

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
CHARGE syndrome (CHARGE association, Hall–Hittner syndrome).
1.2 OMIM# of the disease
#214800.
1.3 Name of the analysed genes or DNA/chromosome segments
CHD7.
1.4 OMIM# of the gene(s)
#608892.
1.5 Mutational spectrum
Predominantly heterozygous point mutations (72% nonsense or frame shift, 13% splice site and 10% missense). Less than 5% of the whole-exon deletions or microdeletions of 8q12.1, including CHD7.1, 2, 3
1.6 Analytical methods
Sequencing of all coding exons, including their boundaries of CDH7, MLPA covering most coding exons, including the 5′UTR and the first non-coding exon of CHD7. Array CGH in selected cases.4, 5
Conventional cytogenetics is usually normal. Translocations with breakpoint through CHD7 have been reported incidentally (Jongmans1).
1.7 Analytical validation
Sequence analysis detects >99% of the (point) mutations present in the area that has been investigated, MLPA has an estimated sensitivity of >90% for individual exons, and >95% for deletions covering more probes.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
Prevalence at birth: 1:10 000 (ranges from 1:8500 to 1:15 000 in the literature).6
1.9 If applicable, prevalence in the ethnic group of investigated person
There is no evidence at present for a different prevalence in various ethnic groups.
1.10 Diagnostic setting

Comment:
2. TEST CHARACTERISTICS
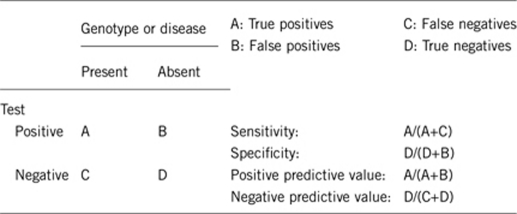
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Depends on the method used. If only CHD7 sequencing is performed, deletions are missed less than 5% due to whole-exon or whole-gene deletions.5, 7 If sequencing is combined with MLPA, 100%.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Almost 100% (some variants may erroneously be interpreted as pathogenic).
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Depends on the clinical criteria used. In over 95% of the patients who fulfil the criteria of Blake or Verloes8, 9 a mutation is found.1 In those who are suspected for CHARGE syndrome in 60–70% a mutation is found (CHARGE syndrome sometimes can be excluded if a patient does not fulfil the clinical criteria and does not carry a mutation or deletion of CHD7).
Some conditions can mimic CHARGE syndrome: 22q11 deletion syndrome, VACTERL association, chromosomal disorders (eg, deletions 3p12p21.210), disorders caused by teratogens (eg, maternal diabetes, Accutane), and Kallmann syndrome.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
The clinical variability of the syndrome is considerable. If the diagnosis is based on the Blake or Verloes criteria, some people with CHARGE will be missed. The clinical specificity is over 95%, since less than 5% of the patients with a CHD7 mutation do not completely fulfil these criteria. However, it should be taken into account that the mild end of the phenotypic spectrum is not completely known yet. For example, CHD7 mutations are also found in patients diagnosed with Kallmann syndrome or hypogonadotropic hypogonadism with minimal additional features of CHARGE syndrome.11, 12
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
100%, but high clinical variability (see also 2.4).
2.6 Negative clinical predictive value
(probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
100%.
Index case in that family had not been tested:
This depends on the a priori chance of the index to find a mutation, which varies between 60–90%. There is always a residual risk, but complete analysis (sequencing and MLPA) will reduce this by 90–95%.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
CHARGE syndrome can be diagnosed clinically but not by solely using the CHARGE acronym (C=coloboma, H=heart defect, A=choanael Atresia, R=retardation of growth and development, and E=ear abnormalities) or the major criteria (coloboma of the eyes, choanal atresia, characteristic ear malformations including deafness, and cranial nerve abnormalities). Scanning of the temporal bones often elicits abnormalities in the semi circular canals, which brings more specificity to the diagnosis.13
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Gene testing is still expensive, and for that reason many parents, particularly those with older children, have not had their child tested.14 Either gene testing or clinical criteria can miss some individuals with CHARGE syndrome. However, gene testing may be important in patients who do not have the classical CHARGE characteristics and may be at risk for the long-term complications of CHARGE syndrome.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
If the test result is positive, yes; see 3.1.4.
If the test result is negative (please describe):
If the test result is negative, depends on clinical manifestations.
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
No special options.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Not necessarily, but if a mutation is found in the patient and not in the parents, recurrence risk can be given with more certainty and prenatal diagnosis can be offered. Moreover, in that situation sibs of the patient do not have an increased risk for having children with CHARGE syndrome. If the mutation is found in mosaic or non-mosaic form in one of the parents, recurrence risk is increased (up to 50%) and prenatal or pre-implantation diagnosis should be discussed.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Family members may have very mild characteristics of CHARGE syndrome and therefore should be tested. Somatic mosaicism has been described in parents. So in case of child wish, parents should be tested to obtain a more accurate recurrence risk.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes. If an index patient has CHARGE syndrome other family members can be genetically tested.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes. If an index patient has CHARGE syndrome, then subsequent pregnancies can be screened genetically and by ultrasound.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? There is a lot of discussion in Hartshorne et al12 that might be helpful, including reasons for testing or not testing given by parents and a discussion of the ethical issues involved.
Acknowledgments
This work was supported by EuroGentest, an EU-FP6-supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2).
The authors declare no conflict of interest.
References
- Jongmans MC, Admiraal RJ, van der Donk KP, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43:306–314. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, Safiullah AM, Fernbach SD, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78:303–314. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Layman WS, MArtin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet Part A. 2010;152A:674–686. doi: 10.1002/ajmg.a.33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CMA, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Bergman JEH, de Wijs I, Hoefsloot LH, Jongmans MCJ, van Ravenswaaij CMA. Exon copy number alterations of the CHD7 gene are not a major cause of CHARGE syndrome. Eur J Med Genet. 2008;51:417–425. doi: 10.1016/j.ejmg.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Issekutz K, Graham J, Prasad C, Smith IM, Blake KD. Behavioural profiles and symptoms of autism in CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet. 2005;133A:309–317. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- Wincent J, Schulze A, Schoumans J. Detection of CHD7 deletions by MLPA in CHARGE syndrome patients with a less typical phenotype. Eur J Med Genet. 2009;52:271–272. doi: 10.1016/j.ejmg.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Blake KD, Davenport SLH, Hall BD, et al. CHARGE association: an update and review for the primary pediatrician. Clin Genet. 1998;37:159–173. doi: 10.1177/000992289803700302. [DOI] [PubMed] [Google Scholar]
- Verloes A. Updated diagnostic criteria for CHARGE syndrome; a proposal. Am J Med Genet. 2005;133A:306–308. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- Wieczorek D, Bolt J, Schwechheimer K, Gillessen-Kaesbach G. A patient with interstitial deletion of the short arm of chromosome 3 (pterrarrp21.2∷p12rarrqter) and a CHARGE-like phenotype. Am J Med Genet. 1997;69:413–417. doi: 10.1002/(sici)1096-8628(19970414)69:4<413::aid-ajmg15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Kim HG, Kurth I, Lan F, et al. Mutations in CHD7, encoding a chromatin-remodelling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MCJ, van Ravenswaaij CMA, Pitteloud N, et al. CHD7 mutations in patients initially diagnosed with Kallmann Syndrome - the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71. doi: 10.1111/j.1399-0004.2008.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007;15:389–399. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- Hartshorne TS, Stratton KK, van Ravenswaaij-Arts CM. Prevalence of genetic testing in CHARGE syndrome. J Genet Couns. 2011;20:49–57. doi: 10.1007/s10897-010-9328-7. [DOI] [PubMed] [Google Scholar]
- Dobbelsteyn C, Peacocke SD, Blake K, Crist W, Rashid M. Feeding difficulties in children with CHARGE syndrome: prevalence, risk factors, and prognosis. Dysphagia. 2008;23:127. doi: 10.1007/s00455-007-9111-6. [DOI] [PubMed] [Google Scholar]
- MacCuspie J, Blake K, Corsten G. Botulinum toxin injections into salivary glands to decrease oral secretions in CHARGE syndrome: prospective case study. Am J Med Genet. 2010. [DOI] [PubMed]
- Forward K, Cummings EA, Blake KD. Risk factors for poor bone health in adolescent and adults with CHARGE syndrome. Am J Med Genet. 2007;143A:839–845. doi: 10.1002/ajmg.a.31670. [DOI] [PubMed] [Google Scholar]