

Abstract
The potency of DNA vaccines may be affected by the efficiency of intracellular processing and MHC class I presentation of encoded antigens. Since a single-chain trimer (SCT) composed of peptide, β2-microglobulin (β2m), and MHC class I heavy chain has been shown to bypass antigen processing and lead to stable presentation of peptides, we investigated the efficacy of a DNA vaccine encoding a SCT composed of an immunodominant CTL epitope of human papillomavirus type 16 (HPV-16) E6 antigen, β2m, and H-2Kb MHC class I heavy chain (pIRES-E6-β2m-Kb). Transfection of 293 cells with pIRES-E6-β2m-Kb can bypass antigen processing and lead to stable presentation of E6 peptide. Furthermore, C57BL/6 mice vaccinated with pIRES-E6-β2m-Kb exhibited significantly increased E6 peptide-specific CD8+ T-cell immune responses compared to mice vaccinated with DNA encoding wild-type E6. Most importantly, 100% of mice vaccinated with pIRES-E6-β2m-Kb DNA were protected against a lethal challenge of E6-expressing TC-1 tumor cells. In contrast, all mice vaccinated with wild-type E6 DNA or control plasmid DNA grew tumors. Our data indicate that a DNA vaccine encoding a SCT can lead to stable enhanced MHC class I presentation of encoded antigenic peptide and may be useful for improving DNA vaccine potency to control tumors or infectious diseases.
Keywords: DNA vaccine, immunotherapy, HPV-16 E6, single-chain trimer, tumor immunology
Introduction
DNA vaccines have become potentially important immunotherapeutic agents for combating cancers (for a review see Boyd et al1). Compared to live viral or bacterial vectors, naked plasmid DNA vaccines are safe and can be administered repeatedly. Other benefits of DNA vaccines include easy preparation on a large scale with high purity and high stability relative to proteins and other biological agents.2,3 One concern involved with naked DNA vaccines is their limited potency, as naked DNA does not have the inherent ability to replicate in vivo. Therefore, attempts have been made to improve the potency of DNA vaccines by targeting DNA to professional antigen-presenting cells (APCs) and by modifying the properties of DNA-transfected APCs (for reviews see Boyd et al1 and Hung and Wu4).
One important strategy for improving DNA vaccine potency is enhancement of MHC class I presentation of encoded antigen by APCs. The potency of DNA vaccines depends on the efficient expression and presentation of the encoded antigen of interest, which is processed in the cytoplasm and presented by MHC class I molecules on the surface of APCs, leading to a cellular and/or humoral immune response against the antigen in immunized individuals. MHC class I antigen processing involves cutting of protein antigen into peptide fragments by a proteasome, transport by TAP into the endoplasmic reticulum lumen, loading onto newly assembled MHC class I heavy chain and β2 microglobulin (β2m), and delivery of the MHC:peptide complex to the cell surface. Each of these steps is under extensive regulation and affects the robustness of immune response generated (for a review, see Pamer and Cresswell5).
We attempted to circumvent antigen processing and elicit stable MHC class I presentation of the antigenic peptide encoded by our DNA vaccine by employing a single-chain construct encoding MHC class I linked to peptide.6–8 By linking together the antigen peptide, β2m, and MHC class I heavy chain into a single-chain trimer (SCT), as illustrated in Figure 1a, it has been shown that stable cell surface expression of the chimeric protein can be detected in DNA-transfected cells using antibodies specific to the antigenic peptide within the trimer. This indicates that the peptide in such SCT must be in an orientation similar to noncovalently attached peptides.8 A major advantage of this approach is that normal antigen processing, which might be regulated by cellular machinery, leading to uncertainties in immunogenicity, may be bypassed. Furthermore, stability is conferred to the MHC:peptide complex by the covalent bonding between the three molecules of the trimer. Also, the antigenic peptide can be selected so that the most immunogenic epitope may be used, which is not possible in conventional antigen-coding DNA vaccines, where proteasome degradation and peptide loading onto MHC class I molecules determines the nature of presented epitopes.
Figure 1.
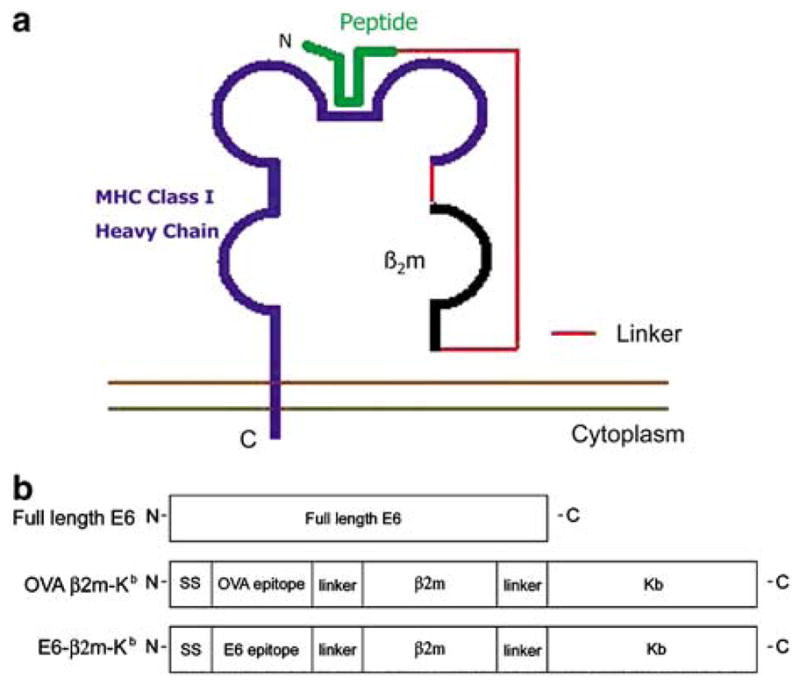
Diagrams depicting structure of our SCT and structures of chimeric DNA constructs. (a) Diagram of a peptide:β2m:MHC SCT on cell surface. (b) Diagram of full-length E6, an SCT encoding OVA linked to β2m and an H2-Kb-restricted MHC class I molecule, and an SCT encoding E6 aa49–57 linked to β2m and an H2-Kb-restricted MHC class I molecule. Each was cloned into the pIRES vector to make the DNA vaccines used in the study.
A DNA vaccine encoding SCT of MHC class I linked to the immunodominant CTL epitope of a tumor antigen has not been tested for the ability to protect against antigen-expressing tumors. For this study, we constructed a DNA vaccine encoding an SCT containing aa49–57 of the human papillomavirus-16 (HPV-16) E6 protein (pIRES-E6-β2m-Kb) and tested its ability to induce protective immunity against the HPV-transformed TC-1 murine tumor model.9 HPV infection, particularly high-risk HPV type-16, is causally linked to the development of cervical cancer and has been linked to other cancers (for a review, see zur Hausen10). HPV oncoprotein E6 is responsible for malignant transformation of HPV-infected cells and is consistently expressed in HPV-associated tumor cells. The E6 protein therefore represents an ideal target antigen for HPV vaccine development. E6 aa48–57 was recently identified as a peptide sequence containing an H2-Kb-restricted immunodominant CTL epitope of the E6 protein in C57BL/6 mice,11 and further studies have indicated that E6 aa50–57 and E6 aa49–57 also function as CTL epitopes of E6 in C57BL/6 mice. We found that vaccination of mice with pIRES-E6-β2m-Kb resulted in stronger E6-specific cellular immune responses and antitumor effects than vaccination of mice with DNA encoding full-length E6 protein. Our findings thus suggest that a DNA vaccine encoding a SCT specific for an HPV antigen has potential clinical application for the control of HPV infection and HPV-related cancers.
Results
E6 peptide-specific CD8+ T cells can be activated by 293 cells transfected with pIRES-E6-β2m-Kb
To determine whether pIRES-E6-β2m-Kb can bypass processing and directly present on the surface of transfected cells, we performed an in vitro transfection experiment. The 293 cells, known to be deficient in Kb molecules, were transfected with pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-Kb. These transfected 293 cells were subsequently used to stimulate H2-Kb-restricted HPV-16 E6 aa49–57 peptide-specific CTLs. As shown in Figure 2, pIRES-E6-β2m-Kb-transfected 293 cells were able to stimulate a high frequency of E6-specific IFN-γ-secreting CD8+ T cells (42.24% of total T cells). In contrast, pIRES-OVA-β2m-Kb- and pIRES-E6-transfected 293 cells and nontransfected 293 cells generated only 1.52, 0.93, and 0.84% E6-specific IFN-γ-secreting CD8+ T cells. Since 293 cells are devoid of Kb, they cannot naturally present Kb-restricted E6 aa49–57 through Kb on the cell surface. Therefore, this suggests that pIRES-E6-β2m-Kb-transfected 293 cells can stimulate E6-specific CD8+ T cells through direct expression of the E6-β2m-Kb SCT on the cell surface.
Figure 2.

Intracellular cytokine staining followed by flow cytometry analysis to demonstrate activation of E6-specific CTL by 293 cells transfected with the various DNA constructs. (a) The 293 cells were either untreated or transfected with pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb using Lipofectamine 2000. The 293 cells were then incubated with an E6-specific CD8+ T-cell line. IFN-γ+ CD8+ T cells were analyzed using flow cytometry. The numbers in the upper right corner of the plots represent the percentage of activated IFN-γ+ E6-specific CD8+ T cells out of total number E6-specific CD8+ T cells. (b) Summary of HPV-16 E6 aa49–57 peptide-specific CTL activation by transfected or nontransfected 293 cells analyzed by intracellular IFN-γ staining. Data are expressed as the mean number of percentage of IFN-γ+ CD8+ T cells out of total CD8+ T cells±s.e.m. The data shown here are from one representative experiment of two performed.
Vaccination with pIRES-E6-β2m-Kb enhances the E6 peptide-specific CD8+ T-cell responses in vaccinated mice
To assess the immunogenicity of our E6-β2m-Kb SCT, we vaccinated mice with pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb, and then performed intracellular cytokine staining with flow cytometry analysis to characterize E6-specific CD8+ T-cell precursors using E6 aa49–57 peptide as a stimulant. As shown in Figure 3, splenocytes isolated from pIRES-E6-β2m-Kb-immunized mice generated a higher frequency of E6-specific IFN-γ-secreting CD8+ T-cell precursors (871±36/3.0 × 105 splenocytes) compared to mice immunized with pIRES-E6 (15±2.5/3.0 × 105 splenocytes), pIRES-OVA-β2m-Kb (9±6/3.0 × 105 splenocytes), or nonimmunized mice (4±1/3.0 × 105 splenocytes).
Figure 3.

Intracellular cytokine staining with flow cytometry analysis to determine IFN-γ-secreting, E6-specific CD8+ T-cell precursors. Mice (five per group) were either naive or immunized with pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb. Splenocytes were collected and cultured. (a) Representative figure of the flow cytometry data from splenocytes harvested from mice vaccinated with pIRES-E6-β2m-Kb and stimulated overnight with the E6 aa49–57 or no peptides. The numbers shown in the upper right corner of the plots represent the total number of E6-specific CD8+ T cells out of 3 × 105 splenocytes from the vaccinated mice. (b) Bar graph depicting the number of E6-specific IFN-γ-secreting CD8+ T-cell precursors/3 × 105 splenocytes (mean±s.e.m.) generated by in vitro stimulation.
This result is in agreement with the previous report that full-length E6 peptide is itself not immunogenic even though it contains the immunodominant epitope.11 To investigate a possible reason for poor immunogenicity of pIRES-E6, we performed in vitro transfection of 293-Kb cells with the various DNA constructs. We then used these transfected 293-Kb as stimulators, incubating them with an H2-Kb-restricted E6-specific CD8+ T-cell line. In contrast to pIRES-E6-β2m-Kb-transfected 293-Kb cells, pIRES-E6-transfected cells failed to stimulate E6-specific CD8+ T cells, as shown in Figure 4, suggesting that the E6 epitope is poorly processed and presented through Kb in cells transfected with pIRES-E6.
Figure 4.

Intracellular cytokine staining followed by flow cytometry analysis to demonstrate activation of E6-specific CTL by 293-Kb cells transfected with the various DNA constructs. The 293-Kb cells were either untreated or transfected with pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb using Lipofectamine 2000. IFN-γ+ CD8+ T cells were analyzed using flow cytometry. (a) Representative figure of flow cytometry data. The numbers in the upper right corner of the plots represent the percentage of activated IFN-γ+ E6-specific CD8+ T cells out of the total number of E6-specific CD8+ T cells. (b) Summary of HPV-16 E6 aa49–57 peptide-specific CTL activation by transfected or untransfected 293-Kb cells analyzed by intracellular IFN-γ staining. Data are expressed as the mean percentage of IFN-γ+ CD8+ T cells out of the total number of CD8+ T cells±s.e.m. The data shown here are from one representative experiment of two performed.
pIRES-E6-β2m-Kb vaccination prevents the growth of E6-expressing TC-1 tumors in mice
Given the immunogenicity of pIRES-E6-β2m-Kb, we next explored whether or not it could elicit an effective protective antitumor effect against E6-expressing tumors. Tumor cells were injected subcutaneously 1 week after DNA vaccination. During a 7-week follow-up period, all the mice receiving pIRES-E6-β2m-Kb remained tumor free, whereas 80% of pIRES-E6-immunized mice developed tumors by the end of the third week. All the nonimmunized and pIRES-OVA-β2m-Kb-immunized mice exhibited tumor growth by the second week (Figure 5a). This result suggests that vaccination with pIRES-E6-β2m-Kb can generate significant protective effects against E6-expressing tumors.
Figure 5.

In vivo tumor protection and antibody depletion experiments to demonstrate the antitumor effect generated by pIRES-E6-β2m-Kb against E6-expressing TC-1 tumors and the contribution of lymphocyte subsets to tumor protection. (a) Mice (five per group) were immunized with pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb. At 1 week after vaccination, mice were challenged subcutaneously with 5 × 104 TC-1 cells/mouse and monitored for evidence of tumor growth by palpation and inspection twice a week. (b) In vivo antibody depletion experiments to determine the contribution of lymphocyte subsets to the tumor protection of the pIRES-E6-β2m-Kb DNA vaccine. CD4, CD8, and NK1.1 depletions were initiated 1 week before tumor challenge.
CD8+ T cells, but not CD4+ T cells or NK cells, are required for protective antitumor effect of pIRES-E6-β2m-Kb
To determine the subsets of lymphocytes that are important for the protection against E6-expressing tumor cells, we performed in vivo antibody depletion experiments. As shown in Figure 5b, all of the mice depleted of CD8+ T cells grew tumors within 1 week of tumor challenge. In contrast, all of the nondepleted, NK- or CD4+ T-cell-depleted mice remained tumor-free 6 weeks after tumor challenge. These results indicate that CD8+ T cells are essential for the protective antitumor immunity generated by the pIRES-E6-β2m-Kb DNA vaccine.
Treatment with pIRES-E6-β2m-Kb DNA eradicates established E6-expressing tumors in the lungs
The therapeutic potential of each vaccine was assessed by performing an in vivo tumor treatment experiment using a previously described lung hematogeneous spread model.12 Mice were challenged with TC-1 tumor cells via tail vein injection followed by treatment with DNA. As shown in Figure 6, C57BL/6 mice treated with pIRES-E6-β2m-Kb DNA exhibited the lowest mean number of pulmonary tumor nodules (14±2), significantly lower than mice treated with pIRES-E6 DNA (65±6) or pIRES-OVA-β2m-Kb DNA (67±6) (one-way analysis of variance (ANOVA), P<0.01). These data indicated that treatment with pIRES-E6-β2m-Kb DNA generated the most potent antitumor effect against E6-expressing tumors compared to treatment with pIRES-E6 or pIRES-OVA-β2m-Kb DNA.
Figure 6.

In vivo tumor treatment experiments to demonstrate the therapeutic effect of pIRES-E6-β2m-Kb in controlling pulmonary tumor cell growth. Mice (five per group) were challenged with 1 ×104 tumor cells 3 days prior to DNA vaccination. The mean number of lung tumor nodules generated in each vaccinated group was determined on day 30 after tumor challenge.
Discussion
In this study, we investigated the effectiveness of a DNA vaccine encoding E6-β2m-Kb for controlling HPV-16 E6-expressing tumors and explored the possible mechanism of its antitumor effect. Transfection of 293 cells with pIRES-E6-β2m-Kb resulted in presentation of E6-β2m-Kb and activation of E6-specific CD8+ T cells. We also demonstrated that vaccination with pIRES-E6-β2m-Kb was capable of generating a significant E6-specific CD8+ T-cell response in vaccinated C57BL/6 mice. This immune response was able to protect vaccinated mice against challenge with an E6-expressing tumor cell line that naturally presents E6 aa49–57 peptide on its cell surface, and we determined that CD8+ T cells are the main effector cells for this tumor-protecting immunity.
We demonstrated that pIRES-E6-β2m-Kb-transfected 293 cells could stimulate E6-specific CTLs. As 293 cells do not express Kb molecules and because the E6 peptide-specific CD8+ T-cell line is Kb restricted, our results suggest that transfection of 293 cells with our E6-β2m-Kb SCT leads to cell surface presentation of the trimer while bypassing normal MHC class I antigen processing pathways. The high percentage of IFN-γ+ CD8+ T cells stimulated by pIRES-E6-β2m-Kb-transfected 293 cells also suggests high-level surface expression of the SCT. This is a particularly useful strategy for antigens, such as HPV-16 E6 protein, whose immunodominant epitopes are not efficiently processed and presented by the MHC class I pathway11 (also see Figures 2 and 3). Our data are consistent with a previous study by Yu et al,8 showing that the covalently bound peptide of an SCT resides stably in the peptide binding groove of the MHC class I molecule, excluding loading of other peptides.
Direct delivery of DNA encoding SCT to professional APCs is probably important for SCT-encoding DNA vaccines to work efficiently. Intradermal administration of DNA vaccines via gene gun has been shown to deliver efficiently DNA into professional APCs (Langerhans cells).13 In comparison, intramuscular injection of DNA vaccines likely delivers DNA to muscle cells. Since muscle cells do not have sufficient costimulatory molecules for the activation of antigen-specific T cells, such as B7.1 and B7.2, presentation of the SCT by muscle cells will not likely result in a significant activation of antigen-specific, CD8+ T cells.
A DNA vaccine encoding SCT could also be used in combination with other strategies to further enhance immunogenicity of DNA vaccines. The SCT strategy enhances immunogenicity of DNA vaccines by bypassing antigen-processing pathways to improve MHC class I presentation of encoded antigenic peptide on the surface of professional APCs such as dendritic cells (DC). We have previously coadministered DNA encoding antiapoptotic proteins with DNA encoding antigen to prolong the life of DCs in vivo.14,15 As these two strategies modify the properties of DCs through different pathways, it should be feasible to coadminister DNA encoding antiapoptotic proteins with DNA encoding SCT to further enhance antigen-specific immune responses to vaccination.
Different approaches to link MHC class I heavy chain and β2m to peptide to make a functional single-chain molecule capable of stimulating antigen-specific T cells have been reported,6,8,16–20 although some are applicable only to certain MHC class I:peptide complexes. An SCT construct in the format used in this study, the ‘peptide-spacer-β2m-spacer-heavy chain’ format, has been reported to be applicable to a wider range of MHC class I:peptide complexes.8,19,21 For this reason, we chose this format for the construction of our E6-β2m-Kb SCT, and our data are consistent with these prior reports.
In this study, we have shown that vaccination with an E6 DNA vaccine encoding SCT can generate a better therapeutic antitumor effect compared to a DNA vaccine encoding wild-type E6 (Figure 6). While the tumor dose we used for our in vivo tumor treatment experiment (104 TC-1 tumor cells) may not form a grossly visible lung tumor 3 days after tumor challenge, this tumor dose has been shown to generate multiple grossly visible lung tumors in 4 weeks and eventually kill the challenged mice if the mice were left untreated.12 We used this experimental design to demonstrate the ability of our DNA vaccine to control ‘minimal residual disease’. Minimal residual disease (such as tumor spread through metastasis) is a major concern for cancer patient management after patients receive initial treatment such as surgery or radiotherapy. The lung hematogeneous spread tumor model represents a model of minimal residual disease in the challenged mice. Our data suggest that an E6 DNA vaccine employing an SCT may potentially generate therapeutic effects against minimal residual disease of HPV-associated malignancies.
One limitation of an MHC class I SCT vaccination strategy is its restriction to presentation of a single antigenic epitope. It is known that expression of tumor-associated antigens or epitopes of antigens can vary between tumors in different hosts or between tumors within a single host. Therefore, therapeutic tumor vaccines targeting multiple tumor-associated antigens or multiple epitopes of a single antigen would likely be more effective than therapeutic vaccines targeting a single epitope. Although a DNA vaccine encoding a SCT is restricted to targeting a single antigenic epitope, multiple SCT-encoding DNA vaccines could be coadministered to elicit an immune response specific for multiple epitopes of the same or different tumor-associated antigens. Thus, DNA vaccines encoding SCT could potentially be used to elicit immune responses against multiple antigens for immunotherapy of cancers with well-defined tumor-specific antigens such as cervical cancer or melanoma.
With the continuing identification of new cancer-related antigens and their epitopes, this SCT strategy may serve as a general technique for the development of effective DNA vaccines against neoplasia. Since SCT can be directly expressed on the cell surface without processing, this allows control over which peptide is presented by a specific MHC class I molecule. Thus, the most immunodominant epitope can always be chosen, as we did in this study. It may also be possible to further enhance antitumor immunity by introducing mutations into the peptide or MHC class I molecule of an SCT. For example, mutation of heavy chain intended to better accommodate the linker between the peptide and MHC molecule was shown to enhance immune presentation,19 and altered peptide (such as heteroclitic peptide), which stabilizes MHC–peptide–TCR complexes strengthens antitumor immunity.22 Most recently, SCT technology has been extended to human MHC class I molecules. SCT composed of either an HLA-A2 or an HLA-B27 molecule linked to peptide can be detected by peptide-specific CTL and antibodies (Hansen et al, personal communication).
In conclusion, our results demonstrate that a DNA vaccine encoding an SCT consisting of MHC class I linked to a CTL epitope of a tumor antigen effectively induces protective antitumor immunity through direct presentation of the CTL epitope on the cell surface by the linked MHC class I molecule. This strategy may serve as a convenient means for development of DNA vaccines to control other tumors and infectious diseases.
Materials and methods
Plasmid DNA constructs and DNA preparation
The plasmid encoding SCT OVA-β2m-Kb (pIRES-OVA-β2m-Kb) has been described previously.8 For the generation of E6 aa49–57-β2m-Kb-expressing plasmid, an insert containing the immunodominant E6 aa49–57 epitope11 and flanking AgeI/NheI restriction enzyme sites was made by annealing two single-stranded oligo-nucleotides (5′-CCGGTTTGTATGCTGTATATGACTTTGCTTTT CGGGATTTAGGAGGAGGTG-3′ and 5′-CTAGCACCT CCTCCTAAATCCCGAAAAGCAAAGTCATATAAGCA TACAAA-3′). It was then cloned into pIRES-OVA-Kb using AgeI/NheI sites to replace the OVA epitope, generating pIRES-E6-β2m-Kb.
Full-length E6 protein, previously cloned in pcDNA plasmid vector (pcDNA-E6),11 was subcloned into pIRES using NotI/BamHI sites to generate pIRES-E6. Figure 1b shows the different constructs used in this study. For the generation of pcDNA3-Kb DNA, DNA encoding Kb was first amplified by RT-PCR with a Superscript One-Step RT-PCR system (Invitrogen, Carlsbad, CA, USA) using cDNA derived from the TC-1 cell line as a template and a set of primers, 5′-AAAGAATTCATGGTACCGTGCA CGCTGCTC-3′ and 5′-TTTGGATCCTCACGCTAGAGA ATGAGGGTC-′. The amplified product was then cloned into the EcoRI/BamHI cloning sites of the pcDNA3 vector. All plasmid constructs were confirmed by DNA sequencing. The DNA was amplified in Escherichia coli DH5α and purified as described previously.23 DNA for vaccination was prepared using an endotoxin-free kit (Qiagen, Valenci, CA, USA).
Cells
The HPV-16 E6-expressing murine tumor model, TC-1, has been described previously.9 Briefly, TC-1 cells were obtained by cotransformation of primary C57BL/6 mice lung epithelial cells by HPV-16 E6, E7, and an activated ras oncogene. The 293 cell line (ATCC Cat. No. CRL-1573) is a human kidney cell line, and 293-Kb cells were generated by the stable transfection of pcDNA3-Kb into 293 cells. Both cell lines were cultured in DMEM containing 10% fetal bovine serum. The H-2Kb-restricted HPV-16 E6 aa49–57 peptide-specific T-cell line was described previously.11 Briefly, the T-cell line was stimulated with an irradiated HPV-16 E6-expressing tumor cell line, TC-1, in the presence of IL-2 (20 U/ml) once a week. The specificity of the CTL lines was characterized by staining for CD8 and by determining the ability of the T cells to kill E6 peptide-pulsed EL-4 cells.
In vitro transfection
The 293 or 293-Kb cells were seeded in six-well plate at a density of 5 × 105 cells/well the day before transfection. Plasmid (5 μg) was used to transfect each well using Lipofectamine 2000 (Invitrogen). The transfected cells were used 24 h later.
Transfected or untransfected 293 or 293-Kb cells were washed with complete RPMI-1640 containing 10% fetal bovine serum, and coincubated with HPV-16 E6 aa49–57 peptide-specific T cells (with a ratio 1:2.5) at the presence of 1 μl/ml of GolgiPlug (BD Pharmingen) at 37°C overnight. Intracellular staining of IFN-γ, flow cytometry and data analysis were performed as described below.
Mice
Female C57BL/6 mice (6–8 weeks old) were purchased from the National Cancer Institute (Frederick, MD, USA) and kept in the oncology animal facility of the Johns Hopkins Hospital (Baltimore, MD, USA). All animal procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals.
DNA vaccination
DNA-coated gold particles were prepared according to a previously described protocol.23 DNA-coated gold particles were delivered to the shaved abdominal region of mice using a helium-driven gene gun (BioRad, Hercules, CA, USA) with a discharge pressure of 400 p.s.i. C57BL/6 mice were immunized with 2 μg of pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb. The mice received three boost vaccinations with the same dose at 1-week intervals.
Intracellular cytokine staining and flow cytometry analysis
Splenocytes were harvested from mice (five per group) 1 week after the last vaccination. Cell surface CD8 marker staining, intracellular IFN-γ staining and flow cytometry analysis were performed as described previously.23 Briefly, 4 × 106 pooled splenocytes from each vaccination group were incubated with 1 μg/ml of E6 aa49–57 peptide in the presence of 1 μl/ml of GolgiPlug (BD Pharmingen) at 37°C overnight. Cells were then washed with FACScan buffer and stained with phycoerythrin-conjugated monoclonal rat anti-mouse CD8 antibody, followed by intracellular cytokine staining with FITC-conjugated IFN-γ using the Cytofix/Cytoperm kit according to the manufacturer’s instructions (BD Pharmingen, San Diego, CA, USA). Analysis was performed on a Becton-Dickinson FACScan with CELLQuest software (Becton Dickinson Immunocytometry System, Mountain View, CA, USA).
In vivo tumor protection experiment
For the tumor protection experiment, C57BL/6 mice (five per group) were immunized via gene gun with 2 μg of pIRES-E6-β2m-Kb, pIRES-E6, or pIRES-OVA-β2m-Kb. Three boosters using the same regimen were given at 1-week intervals. At 1 week after the last vaccination, each mouse was challenged with 5 × 104 TC-1 tumor cells subcutaneously in the right leg and then monitored twice a week.
In vivo tumor treatment experiment
For the tumor treatment experiments, mice (five per group) were challenged with 1 ×104 TC-1 tumor cells/mouse in the tail vein to simulate hematogenous spread of tumors. DNA vaccines were given 3 days after tumor challenge at a dose of 2 μg/mouse, followed by two boosters of the same dose at 1-week intervals. Mice were killed on day 30 after tumor challenge.
In vivo antibody depletion experiment
To study the subsets of lymphocytes that are important for the antitumor effects, a tumor protection experiment was performed as described above, coupled with in vivo antibody depletion of CD4 T cells, CD8 T cells, and NK cells, using the method described previously.9 Briefly, mAb GK1.5, mAb 2.43, and mAb PK136 (Harlan, Indianapolis, IA, USA) were used for CD4 depletion, CD8 depletion, and NK1.1 depletion, respectively. We have confirmed that more than 95% depletion of the CD8 or CD4 T cells and more than 90% of NK cells were achieved with normal levels of the other subsets. Mice (five per group) were monitored twice a week and killed on day 42 after tumor challenge.
Statistical analysis
All data expressed as means±s.e.m. are representative of at least two different experiments. Data for intracellular cytokine staining with flow cytometry analysis were evaluated by ANOVA. Comparisons between individual data points were made using a Student’s t-test. For statistical analysis of the tumor protection and antibody depletion experiments, we used Kaplan–Meier analysis.
Acknowledgments
We thank Dr Richard Roden for helpful discussions. We would also like to thank Drs Ralph Hruban and Ken-Yu Lin for critical review of this paper. This work was supported by grants from the National Institute of Allergy and Infectious Disease, the National Cancer Institute, and the American Cancer Society.
References
- 1.Boyd D, Hung CF, Wu TC. DNA vaccines for cancer. IDrugs. 2003;6:1155–1164. [PubMed] [Google Scholar]
- 2.Donnelly JJ, Ulmer JB, Shiver JW, Liu MA. DNA vaccines. Annu Rev Immunol. 1997;15:617–648. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 3.Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology, application, and optimization*. Annu Rev Immunol. 2000;18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 4.Hung CF, Wu TC. Improving DNA vaccine potency via modification of professional antigen presenting cells. Curr Opin Mol Ther. 2003;5:20–24. [PubMed] [Google Scholar]
- 5.Pamer E, Cresswell P. Mechanisms of MHC class I-restricted antigen processing. Annu Rev Immunol. 1998;16:323–358. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 6.Mottez E, et al. Cells expressing a major histocompatibility complex class I molecule with a single covalently bound peptide are highly immunogenic. J Exp Med. 1995;181:493–502. doi: 10.1084/jem.181.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uger RA, Barber BH. Creating CTL targets with epitope-linked beta 2-microglobulin constructs. J Immunol. 1998;160:1598–1605. [PubMed] [Google Scholar]
- 8.Yu YY, et al. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J Immunol. 2002;168:3145–3149. doi: 10.4049/jimmunol.168.7.3145. [DOI] [PubMed] [Google Scholar]
- 9.Lin K-Y, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 10.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 11.Peng S, et al. Development of a DNA vaccine targeting HPV-16 oncogenic protein E6. J Virol. 2004;78:8468–8476. doi: 10.1128/JVI.78.16.8468-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji H, et al. Antigen-specific immunotherapy for murine lung metastatic tumors expressing human papillomavirus type 16 E7 oncoprotein. Int J Cancer. 1998;78:41–45. doi: 10.1002/(sici)1097-0215(19980925)78:1<41::aid-ijc8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 13.Porgador A, et al. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med. 1998;188:1075–1082. doi: 10.1084/jem.188.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim TW, et al. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. J Clin Invest. 2003;112:109–117. doi: 10.1172/JCI17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim TW, et al. Enhancing DNA vaccine potency by combining a strategy to prolong dendritic cell life with intracellular targeting strategies. J Immunol. 2003;171:2970–2976. doi: 10.4049/jimmunol.171.6.2970. [DOI] [PubMed] [Google Scholar]
- 16.Chung DH, et al. NK and CTL recognition of a single chain H-2Dd molecule: distinct sites of H-2Dd interact with NK and TCR. J Immunol. 1999;163:3699–3708. [PubMed] [Google Scholar]
- 17.Lee L, et al. Functional cell surface expression by a recombinant single-chain class I major histocompatibility complex molecule with a cis-active beta 2-microglobulin domain. Eur J Immunol. 1994;24:2633–2639. doi: 10.1002/eji.1830241110. [DOI] [PubMed] [Google Scholar]
- 18.Lone YC, et al. In vitro induction of specific cytotoxic T lymphocytes using recombinant single-chain MHC class I/peptide complexes. J Immunother. 1998;21:283–294. doi: 10.1097/00002371-199807000-00006. [DOI] [PubMed] [Google Scholar]
- 19.Lybarger L, et al. Enhanced immune presentation of a single-chain major histocompatibility complex class I molecule engineered to optimize linkage of a C-terminally extended peptide. J Biol Chem. 2003;278:27105–27111. doi: 10.1074/jbc.M303716200. [DOI] [PubMed] [Google Scholar]
- 20.Anjuere F, Horvath C, Cerottini JC, Luescher IF. Induction of CTL in vivo by major histocompatibility complex class I–peptide complexes covalently associated on the cell surface. Eur J Immunol. 1995;25:1535–1540. doi: 10.1002/eji.1830250610. [DOI] [PubMed] [Google Scholar]
- 21.Greten TF, et al. Peptide–beta2-microglobulin–MHC fusion molecules bind antigen-specific T cells and can be used for multivalent MHC–Ig complexes. J Immunol Methods. 2002;271:125–135. doi: 10.1016/s0022-1759(02)00346-0. [DOI] [PubMed] [Google Scholar]
- 22.Slansky JE, et al. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC–peptide–TCR complex. Immunity. 2000;13:529–538. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 23.Chen C-H, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res. 2000;60:1035–1042. [PubMed] [Google Scholar]