexes, although the differences were greater for males than for females. Conditional deletion of Igf-1 in skeletal muscle and bone resulted in decreased femoral size, increased apoptosis, and decreased bone-formation rate.(57)
When the Igf-1R was disrupted in osteoblasts (ΔIgfr mice), no alterations in body size, weight, or femoral length by 6 weeks of age were found. However, ΔIgfr mice showed significant reductions in distal metaphyseal trabecular BV/TV, Tb.Th, TB.N, and MAR as well as increased osteoid volume and osteoid surface.(58) This study was crucial to establishing the role of Igf-1 in bone mineralization. Mice bearing Igf1i or Igf1r gene deletions also have been useful for determining if the known anabolic factors for bone require expression of Igf-1. Mice with an Igf1r depletion in osteoblasts showed decreased endosteal bone formation in response to PTH compared with controls. This defect was demonstrated in bone marrow stromal cells derived from these animals through a decreased number of alkaline phosphatase colonies and decreased mineralization in response to PTH.(59) These findings also have been replicated in Igf1 knockout mice.(60) It should be noted that tissue-specific effects of Igf-1 have been hard to quantify because they are not easily separable from the endocrine reservoir of Igf-1. Thus, in the previous example of ΔIgfr mice, disruption of the Igf-1r will impair both autocrine/paracrine and endocrine Igf-1 effects on osteoblasts.
Tissue-specific expression of Igfbps leads to decreased Igf-1 bioavailability and generally shows phenotypes similar to the global Igfbp transgenics, namely, reduced body weight and length. Igfbp-4 overexpression in osteoblasts resulted in decreased femoral cortical density, cortical thickess, and periosteal circumference in both males and females by 6 weeks of age.(61) Mice expressing Igfbp-5 under the osteocalcin promoter demonstrated decreased BMD, trabecular bone volume, and bone formation,(62) as well as mineralization defects indicated by reduced mineral/matrix ratio in cortical bone and reduced collagen maturity in secondary ossification centers.(63) As was stated previously, local expression of Igfbps does not distinguish between autocrine/paracrine and endocrine Igf-1 effects but rather blocks the Igf-1 axis in a cell-specific manner. In contrast, Igf-1 overexpression in osteoblasts under the osteocalcin promoter (OC-Igf1 transgenic mice) resulted in no change in serum Igf-1 levels or body weights up to 16 weeks but significant increases in cortical and trabecular BMD as well as trabecular bone volume and trabecular thickness.(64) In these OC-Igf1 mice, histomorphometric parameters were no different from those of control mice at the later age (24 weeks), but increases in bone-formation rate were apparent at 6 weeks of age, indicating that increased bone accrual is accomplished largely during puberty.
As a point of comparison with the previously discussed HIT mouse mode, the KO-HIT mouse model lacks tissue Igf-1 in all tissues, and the sole source of Igf-1 production is through a transgene expressed in the liver. Thus serum Igf-1 levels are identical to HIT mice and threefold higher than control mice. Differences between HIT and KO-HIT mice therefore represent the consequences of tissue Igf1 gene ablation from birth. Interestingly, body weight and length were not increased in KO-HIT mice, as they were in HIT mice, and morphologic analyses revealed that KO-HIT mice had skeletal properties similar to controls starting at 8 weeks of age.(52) However, early in development (before 8 weeks), significant reductions in femoral length, Tt.Ar, Ct.Ar, and robustness were evident. By 16 weeks, KO-HIT traits were normalized to control levels (eg, femoral length, Tt.Ar., and robustness) or exceeded control levels in a manner similar to HIT mice (ie, Ct.Ar). These findings are in agreement with another study in which liver Igf-1 was reexpressed in Igf1 null mice resulting in a partial postnatal restoration of serum Igf-1 levels (∼50% of normal) and body weight (∼70% of normal).(65) Thus tissue Igf-1 appears crucial for early postnatal and pubertal development of cortical bone, but serum Igf-1 can permit “catchup” growth postpubertally (Table 1). Whether reexpression of normal levels of serum Igf-1 sufficient for normal growth in the absence of tissue Igf-1 remains unknown.
Final Considerations
Throughout this article, the skeletal phenotypes of Igf-1 mouse models have been presented without context of their genetic backgrounds. This is an important consideration, and for each model discussed in this article, genetic backgrounds are given (where available) in Table 1. In addition, Table 1 offers a more extensive list of mouse models relating to the GH/Igf-1 axis as well as summaries of their skeletal phenotypes. Numerous studies indicate that differences in genetic background, whether complete(66,67) or even partial,(68–71) can result in significant alterations in skeletal properties and mechanical function. Therefore, any assessment of genetically engineered mice must consider the possibility that gene interactions from genetic background effects may determine at least partially the phenotype. As indicated in Table 1, the majority of Igf-1 mouse models have been created on a few select backgrounds (ie, CD-1, C57BL/6, and FVB/N) or on mixed backgrounds. Nevertheless, a review of the existing data from the different genetic backgrounds used indicates that the major roles of Igf-1 (eg, growth retardation from global deficiency, periosteal inhibition from reduced serum Igf-1 levels, etc.) exist despite differences in genetic background.
Concluding Remarks
After 60 years of investigation, it is apparent that the GH/IGF axis plays a prominent regulatory role in skeletal development and mineral acquisition. Clinical studies as well as animal models have taught us that global loss of IGF-1 affects growth and skeletal gains at all ages, resulting in short stature and slender and weaker bones. With the development of a tissue-specific gene approach, we have advanced our knowledge regarding IGF-1's mode of action. We now know that loss of serum IGF-1 affects mainly postpubertal bone accrual. Longitudinal studies have demonstrated the importance of serum IGF-1 in transverse bone growth and periosteal bone apposition, as well as in bone adaptation to increases in body weight. We have learned that loss of tissue IGF-1 affects early postnatal and prepubertal growth, but there is some compensation when serum IGF-1 levels increase postpubertally. These studies also clearly demonstrate that the IGF-1 axis in osteoblasts is a strong determinant of bone mineralization.
Future Directions
Notwithstanding our extensive knowledge of IGF-I action on the growing skeleton, its role in skeletal homeostasis during aging is still unclear. Moreover, our understanding of IGF-1 interactions with steroid hormones, insulin, PTH, sclerostin, and the Wnt pathway and their effects on the skeleton during growth and aging is incomplete. Other open questions regarding IGF-1 action include its role in osteocyte function and in osteoclastogenesis. Lastly, perhaps the most important question is: How and when should we intervene through manipulation of the GH/IGF axis to obtain a more robust, mechanically fit skeleton.
Acknowledgments
Financial support was received from funding agencies in the United States (NIH Grants AR054919 and AR055141 to SY and AG-02331 to DC).
Disclosures
All the authors state that they have no conflicts of interest.
References
- 1.Li S, Crenshaw EB, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit1. Nature. 1990;347:528–533. doi: 10.1038/347528a0. [DOI] [PubMed] [Google Scholar]
- 2.van Buul-Offers S, Smeets T, Van den Brande JL. Effects of growth hormone and thyroxine on the relation between tibial length and the histological appearance of the proximal tibial epiphysis in snell dwarf mice. Growth. 1984;48:166–175. [PubMed] [Google Scholar]
- 3.Smeets T, van Buul-Offers S. A Morphological study of the development of the tibial proximal Epiphysis and growth plate of normal and dwarfed snell mice. Growth. 1983;47:145–159. [PubMed] [Google Scholar]
- 4.van Buul S, Van den Brande J. The snell-dwarf mouse: I. general growth pattern, before and during growth hormone and thyroxine therapy. Acta Endocrinol (Copenh). 1978;89:632–645. [PubMed] [Google Scholar]
- 5.Heiman ML, Tinsley FC, Mattison JA, Hauck S, Bartke A. Body composition of prolactin-, growth hormone–, and thyrotropin-deficient ames dwarf mice. Endocrinology. 2003;20:149–154. doi: 10.1385/ENDO:20:1-2:149. [DOI] [PubMed] [Google Scholar]
- 6.Snell GD. Dwarf, a new mendelian recessive character of the house mouse. Proc Natl Acad Sci USA. 1929;15:733–734. doi: 10.1073/pnas.15.9.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha YN, Salocks CB, Vanderlaan WP. Pituitary and serum concentrations of prolactin and GH in snell dwarf mice. Proc Soc Exp Biol Med. 1975;150:207–210. doi: 10.3181/00379727-150-39003. [DOI] [PubMed] [Google Scholar]
- 8.Sornson MW, Wu W, Dasen JS, et al. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature. 1996;384:327–333. doi: 10.1038/384327a0. [DOI] [PubMed] [Google Scholar]
- 9.Udy GB, Towers RP, Snell RG, et al. Requirement of STAT5b for sexual dimorphism of body growth Rates and liver gene expression. Proc Natl Acad Sci USA. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J Clin Endocrinol Metab. 2004;89:1031–1044. doi: 10.1210/jc.2003-031033. [DOI] [PubMed] [Google Scholar]
- 11.Bachrach LK, Marcus R, Ott SM, et al. Bone mineral, histomorphometry, and body composition in adults with growth hormone receptor deficiency. J Bone Miner Res. 1998;13:415–421. doi: 10.1359/jbmr.1998.13.3.415. [DOI] [PubMed] [Google Scholar]
- 12.Kofoed EM, Hwa V, Little B, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–1147. doi: 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- 13.Hwa V, Haeusler G, Pratt KL, et al. Total absence of functional acid labile subunit, resulting in severe insulin-like growth factor deficiency and moderate growth failure. J Clin Endocrinol Metab. 2006;91:1826–1831. doi: 10.1210/jc.2005-2842. [DOI] [PubMed] [Google Scholar]
- 14.Hwa V, Camacho-Hubner C, Little BM, et al. Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13-intron 13 junction of the STAT5b gene. Horm Res. 2007;68:218–224. doi: 10.1159/000101334. [DOI] [PubMed] [Google Scholar]
- 15.Vidarsdottir S, Walenkamp MJ, Pereira AM, et al. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J Clin Endocrinol Metab. 2006;91:3482–3485. doi: 10.1210/jc.2006-0368. [DOI] [PubMed] [Google Scholar]
- 16.Powell-Braxton L, Hollingshead P, Giltinan D, Pitts-Meek S, Stewart T. Inactivation of the IGF-I gene in mice results in perinatal lethality. Ann N Y Acad Sci. 1993;692:300–301. doi: 10.1111/j.1749-6632.1993.tb26240.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang YM, Nishida S, Sakata T, et al. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology. 2006;147:4753–4761. doi: 10.1210/en.2006-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He J, Rosen CJ, Adams DJ, Kream BE. Postnatal growth and bone mass in mice with IGF-I haploinsufficiency. Bone. 2006;38:826–835. doi: 10.1016/j.bone.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 19.Mohan S, Baylink DJ. Impaired skeletal growth in mice with haploinsufficiency of IGF-I: genetic evidence that differences in IGF-I expression could contribute to peak bone mineral density differences. J Endocrinol. 2005;185:415–420. doi: 10.1677/joe.1.06141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bikle D, Majumdar S, Laib A, et al. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res. 2001;16:2320–2329. doi: 10.1359/jbmr.2001.16.12.2320. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD. Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res. 2006;21:1350–1358. doi: 10.1359/jbmr.060610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363–1367. doi: 10.1056/NEJM199610313351805. [DOI] [PubMed] [Google Scholar]
- 23.Woods KA, Camacho-Hubner C, Bergman RN, Barter D, Clark AJ, Savage MO. Effects of insulin-like growth factor I (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J Clin Endocrinol Metab. 2000;85:1407–1411. doi: 10.1210/jcem.85.4.6495. [DOI] [PubMed] [Google Scholar]
- 24.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 25.Holzenberger M, Dupont J, Ducos B, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 26.Walenkamp MJ, Karperien M, Pereira AM, et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–2864. doi: 10.1210/jc.2004-1254. [DOI] [PubMed] [Google Scholar]
- 27.Walenkamp MJ, de Muinck Keizer-Schrama SM, de Mos M, et al. Successful long-term growth hormone therapy in a girl with haploinsufficiency of the insulin-like growth factor-I receptor due to a terminal 15q26.2->qter deletion detected by multiplex ligation probe amplification. J Clin Endocrinol Metab. 2008;93:2421–2425. doi: 10.1210/jc.2007-1789. [DOI] [PubMed] [Google Scholar]
- 28.Fang P, Schwartz ID, Johnson BD, et al. Familial short stature caused by haploinsufficiency of the insulin-like growth factor I receptor due to nonsense-mediated messenger ribonucleic acid decay. J Clin Endocrinol Metab. 2009;94:1740–1747. doi: 10.1210/jc.2008-1903. [DOI] [PubMed] [Google Scholar]
- 29.Ester WA, van Duyvenvoorde HA, de Wit CC, et al. Two short children born small for gestational age with insulin-like growth factor 1 receptor haploinsufficiency illustrate the heterogeneity of its phenotype. J Clin Endocrinol Metab. 2009;94:4717–4727. doi: 10.1210/jc.2008-1502. [DOI] [PubMed] [Google Scholar]
- 30.Abuzzahab MJ, Schneider A, Goddard A, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003;349:2211–2222. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- 31.Kruis T, Klammt J, Galli-Tsinopoulou A, et al. Heterozygous mutation within a kinase-conserved motif of the insulin-like growth factor I receptor causes intrauterine and postnatal growth retardation. J Clin Endocrinol Metab. 2010;95:1137–1142. doi: 10.1210/jc.2009-1433. [DOI] [PubMed] [Google Scholar]
- 32.Suh Y, Atzmon G, Cho MO, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inagaki K, Tiulpakov A, Rubtsov P, et al. A Familial insulin-like growth factor-I receptor mutant leads to short stature: clinical and biochemical characterization. J Clin Endocrinol Metab. 2007;92:1542–1548. doi: 10.1210/jc.2006-2354. [DOI] [PubMed] [Google Scholar]
- 34.Walenkamp MJ, van der Kamp HJ, Pereira AM, et al. A Variable degree of intrauterine and postnatal growth retardation in a family with a missense Mutation in the insulin-like growth factor I receptor. J Clin Endocrinol Metab. 2006;91:3062–3070. doi: 10.1210/jc.2005-1597. [DOI] [PubMed] [Google Scholar]
- 35.Araki E, Lipes MA, Patti ME, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 36.DeMambro VE, Kawai M, Clemens TL, et al. A novel spontaneous mutation of Irs1 in mice results in hyperinsulinemia, reduced growth, low bone mass and impaired adipogenesis. J Endocrinol. 2010;204:241–253. doi: 10.1677/JOE-09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simonska-Cichocka E, Gumprecht J, Zychma M, et al. The polymorphism in insulin receptor substrate-1 gene and birth weight in neonates at term. Endokrynol Pol. 2008;59:212–216. [PubMed] [Google Scholar]
- 38.Silha JV, Gui Y, Modric T, et al. Overexpression of the acid-labile subunit of the IGF ternary complex in transgenic mice. Endocrinology. 2001;142:4305–4313. doi: 10.1210/endo.142.10.8427. [DOI] [PubMed] [Google Scholar]
- 39.Ben Lagha N, Seurin D, Le Bouc Y, et al. Insulin-like growth factor binding protein (IGFBP-1) involvement in intrauterine growth retardation: study on IGFBP-1 overexpressing transgenic mice. Endocrinology. 2006;147:4730–4737. doi: 10.1210/en.2006-0171. [DOI] [PubMed] [Google Scholar]
- 40.Eckstein F, Pavicic T, Nedbal S, et al. Insulin-like growth factor-binding protein-2 (IGFBP-2) overexpression negatively regulates bone size and mass, but not density, in the absence and presence of growth hormone/IGF-I excess in transgenic mice. Anat Embryol (Berl). 2002;206:139–148. doi: 10.1007/s00429-002-0282-5. [DOI] [PubMed] [Google Scholar]
- 41.Silha JV, Mishra S, Rosen CJ, et al. Perturbations in bone formation and resorption in insulin-like growth factor binding protein-3 transgenic mice. J Bone Miner Res. 2003;18:1834–1841. doi: 10.1359/jbmr.2003.18.10.1834. [DOI] [PubMed] [Google Scholar]
- 42.Salih DA, Mohan S, Kasukawa Y, et al. Insulin-like growth factor-binding protein-5 induces a gender-related decrease in bone mineral density in transgenic mice. Endocrinology. 2005;146:931–940. doi: 10.1210/en.2004-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathews LS, Hammer RE, Behringer RR, et al. Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology. 1988;123:2827–2833. doi: 10.1210/endo-123-6-2827. [DOI] [PubMed] [Google Scholar]
- 44.Faivre L, Gosset P, Cormier-Daire V, et al. Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: report of two families and review of the literature. Eur J Hum Genet. 2002;10:699–706. doi: 10.1038/sj.ejhg.5200879. [DOI] [PubMed] [Google Scholar]
- 45.Kant SG, Kriek M, Walenkamp MJ, et al. Tall stature and duplication of the insulin-like growth factor I receptor gene. Eur J Med Genet. 2007;50:1–10. doi: 10.1016/j.ejmg.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 46.Bouxsein ML, Rosen CJ, Turner CH, et al. Generation of a new congenic mouse strain to test the relationships among serum insulin-like growth factor I, bone mineral density, and skeletal morphology in vivo. J Bone Miner Res. 2002;17:570–579. doi: 10.1359/jbmr.2002.17.4.570. [DOI] [PubMed] [Google Scholar]
- 47.Rosen CJ, Ackert-Bicknell CL, Adamo ML, et al. Congenic mice with low serum IGF-I have increased body fat, reduced bone mineral density, and an altered osteoblast differentiation program. Bone. 2004;35:1046–1058. doi: 10.1016/j.bone.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 48.Yakar S, Rosen CJ, Bouxsein ML, et al. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. Faseb J. 2009;23:709–719. doi: 10.1096/fj.08-118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yakar S, Canalis E, Sun H, et al. Serum IGF-1 determines skeletal strength by regulating subperiosteal expansion and trait interactions. J Bone Miner Res. 2009;24:1481–1492. doi: 10.1359/JBMR.090226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Courtland HW, Demambro V, Maynard J, et al. Sex-specific regulation of body size and bone slenderness by the acid labile subunit. J Bone Miner Res. 2010;25:2059–2068. doi: 10.1002/jbmr.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Domene HM, Hwa V, Argente J, et al. Human acid-labile subunit deficiency: clinical, endocrine and metabolic consequences. Horm Res. 2009;72:129–141. doi: 10.1159/000232486. [DOI] [PubMed] [Google Scholar]
- 52.Elis S, Courtland HW, Wu Y, et al. Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. J Bone Miner Res. 2010;25:1257–1266. doi: 10.1002/jbmr.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elis S, Courtland HW, Wu Y, Sun H, Rosen CJ, Yakar S. Elevated serum IGF-1 levels synergize PTH action on the skeleton only when the tissue IGF-1 axis is intact. J Bone Miner Res. 2010;25:2051–2058. doi: 10.1002/jbmr.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maheshwari HG, Bouillon R, Nijs J, Oganov VS, Bakulin AV, Baumann G. The impact of congenital, severe, untreated growth hormone (GH) deficiency on bone size and density in young adults: insights from genetic GH-releasing hormone receptor deficiency. J Clin Endocrinol Metab. 2003;88:2614–2618. doi: 10.1210/jc.2002-021120. [DOI] [PubMed] [Google Scholar]
- 55.Bouillon R, Koledova E, Bezlepkina O, et al. Bone status and fracture prevalence in russian adults with childhood-onset growth hormone deficiency. J Clin Endocrinol Metab. 2004;89:4993–4998. doi: 10.1210/jc.2004-0054. [DOI] [PubMed] [Google Scholar]
- 56.Govoni KE, Lee SK, Chung YS, et al. Disruption of insulin-like growth factor-I expression in type IIalphaI collagen-expressing cells reduces bone length and width in mice. Physiol Genomics. 2007;30:354–362. doi: 10.1152/physiolgenomics.00022.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Govoni KE, Wergedal JE, Florin L, Angel P, Baylink DJ, Mohan S. Conditional deletion of insulin-like growth factor-I in collagen type 1alpha2-expressing cells results in postnatal lethality and a dramatic reduction in bone accretion. Endocrinology. 2007;148:5706–5715. doi: 10.1210/en.2007-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang M, Xuan S, Bouxsein ML, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 59.Wang YM, Nishida S, Boudignon BM, et al. IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res. 2007;22:1329–1337. doi: 10.1359/jbmr.070517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bikle DD, Sakata T, Leary C, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17:1570–1578. doi: 10.1359/jbmr.2002.17.9.1570. [DOI] [PubMed] [Google Scholar]
- 61.Zhang M, Faugere MC, Malluche H, Rosen CJ, Chernausek SD, Clemens TL. Paracrine overexpression of IGFBP-4 in osteoblasts of transgenic mice decreases bone turnover and causes global growth retardation. J Bone Miner Res. 2003;18:836–843. doi: 10.1359/jbmr.2003.18.5.836. [DOI] [PubMed] [Google Scholar]
- 62.Devlin RD, Du Z, Buccilli V, Jorgetti V, Canalis E. Transgenic mice overexpressing insulin-like growth factor binding Protein-5 display transiently decreased osteoblastic function and osteopenia. Endocrinology. 2002;143:3955–3962. doi: 10.1210/en.2002-220129. [DOI] [PubMed] [Google Scholar]
- 63.Atti E, Boskey AL, Canalis E. Overexpression of IGF-binding protein 5 alters mineral and matrix properties in mouse femora: an infrared imaging study. Calcif Tissue Int. 2005;76:187–193. doi: 10.1007/s00223-004-0076-2. [DOI] [PubMed] [Google Scholar]
- 64.Zhao G, Monier-Faugere MC, Langub MC, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141:2674–2682. doi: 10.1210/endo.141.7.7585. [DOI] [PubMed] [Google Scholar]
- 65.Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci USA. 2008;105:19378–19383. doi: 10.1073/pnas.0809223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jepsen KJ, Akkus OJ, Majeska RJ, Nadeau JH. Hierarchical relationship between bone traits and mechanical properties in inbred mice. Mamm Genome. 2003;14:97–104. doi: 10.1007/s00335-002-3045-y. [DOI] [PubMed] [Google Scholar]
- 67.Courtland HW, Nasser P, Goldstone AB, Spevak L, Boskey AL, Jepsen KJ. Fourier transform infrared imaging microspectroscopy and tissue-level mechanical testing reveal intraspecies variation in mouse bone mineral and matrix composition. Calcif Tissue Int. 2008;83:342–353. doi: 10.1007/s00223-008-9176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tommasini SM, Hu B, Nadeau JH, Jepsen KJ. Phenotypic integration among trabecular and cortical bone traits establishes mechanical functionality of inbred mouse vertebrae. J Bone Miner Res. 2009;24:606–620. doi: 10.1359/JBMR.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jepsen KJ, Hu B, Tommasini SM, et al. Genetic randomization reveals functional relationships among morphologic and tissue-quality traits that contribute to bone strength and fragility. Mamm Genome. 2007;18:492–507. doi: 10.1007/s00335-007-9017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shao H, Burrage LC, Sinasac DS, et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc Natl Acad Sci USA. 2008;105:19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turner CH, Sun Q, Schriefer J, et al. Congenic mice reveal sex-specific genetic regulation of femoral structure and strength. Calcif Tissue Int. 2003;73:297–303. doi: 10.1007/s00223-002-1062-1. [DOI] [PubMed] [Google Scholar]
- 72.Li S, Crenshaw EB, 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene Pit-1. Nature. 1990;347:528–533. doi: 10.1038/347528a0. [DOI] [PubMed] [Google Scholar]
- 73.Andersen B, Pearse RV, 2nd, Jenne K, et al. The Ames dwarf gene is required for Pit-1 gene activation. Dev Biol. 1995;172:495–503. doi: 10.1006/dbio.1995.8040. [DOI] [PubMed] [Google Scholar]
- 74.Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE. GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet. 1993;4:227–232. doi: 10.1038/ng0793-227. [DOI] [PubMed] [Google Scholar]
- 75.Zhou Y, Xu BC, Maheshwari HG, et al. A Mammalian model for laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc Natl Acad Sci U S A. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180:247–255. doi: 10.1677/joe.0.1800247. [DOI] [PubMed] [Google Scholar]
- 77.Sims NA, Clement-Lacroix P, Da Ponte F, et al. Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest. 2000;106:1095–1103. doi: 10.1172/JCI10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McIlwain DL, Hoke VB, Kopchick JJ, Fuller CR, Lund PK. Differential inhibition of postnatal brain, spinal cord and body growth by a growth hormone antagonist. BMC Neurosci. 2004;5:6. doi: 10.1186/1471-2202-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turner ND, Knapp JR, Byers FM, Kopchick JJ. Physical and mechanical characteristics of tibias from transgenic mice expressing mutant bovine growth hormone genes. Exp Biol Med (Maywood). 2001;226:133–9. doi: 10.1177/153537020122600211. [DOI] [PubMed] [Google Scholar]
- 80.Moerth C, Schneider MR, Renner-Mueller I, et al. Postnatally elevated levels of insulin-like growth factor (IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except kidney weight. Endocrinology. 2007;148:441–451. doi: 10.1210/en.2006-0385. [DOI] [PubMed] [Google Scholar]
- 81.Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet. 2003;40:913–917. doi: 10.1136/jmg.40.12.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao WQ, Shinsky N, Ingle G, Beck K, Elias KA, Powell-Braxton L. IGF-I deficient mice show reduced peripheral nerve conduction velocities and decreased axonal diameters and respond to exogenous IGF-I treatment. J Neurobiol. 1999;39:142–152. doi: 10.1002/(sici)1097-4695(199904)39:1<142::aid-neu11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 83.Rosen CJ, Dimai HP, Vereault D, et al. Circulating and skeletal insulin-like growth factor-I (IGF-I) concentrations in two inbred strains of mice with different bone mineral densities. Bone. 1997;21:217–223. doi: 10.1016/s8756-3282(97)00143-9. [DOI] [PubMed] [Google Scholar]
- 84.DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78–80. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- 85.Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001;229:141–162. doi: 10.1006/dbio.2000.9975. [DOI] [PubMed] [Google Scholar]
- 86.Esposito DL, Li Y, Vanni C, et al. A novel T608R missense mutation in insulin receptor substrate-1 identified in a subject with type 2 diabetes impairs metabolic insulin signaling. J Clin Endocrinol Metab. 2003;88:1468–1475. doi: 10.1210/jc.2002-020933. [DOI] [PubMed] [Google Scholar]
- 87.Peng XD, Xu PZ, Chen ML, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ambrogini E, Almeida M, Martin-Millan M, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010;11:136–146. doi: 10.1016/j.cmet.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.DeMambro VE, Clemmons DR, Horton LG, et al. Gender-specific changes in bone turnover and skeletal architecture in igfbp-2-null mice. Endocrinology. 2008;149:2051–2061. doi: 10.1210/en.2007-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoeflich A, Wu M, Mohan S, et al. Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology. 1999;140:5488–5496. doi: 10.1210/endo.140.12.7169. [DOI] [PubMed] [Google Scholar]
- 91.Ning Y, Schuller AG, Bradshaw S, et al. Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol Endocrinol. 2006;20:2173–2186. doi: 10.1210/me.2005-0196. [DOI] [PubMed] [Google Scholar]
- 92.Parker A, Gockerman A, Busby WH, Clemmons DR. Properties of an insulin-like growth factor-binding protein-4 protease that is secreted by smooth muscle cells. Endocrinology. 1995;136:2470–2476. doi: 10.1210/endo.136.6.7538463. [DOI] [PubMed] [Google Scholar]
- 93.Yakar S, Rosen CJ, Beamer WG, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–781. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ning Y, Schuller AG, Conover CA, Pintar JE. Insulin-like growth factor (IGF) binding protein-4 is both a positive and negative regulator of IGF activity in vivo. Mol Endocrinol. 2008;22:1213–1225. doi: 10.1210/me.2007-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rehage M, Mohan S, Wergedal JE, et al. Transgenic overexpression of pregnancy-associated plasma protein-A increases the somatic growth and skeletal muscle mass in mice. Endocrinology. 2007;148:6176–6185. doi: 10.1210/en.2007-0274. [DOI] [PubMed] [Google Scholar]
- 96.Liao L, Dearth RK, Zhou S, Britton OL, Lee AV, Xu J. Liver-specific overexpression of the insulin-like growth factor-I enhances somatic growth and partially prevents the effects of growth hormone deficiency. Endocrinology. 2006;147:3877–3888. doi: 10.1210/en.2005-1537. [DOI] [PubMed] [Google Scholar]
- 97.Wu Y, Sun H, Yakar S, LeRoith D. Elevated levels of insulin-like growth factor (IGF)-I in serum rescue the severe growth retardation of IGF-I null mice. Endocrinology. 2009;150:4395–4403. doi: 10.1210/en.2009-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qin X, Wergedal JE, Rehage M, et al. Pregnancy-associated plasma protein-A increases osteoblast proliferation in vitro and bone formation in vivo. Endocrinology. 2006;147:5653–5661. doi: 10.1210/en.2006-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]