

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Renal coloboma syndrome (Papillorenal syndrome).
1.2 OMIM# of the disease
120330.
1.3 Name of the analyzed genes or DNA/chromosome segments
Paired box gene 2 (PAX2).
1.4 OMIM# of the gene(s)
167409.
1.5 Mutational spectrum
To date, there have been more than 25 published autosomal dominant mutations identified in the coding region of the PAX2 gene. The majority of these mutations occur in exons 2–4 (encoding the paired domain) and exons 7–9 (encoding the transactivation domain). The most common recurrent mutations are frameshift mutations within a homoguanine stretch (7Gs) in exon 2 (c.76dup, c.76del, c.75_76dup).1, 2, 3 These mutations were originally identified as 619insG, 619delG and 619insGG.
Three genomic rearrangements of the PAX2 gene have been reported to date: A balanced 10;13 chromosome translocation with break points between PAX2 exons 3 and 4,4 a large cytogenetically visible deletion of the PAX2 gene involving 90 other genes5 and a small 200 kb deletion of the entire PAX2 locus identified by high-density array CGH (Raca G and Schimmenti L personal communication).
1.6 Analytical methods
Current test methodology is either bidirectional (Minnesota, USA; Otago, NZ) or unidirectional (Paris, France) Sanger sequencing of all 12 coding exons of the PAX2 gene and adjoining intronic regions. Analysis for exon copy number variation by MLPA is under investigation in Minnesota, USA. Analysis for exon copy number variation by quantitative PCR was investigated in Paris, France. As copy number variation was not detected in more than 40 samples in Paris, this testing is not routinely performed.
1.7 Analytical validation
Full sequencing of six blinded samples that had been previously sequenced (Minnesota, USA).
A mutation positive sample is included in each analysis (Otago, New Zealand).
1.8 Estimated frequency of the disease
(Incidence at birth (‘birth prevalence') or population prevalence)
Because of the rarity of the disease, the precise incidence at birth is not known. In patients with renal hypodysplasia, 7% will have mutations in PAX2.6
1.9 If applicable, prevalence in the ethnic group of investigated person
There is no known ethnic group with a higher incidence of this condition. Affected patients have been identified from Caucasian, Asian and African groups.
1.10 Diagnostic setting

Comment: Requests for testing in all four categories have occurred in each of three clinical laboratories.
2. TEST CHARACTERISTICS
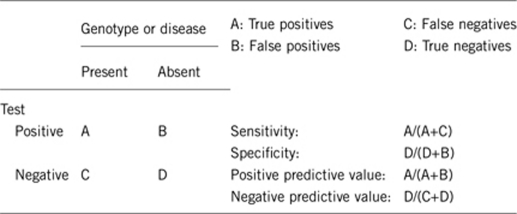
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
Bidirectional Sanger sequencing is expected to detect >99% of missense mutations, frameshift mutations, and splice-site mutations in adjacent intronic regions. These mutations account for all but three mutations reported to date: (a de novo unbalanced 10; 13 translocation)4 and two mutations that result from whole-gene deletions,5 (Raca and Schimmenti, personal communication). This may change as methods to detect genomic duplications and deletions become part of standard mutation detection strategies.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
>99%.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
In a series of patients with optic nerve dysplasia and renal hypodysplasia, analyzed by Dureau et al,7 (9/17) ∼half had point mutations in the PAX2 gene.
In a series of unselected patients with renal hypodysplasia, PAX2 mutations were identified in 7/99 (7%) of patients.6
In a published series of 100 patients with colobomatous eye abnormalities (including iris colobomas), only one patient had a mutation in PAX2.8 This patient had optic nerve and renal findings consistent with renal coloboma syndrome.
In two families, described by Parsa et al,9 with phenotypes consistent with renal coloboma syndome (Papillorenal syndrome), no mutations were identified by sequencing of the coding region of the PAX2 gene and adjacent intronic regions.
In the combined experience of the three labs contributing to this review, 48/208 probands have mutations in the PAX2 gene (23%). Many of these individuals did not have classic findings of renal coloboma syndrome. If this group is limited to individuals with documented evidence of optic nerve abnormality and renal hypodysplasia, mutations were identified in 34/59 cases (57%).
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
The clinical specificity is expected to be >99%. No mutations have been reported in the medical literature in individuals without clinical findings. A single individual carrying an R252X mutation has been found to have normal ophthalmological exam and normal renal function (Paris, France). This individual had a brother with the same mutation presenting with classic renal coloboma syndrome.
2.5 Positive clinical predictive value (life-time risk to develop the disease if the test is positive)
Penetrance is believed to be extremely high. One non-penetrant individual was reported (Heidet, Paris France). Although characterized by high penetrance, renal coloboma syndrome is also characterized by extremely variable expression. With the exception of the above-mentioned case, all known gene positive individuals have had at least optic nerve finding (optic nerve coloboma and optic nerve dysplasia) or kidney finding (renal failure and renal hypodysplasia).
2.6 Negative clinical predictive value (probability not to develop the disease if the test is negative)
Assume an increased risk, based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Interpretation of negative clinical results require caution in interpretation as described below:
Index case in that family had been tested:
If a mutation has been identified in the affected proband, the negative predictive value of the test is 100%.
Index case in that family had not been tested:
PAX2 sequencing has no negative predictive value if the mutation has not been identified in the proband.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: the tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
The most invasive procedure that might be considered in diagnosis is renal biopsy. However, renal biopsy cannot determine whether or not the kidney disease is due to PAX2 mutations. Most of the above procedures would be pursued, regardless of gene testing results, as testing assists in medical management.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Not applicable.
3.1.4 Will disease management be influenced by the result of a genetic test?
3.2 Predictive setting: the tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked).
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
The answers below assume that a family member has clinical findings consistent with renal coloboma syndrome AND a pathogenic mutation in the PAX2 gene.
If the test result is positive (please describe):
If the gene test is positive and the individual is asymptomatic, then the patient and clinicians might be more vigilant in monitoring kidney function. This might lead to lifestyle choices that would minimize the chances of developing renal failure.
If the test result is negative (please describe):
If the test result is negative in the asymptomatic individual, this may relieve the burden of being concerned about developing renal failure.
Individuals with optic nerve dysplasia (coloboma) are at increased risk of retinal detachment. Close clinical observation for changes in vision will improve clinical outcomes if addressed early.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Individuals could choose to have ophthalmological examinations and renal evaluations to look for sub-clinical features of the disease. Individuals could also make lifestyle choices to minimize the chance of kidney failure and retinal detachment.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
A positive gene test in the index patient would clearly establish the presence of an autosomal dominant disease with a 50% risk to each of his/her children.
In index cases with no known family history, gene testing of the parents may demonstrate that one parent carries a mutation, which would place future children at 50% risk. Alternatively, normal genetic testing in both clinically unaffected parents is consistent with a de novo origin for the mutation. Parents in this situation should be reassured that the risk of recurrence is low. Parents should be cautioned that, although the recurrence risk is low, both maternal and paternal germline mosaicism have been reported in the medical literature,10, 11 and mosaicism has been identified on one other occasion in a clinical laboratory (Minnesota, USA). Thus, the risk of recurrence is higher than the general population risk for renal coloboma syndrome. Owing to the rarity of renal coloboma syndrome, the frequency of germline mosaicism is not known.
Individuals at risk in a family with an identified mutation can be identified and managed prospectively to prevent ophthalmological complications of retinal detachment and prevent other complications of renal insufficiency.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
If the genetic test is positive in the index patient, then other family members who are gene negative may not have to undergo renal and ophthalmological evaluations.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
In most cases in which there is a clearly pathogenic mutation (ie, frameshift, nonsense or splice-site mutations), predictive genetic testing is available to asymptomatic family members. In cases with novel missense mutations or novel intronic variations that do not affect conserved splice sites, predictive testing should be accompanied by a careful discussion of the fact that excluding the familial variant may not actually exclude the disease.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, if a clearly pathogenic mutation is identified in the index patient, then prenatal diagnosis is available to pregnancies at 50% risk. However, a positive prenatal test might not be able to predict the severity of the findings in the affected fetus. The absence of a mutation in a fetus at 50% risk would exclude the diagnosis.
In a fetus with renal hypoplasia, renal agenesis, or renal dysgenesis identified by prenatal ultrasound, the finding of a PAX2 mutation may confirm the cause for the renal findings, but may not accurately predict future prognosis.
Some caution should be used in the use of prenatal test results in cases in which the proband carries a novel missense or intronic mutation of uncertain clinical significance.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
One important consideration is that gene-positive individuals might be at risk for having pregnancies with renal agenesis/Potter sequence. This has been reported on at least two occasions in the medical literature.12, 13 This may affect decisions about child bearing and fetal monitoring during pregnancy. This is an important point, as some individuals with relatively mild renal disease may have pregnancies with lethal renal agenesis.
Identification of a pathogenic PAX2 mutation allows couples the option of pursuing either prenatal diagnosis or preimplantation genetic diagnosis.
Acknowledgments
This work was supported by the EuroGentest, an EU-FP6 supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2).
The authors declare no conflict of interest.
References
- Schimmenti L, Eccles M.Renal coloboma syndrome GeneClinicsRetrieved 20 August 2010, from http://www.ncbi.nlm.nih.gov/bookshelf/ br.fcgi?book=gene&part=papr .
- Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR. Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet. 1995;4:2183–2184. doi: 10.1093/hmg/4.11.2183. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, Shim HH, Wirtschafter JD, et al. Homonucleotide expansion and contraction mutations of PAX2 and inclusion of Chiari 1 malformation as part of renal coloboma syndrome. Hum Mutat. 1999;14:369–376. doi: 10.1002/(SICI)1098-1004(199911)14:5<369::AID-HUMU2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Narahara K, Baker E, Ito S, et al. Localisation of a 10q breakpoint within the PAX2 gene in a patient with a de novo t(10;13) translocation and optic nerve coloboma renal disease. J Med Genet. 1997;34:213–216. doi: 10.1136/jmg.34.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetti E, Artifoni L, Salviati L, Pinello L, et al. Renal hypoplasia without optic coloboma associated with PAX2 gene deletion. Nephrol Dial Transplant. 2007;22:2076–2078. doi: 10.1093/ndt/gfm187. [DOI] [PubMed] [Google Scholar]
- Weber S, Moriniere V, Knuppel T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17:2864–2870. doi: 10.1681/ASN.2006030277. [DOI] [PubMed] [Google Scholar]
- Dureau P, Attie-Bitach T, Salomon R, et al. Renal coloboma syndrome. Ophthalmology. 2001;108:1912–1916. doi: 10.1016/s0161-6420(01)00722-9. [DOI] [PubMed] [Google Scholar]
- Cunliffe HE, McNoe LA, Ward TA, Devriendt K, Brunner HG, Eccles MR. The prevalence of PAX2 mutations in patients with isolated colobomas or colobomas associated with urogenital anomalies. J Med Genet. 1998;35:806–812. doi: 10.1136/jmg.35.10.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa CF, Silva ED, Sundin OH, et al. Redefining papillorenal syndrome: an underdiagnosed cause of ocular and renal morbidity. Ophthalmology. 2001;108:738–749. doi: 10.1016/s0161-6420(00)00661-8. [DOI] [PubMed] [Google Scholar]
- Amiel J, Audollent S, Joly D, et al. PAX2 mutations in renal-coloboma syndrome: mutational hotspot and germline mosaicism. Eur J Hum Genet. 2000;8:820–826. doi: 10.1038/sj.ejhg.5200539. [DOI] [PubMed] [Google Scholar]
- Cheong HI, Cho HY, Kim JH, Yu YS, Ha IS, Choi Y. A clinico-genetic study of renal coloboma syndrome in children. Pediatr Nephrol. 2007;22:1283–1289. doi: 10.1007/s00467-007-0525-z. [DOI] [PubMed] [Google Scholar]
- Ford B, Rupps R, Lirenman D, et al. Renal coloboma syndrome: prenatal detection and clinical spectrum in a large family. Am J Med Genet. 2001;99:137–141. doi: 10.1002/1096-8628(2000)9999:999<00::aid-ajmg1143>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Martinovic-Bouriel J, Benachi A, Bonniere M, et al. PAX2 mutations in fetal renal hypodysplasia. Am J Med Genet A. 2010;152A:830–835. doi: 10.1002/ajmg.a.33133. [DOI] [PubMed] [Google Scholar]
Other References
- Chung GW, Edwards AO, Schimmenti LA, Manligas GS, Zhang YH, Ritter R., III Renal coloboma syndrome: report of a novel PAX2 gene mutation. Am J Ophthalmol. 2001;132:910–914. doi: 10.1016/s0002-9394(01)01231-4. [DOI] [PubMed] [Google Scholar]
- Cunliffe HE, McNoe LA, Ward TA, Devriendt K, Brunner HG, Eccles MR. The prevalence of PAX2 mutations in patients with isolated colobomas or colobomas associated with urogenital anomalies. J Med Genet. 1998;35:806–812. doi: 10.1136/jmg.35.10.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devriendt K, Matthijs G, Van Damme B, et al. Missense mutation and hexanucleotide duplication in the PAX2 gene in two unrelated families with renal coloboma syndrome (MIM 120330) Hum Genet. 1998;103:149–153. doi: 10.1007/s004390050798. [DOI] [PubMed] [Google Scholar]
- Dureau P, Attie-Bitach T, Salomon R, et al. Renal coloboma syndrome. Ophthalmology. 2001;108:1912–1916. doi: 10.1016/s0161-6420(01)00722-9. [DOI] [PubMed] [Google Scholar]
- Eccles MR, Wallis LJ, Fidler AE, Spurr NK, Goodfellow PJ, Reeve AE. Expression of the PAX2 gene in human fetal kidney and Wilms' tumor. Cell Growth Differ. 1992;3:279–289. [PubMed] [Google Scholar]
- Fletcher J, Hu M, Berman Y, et al. Multicystic dysplastic kidney and variable phenotype in a family with a novel deletion mutation of PAX2. J Am Soc Nephrol. 2005;16:2754–2761. doi: 10.1681/ASN.2005030239. [DOI] [PubMed] [Google Scholar]
- Higashide T, Wada T, Sakurai M, Yokoyama H, Sugiyama K. Macular abnormalities and optic disk anomaly associated with a new PAX2 missense mutation. Am J Ophthalmol. 2005;139:203–205. doi: 10.1016/j.ajo.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Miyazawa T, Nakano M, Takemura Y, et al. A case of renal-coloboma syndrome associated with mental developmental delay exhibiting a novel PAX2 gene mutation. Clin Nephrol. 2009;72:497–500. doi: 10.5414/cnp72497. [DOI] [PubMed] [Google Scholar]
- Nishimoto K, Iijima K, Shirakawa T, et al. PAX2 gene mutation in a family with isolated renal hypoplasia. J Am Soc Nephrol. 2001;12:1769–1772. doi: 10.1681/ASN.V1281769. [DOI] [PubMed] [Google Scholar]
- Porteous S, Torban E, Cho NP, et al. Primary renal hypoplasia in humans and mice with PAX2 mutations: evidence of increased apoptosis in fetal kidneys of Pax2(1Neu) +/− mutant mice. Hum Mol Genet. 2000;9:1–11. doi: 10.1093/hmg/9.1.1. [DOI] [PubMed] [Google Scholar]
- Quinlan J, Lemire M, Hudson T, et al. A common variant of the PAX2 gene is associated with reduced newborn kidney size. J Am Soc Nephrol. 2007;18:1915–1921. doi: 10.1681/ASN.2006101107. [DOI] [PubMed] [Google Scholar]
- Samimi S, Antignac C, Combe C, Lacombe D, Renaud Rougier MB, Korobelnik JF. Bilateral macular detachment caused by bilateral optic nerve malformation in a papillorenal syndrome due to a new PAX2 mutation. Eur J Ophthalmol. 2008;18:656–658. doi: 10.1177/112067210801800430. [DOI] [PubMed] [Google Scholar]
- Sanyanusin P, Norrish JH, Ward TA, Nebel A, McNoe LA, Eccles MR. Genomic structure of the human PAX2 gene. Genomics. 1996;35:258–261. doi: 10.1006/geno.1996.0350. [DOI] [PubMed] [Google Scholar]
- Sanyanusin P, Schimmenti LA, McNoe LA, et al. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995;9:358–364. doi: 10.1038/ng0495-358. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, Cunliffe HE, McNoe LA, et al. Further delineation of renal-coloboma syndrome in patients with extreme variability of phenotype and identical PAX2 mutations. Am J Hum Genet. 1997;60:869–878. [PMC free article] [PubMed] [Google Scholar]
- Schimmenti LA, Manligas GS, Sieving PA. Optic nerve dysplasia and renal insufficiency in a family with a novel PAX2 mutation, Arg115X: further ophthalmologic delineation of the renal-coloboma syndrome. Ophthalmic Genet. 2003;24:191–202. doi: 10.1076/opge.24.4.191.17229. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, Pierpont ME, Carpenter BL, Kashtan CE, Johnson MR, Dobyns WB. Autosomal dominant optic nerve colobomas, vesicoureteral reflux, and renal anomalies. Am J Med Genet. 1995;59:204–208. doi: 10.1002/ajmg.1320590217. [DOI] [PubMed] [Google Scholar]
- Taranta A, Palma A, De Luca V, et al. Renal-coloboma syndrome: a single nucleotide deletion in the PAX2 gene at exon 8 is associated with a highly variable phenotype. Clin Nephrol. 2007;67:1–4. doi: 10.5414/cnp67001. [DOI] [PubMed] [Google Scholar]