

Abstract
Neuronal network oscillations are a unifying phenomenon in neuroscience research, with comparable measurements across scales and species. Cortical oscillations are of central importance in the characterization of neuronal network function in health and disease and are influential in effective drug development. Whilst animal in vitro and in vivo electrophysiology is able to characterize pharmacologically induced modulations in neuronal activity, present human counterparts have spatial and temporal limitations. Consequently, the potential applications for a human equivalent are extensive. Here, we demonstrate a novel implementation of contemporary neuroimaging methods called pharmaco‐magnetoencephalography. This approach determines the spatial profile of neuronal network oscillatory power change across the cortex following drug administration and reconstructs the time course of these modulations at focal regions of interest. As a proof of concept, we characterize the nonspecific GABAergic modulator diazepam, which has a broad range of therapeutic applications. We demonstrate that diazepam variously modulates θ (4–7 Hz), α (7–14 Hz), β (15–25 Hz), and γ (30–80 Hz) frequency oscillations in specific regions of the cortex, with a pharmacodynamic profile consistent with that of drug uptake. We examine the relevance of these results with regard to the spatial and temporal observations from other modalities and the various therapeutic consequences of diazepam and discuss the potential applications of such an approach in terms of drug development and translational neuroscience. Hum Brain Mapp, 2010. © 2009 Wiley‐Liss, Inc.
Keywords: γ‐amino butyric acid, oscillations, neuroimaging, magnetoencephalography, cortex, pharmacodynamic, gamma, beta, benzodiazipine, diazepam
INTRODUCTION
There are a range of established in vitro and in vivo animal methods for directly observing the effects of neuroactive substrates upon neuronal activity over time [e.g. Cunningham et al., 2003; Takita et al., 2007]. The potential for an equivalent noninvasive human method, which is able to resolve the temporal characteristics of spatially discrete neuronal activity as a result of neurochemical manipulation, is extensive. The use of functional neuroimaging to investigate drug responses has seen modest progress, largely due to the temporal limitations of the prevalent neuroimaging techniques. Consequently, the focus has been upon spatial reconstruction, whilst temporally oriented methods have been generally overlooked [Mehta and Richell, 2005].
Recent developments in magnetoencephalography (MEG) facilitate the accurate reconstruction of spatial and temporal characteristics of brain activity, concordant with functional magnetic resonance imaging (fMRI) [Singh et al., 2002] and invasive surgical recordings [Mamelak et al., 2002], respectively. In this work, we describe a novel method called “pharmaco‐MEG” which can spatially characterize oscillatory modulations as a consequence of drug action and reconstruct the temporal profile at regions of interest (ROI). As a proof of concept, we demonstrate its application to the whole cortex characterization of a well‐described GABAergic modulator of the benzodiazepine family.
γ‐Amino butyric acid (GABA) is the predominant inhibitory neurotransmitter in the mammalian brain [Whiting, 2006]. GABAA is the principal receptor subtype in the human brain and has a diverse subunit composition that interchangeably incorporates binding sites for many neuroactive substrates that mediate a variety of neuronal mechanisms [Nutt and Malizia, 2001; Wafford and Ebert, 2006]. The most clinically useful of the GABAA modulators are those that act upon the benzodiazepine‐binding sites, as reflected by their wide use in clinical practice [Alldredge et al., 2001]. The benzodiazepines have a diverse range of therapeutic applications: diazepam for example, has an affinity for several subunits and is prescribed as an anxiolytic, muscle relaxant, sedative, and anticonvulsant [Giusti and Arban, 1993]; whereas zolpidem is α1 subunit specific and is an effective sedative without the other effects at typical therapeutic doses [Lavoie et al., 1997].
A number of studies have used EEG and MEG as tools to study the effects of the benzodiazepines upon neuronal activity [Baker and Baker, 2003; Fingelkurts et al., 2004; Greenblatt et al., 1989, 1992, 2004; Jensen et al., 2005; Lucchesi et al., 2003; Romano‐Torres et al., 2002]. Several studies make task‐dependant observations using, for example, mismatch negativity [Kasai et al., 2002], motor tasks [Baker and Baker, 2003], median nerve stimulation [Haueisen et al., 2000], and eye‐closure [Ahveninen et al., 2007]. Whilst others focus upon intrinsic oscillatory changes either regionally [Greenblatt et al., 1989, 2004] or focally [Jensen et al., 2005]. However, while oscillations are a fundamental electrophysiological feature and temporal resolution is the principal advantage of these techniques, the continuous temporal profile of drug‐induced oscillatory change, until now, has not been demonstrated. The elevation in beta power as a result of benzodiazepine administration is a phenomenon that is well recognized in the EEG literature. In fact, the dominance of such features in a clinical setting often precludes an informative assessment. This elevation in cortical beta is a common feature in patients prescribed GABAA modulatory medications [Giusti and Arban, 1993] or where alcohol consumption is a regular factor [Rangaswamy et al., 2002].
Several studies have investigated the spatial distribution of benzodiazepine receptors, using autoradiography [Zezula et al., 1988], immunohistochemistry [Rudolph et al., 2001], or PET [Chang et al., 2005; Fedi et al., 2006]. Whilst these studies are able to determine the profile of binding across the cortex, their functional significance remains unclear. Ideally, one would use a technique that is able to obtain a whole cortex spatial image of neuronal modulation and reconstruct the temporal profile of all cortical regions in parallel.
Here, we use contemporary MEG techniques to determine the spatial distribution of changes in intrinsic neuronal oscillations following the administration of a low dose of diazepam. We then use group imaging to resolve the profile that is robust across the participant group and “virtual electrodes” (VEs) to reconstruct the temporal profile of oscillatory change in ROIs over the period of drug uptake. We present the pharmaco‐MEG profile of diazepam and discuss our findings in the context of the spatial distribution compared to receptor‐binding studies and the frequency and temporal profiles compared to other electrophysiology studies. We consider the spatiotemporal and pharmacodynamic profile with regard to the therapeutic implications of diazepam and explore the potential advantages of this approach in terms of drug development, neuronal network investigation, and translational research.
MATERIALS AND METHODS
Data Acquisition
A total of eight male subjects (seven right handed), with a mean age of 37 (range, 30–48) participated in the study; each of whom gave informed consent in accordance with the local ethics approval for the study. Participants were placed into a 275 channel MEG system in a supine position, for a duration of 60 min. At the start of the recording a low dose of diazepam (5 mg) was administered orally in a polysaccharide cased tablet, allowing delivery of the drug after 10–15 min [Moolenaar et al., 1980]. MEG data were acquired in a continuous 60‐min recording; at a sampling rate of 600 Hz, with head position determined continuously throughout the experiment via three‐head‐localization coils. Following the MEG recording, the three‐dimensional coil location, with respect to the scalp, was determined using a Polhemus Isotrack digitization system. Subsequently, data were coregistered with the individual's MRI (see Adjamian et al. [ 2004a] for details). The 60‐min recording period was divided into six identical epochs of 10 min. During each 10‐min period, participants were verbally instructed to perform a simple 30‐s isometric contraction of the right hand to localize the precentral gyrus hand area, a 30‐s period of “eye closure” and two brief (2 min) visual observation and discrimination tasks (results not reported here); these tasks served to maintain and confirm attention throughout and to provide verifiable anatomical localizations (see Placebo Control Experiments). Interleaved between the 5 min of tasks were 5 min of passive eyes‐open relaxation, which were used in the analyses described here.
Data Analysis
The 5 min of passive eyes‐open data from each of the six periods were used to determine the spatial distribution of intrinsic neuronal activity change. The initial period was considered as the baseline period, whilst the subsequent periods were regarded as “drug‐active” periods. Data were manually screened for eye blink and movement artifacts, which were routinely removed before SAM analysis. SAM analysis was conducted (see Synthetic Aperture Magnetometry Analysis), comparing each of the five drug‐active periods separately to the baseline period, thereby providing spatial reconstructions of the averaged cortical oscillatory changes at 10‐min intervals over the entire 60‐min duration. The SAM method requires that the frequency band of interest is predetermined. Therefore, we analyzed activity using the classical θ (4–7 Hz), α (7–14 Hz), β (15–25 Hz), and γ (30–80 Hz) frequency bands, in addition to analyzing in 10‐Hz frequency bands between 1 and 100 Hz to improve the observation of activity distributed between the classical frequency bands. Subsequent to the individual SAM analyses, group SAM [Singh et al., 2002] analysis was applied, which generates an image of the spatial distribution of activity that is consistent across the group (see Group SAM Analysis). These loci were used to determine ROIs, from which the temporal profile of oscillatory change was reconstructed in each individual (see Virtual Electrode Analysis). This approach means that each participant acts as their own individual control.
Synthetic Aperture Magnetometry Analysis
Analyses of oscillatory power change in this study were conducted at the source level, at ROIs determined using the SAM method [Hillebrand et al., 2005]. In brief, SAM uses a linearly constrained minimum variance beamformer algorithm [Van Veen et al., 1997] to estimate the electrical activity in each part of a predefined source space through a weighted sum of the sensor channels. A spatial filter is computed for each source location based on the minimization of the output power and with the constraint that the filter has unity gain at each location; effectively making the beamformer a special case of the generalized linear inverse (or Wiener) solution (Eq. 1). The underlying source current covariance matrix is diagonal, and the amplitude of its elements is estimated from the data, rather than sensor‐source space geometry as used in weighted minimum norm solutions. The generalized linear inverse, which gives the estimate of the distribution of underlying neuronal currents j, is given by
| (1) |
with b the measured field vector (1 × N), C D the data covariance matrix (N × N), C j the source current covariance matrix (M × M), L the lead field matrix (N × M), N the number of channels, and M the number of elements in the source space. In the beamformer formulation, the source current covariance matrix C j is assumed to be diagonal and the variance component σ corresponding to source space element i with lead field l i is estimated from:
| (2) |
Various parameters can be computed based on the beamformer output (Eq. 1) for a target location, such as the Neural Activity Index [Van Veen et al., 1997]. A volumetric spatial image of brain activity is created by estimating such an index of neuronal activity sequentially. Furthermore, active and passive experimental states can be defined for each target voxel and statistically comparisons made of the oscillatory power in predetermined frequency ranges between these states, forming so‐called Statistical Parametric Maps that highlight brain regions that were differentially activated between the active and passive states. Here, our active and periods were defined as the drug‐active (10–60‐min postdiazepam) and drug inactive (0–10‐min postdiazepam) periods, respectively. A pseudo‐t statistic for the entire brain was computed for the θ (4–7 Hz), α (7–14 Hz), β (15–25 Hz), and γ (30–80 Hz) for each drug active versus drug passive period. This provided a complete profile of oscillatory change across the duration of the experiment for each individual. Following the identification of robust activity across the group (see Group SAM Analysis), these loci were used for further analysis using VE reconstruction of activity at these ROIs (see Virtual Electrode Analysis).
Group SAM Analysis
In order for pharmaco‐MEG to be implemented as a successful tool for the development of pharmacological models, it is necessary to establish those changes in neuronal activity that are consistently present across the participant group. Group SAM analysis achieves this by normalizing individuals' anatomical MRIs to a template MRI [Singh et al., 2002], with the same normalization parameters subsequently applied to the corresponding SAM images of neuronal activity change. This determines the spatial distribution of oscillatory power change, in predetermined frequency bands, that are robust across individuals. Here, we used group SAM analyses to determine loci and frequency bands that showed robust modulation over the period of drug uptake. Subsequently, the corresponding coordinates of peak SAM activity in each ROI and frequency were explored using VE analysis.
Virtual Electrode Analysis
The VE approach makes use of the same constrained spatial filter as for SAM analysis, with the beamformer weights for each location computed for a predetermined set of coordinates. Here, our coordinates were determined for each individual SAM peak based upon corresponding group‐SAM analysis. These anatomically discrete fluctuations in a specific frequency band were visualized by band‐pass filtering the VE data at each location to the relevant frequency band (θ, α, β, or γ) to generate a ROI‐specific envelope of oscillatory power during drug uptake. Root‐mean‐square (RMS) values were computed for each envelope, normalized to maximum for each participant, and presented as individual and corrected group means for each ROI.
Placebo–Control Studies
Blinded placebo–control recordings were conducted in four participants. SAM analysis of drug‐active versus drug‐passive periods revealed no observable modulation of intrinsic oscillatory power at ROI either within or between participants over the duration of the study consistent with previous study recordings [e.g. Hall et al., 2004]. To directly compare the envelopes of oscillatory power in the diazepam and placebo conditions, we used specific tasks to identify ROIs in visual and motor cortex corresponding to observed effects (see Results). Specifically, we used SAM analysis to identify primary motor cortex (M1) using finger movement‐related beta oscillations (e.g. Jurkiewicz et al. [ 2006]) and visual cortex using eye‐closure‐induced alpha oscillations (e.g. Ahveninen et al. [ 2007]) and visual stimulus (grating) induced gamma oscillations (e.g. Hall et al. [ 2005]) At these loci, the motor beta, visual alpha, and visual gamma oscillations following administration of either diazepam or placebo were compared by reconstructing the envelope of the VE power using the method described in previous sections (Fig. 5).
Figure 5.
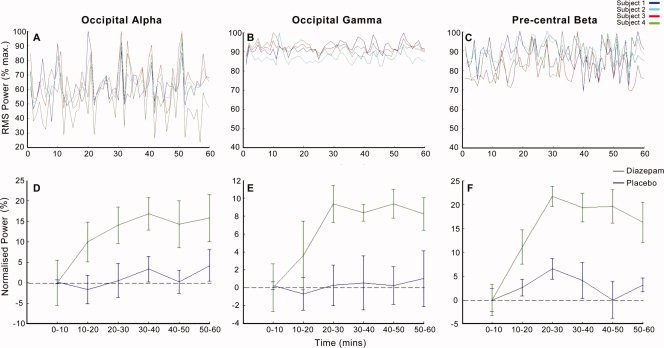
Placebo‐control Data. Envelopes of RMS oscillatory power during the 60‐min postplacebo administration at functionally derived SAM peaks. Data show alpha power in the visual cortex determined from eye‐closure induced alpha SAM peak (A and D), gamma power in visual cortex determined from visually induced gamma SAM peak (B and E), and beta power in motor cortex determined from finger movement induced beta (C and F). The top panel shows data (1 sample/minute) for four subjects' placebo data, while the bottom panel shows group mean over each 10 min period ±1.96 SEM for placebo versus diazepam at the same loci.
RESULTS
Consistent with the clinical EEG observations, there were large scale increases in oscillatory activity, dominated by the beta band. However, SAM revealed there to be a broad range of oscillatory changes across several regions of the cortex. Supplementary to beta frequency increases, there were increases in both the alpha and gamma frequencies and decreases in theta frequency. SAM analyses demonstrated that the profile of oscillatory change showed some variability, with a small number of loci that were robustly (between drug active periods) present only at the individual level (Fig. 1); whilst the majority of loci were robustly represented across the participant cohort.
Figure 1.
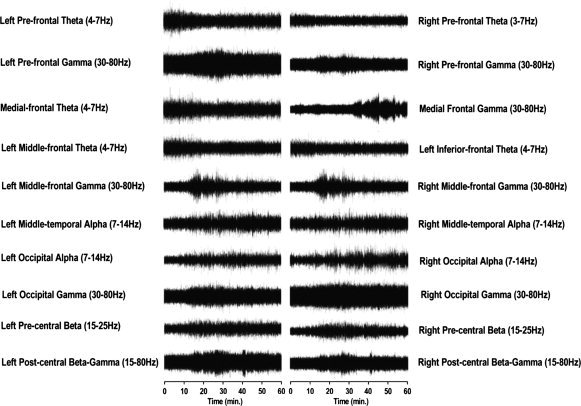
Virtual electrode reconstruction of the neuronal activity at peak loci in a representative individual. Traces reflect band pass filtered oscillatory power at peak frequencies over 60 min at loci of maximal power change, as determined from individual SAM analysis in a representative individual.
Group SAM analysis confirmed the profile of neuronal oscillatory change, although widespread, could be characterized in terms of region and frequency, with reliability across the group (Fig. 2). These analyses revealed increases in α, β, and γ power in frontal, temporal, precentral, postcentral, and occipital areas and decreases in frontal theta power. These changes typically reached their maxima between 20 and 40 min after drug intake (Fig. 3), consistent with the rapid absorption and bioavailability of oral diazepam [Moolenaar et al., 1980], after which oscillations reduced to a level still substantially elevated with respect to the baseline, consistent with rapid initial elimination rate, and long half‐life [Yamazaki et al., 2007]. Results from the visual discrimination tasks showed no significant change in participants' responses to stimuli over the duration of the experiment, suggesting that the observed results were not a consequence of attentional modulation. The results are summarized in Table I and described in detail below.
Figure 2.
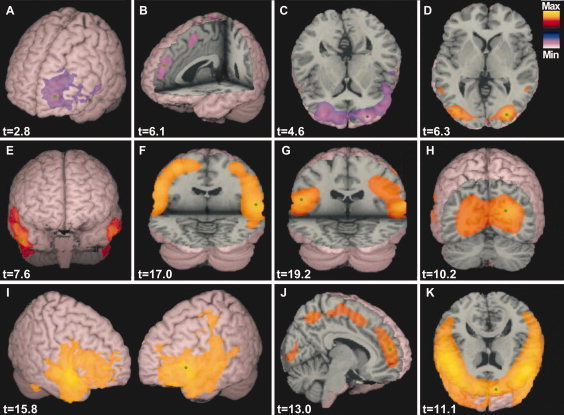
Group SAM images of oscillatory power change in θ (4–7 Hz), α (7–14 Hz), β (15–30 Hz), and γ (30–80 Hz) frequency ranges in response to diazepam administration. Images displayed are a comparison of the initial baseline period (0–10 min) and the final drug active period (50–60 min). Hot colors reflect an increase and cool colors a decrease in synchronous power, with pseudo‐t values displayed for the peak loci of each image (green dot); images are thresholded at 50% of maximum. The panel shows: unilateral decrease in theta in left middle frontal cortex (A), a decrease in medial frontal theta (B), bilateral decrease in prefrontal theta (C), bilateral increase in occipital alpha (D), bilateral increase in alpha in middle temporal cortex (E), bilateral increase in beta power in the pre‐central gyrus (F), bilateral increase in β–γ in the postcentral gyrus (G), bilateral increase in occipital gamma (H), bilateral increase in inferior frontal gamma (I), increase in medial frontal gamma (J), and a bilateral increase in prefrontal gamma (K). For further details see Table I.
Figure 3.
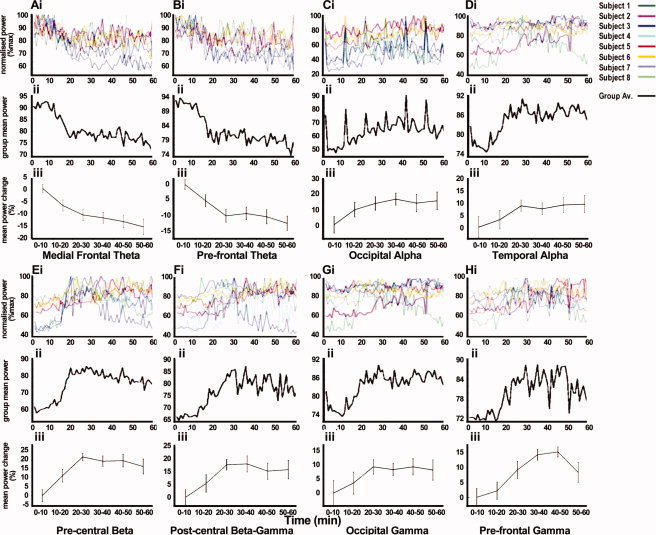
Pharmacodynamic envelopes of oscillatory power change at ROIs. RMS power in the θ, α, β, and γ frequency bands at peak loci. Data are displayed as 1 sample/minute for each individual, normalized to the maximal power (i), the group‐mean power displayed as 1 sample/minute (ii) and as the group‐mean power change over each 10 min period ±1.96 SEM, normalized to the baseline period (iii). Data are presented from loci that are consistent across participants and where bilateral or midline results are observed; all data shown are from the left hemisphere as an example. The data shown demonstrate the pharmacodynamic profiles of: medial frontal theta (A), prefrontal theta (B), occipital alpha (C), temporal alpha (D), precentral beta (E), postcentral β–γ (F), occipital gamma (G), and prefrontal gamma (H).
Table I.
Locations of the peaks of activation in each of the frequency bands identified in the group SAM images, amplitude of power change (pseudo‐t), and Talairach coordinates
| Frequency (Hz) | Cortical location | Power change (t) | Talairach coordinates (x, y, z) | ||
|---|---|---|---|---|---|
| Theta (4–7 Hz) | Superior frontal gyrus (left) | 4.6 | 15.1 | 51.2 | 36.0 |
| Superior frontal gyrus (right) | 3.5 | −16.1 | 64.3 | 24.0 | |
| Medial frontal gyrus (central) | 6.1 | 3.0 | 52.2 | 36.0 | |
| Middle frontal gyrus (left) | 2.8 | −39.2 | 52.2 | 31.0 | |
| Alpha (7–14 Hz) | Inferior occipital gyrus (left) | 5.7 | −33.1 | −93.4 | −18.0 |
| Inferior occipital gyrus (right) | 6.3 | 30.1 | −90.4 | −15.0 | |
| Middle temporal gyrus (left) | 2.9 | −57.2 | 6.0 | −12.0 | |
| Middle temporal gyrus (right) | 3.0 | 63.2 | 6.0 | −24.0 | |
| Beta (15–25 Hz) | Precentral gyrus (left) | 14.2 | −54.2 | −6.0 | 30.0 |
| Precentral gyrus (right) | 17.0 | 57.2 | −7.0 | 20.0 | |
| Beta/gamma (15–80 Hz) | Postcentral gyrus (left) | 19.2 | 60.2 | −12.2 | 18.0 |
| Postcentral gyrus (right) | 16.8 | 63.2 | −24.1 | 23.0 | |
| Gamma (30–80 Hz) | Superior frontal gyrus (left) | 11.1 | −23.1 | 55.2 | 15.0 |
| Superior frontal gyrus (right) | 12.4 | 18.1 | 54.2 | 18.0 | |
| Medial frontal gyrus (central) | 13.0 | 6.0 | 51.2 | 15.0 | |
| Inferior frontal gyrus (left) | 15.8 | −48.2 | 27.1 | 6.0 | |
| Inferior frontal gyrus (right) | 10.6 | 57.2 | 18.1 | −6.0 | |
| Middle occipital gyrus (left) | 9.4 | −24.1 | −84.3 | 0.0 | |
| Middle occipital gyrus (right) | 10.2 | 24.1 | −84.3 | 0.0 | |
Sensorimotor Cortex
The administration of diazepam elicited large power increases in the beta band (15–25 Hz) in the precentral gyrus of all participants (Fig. 2F). The peak locus was typically in the hand region of the motor cortex, verified by isometric contraction (Fig. 4). A gradual beta power increase was observed, accompanied by a transient increase in gamma power that coincided with maximal beta power at around 20–30 min (Fig. 3E). These data show a mean beta power after increase after 60 min of 16.3% ± 4.21% (P < 0.0005). A SAM peak in the postcentral gyrus was observed in broad‐band beta and gamma (15–80 Hz) frequencies (Fig. 2G), which typically reached maximum at around 30 min (Fig. 3F). These data show a mean β–γ power increase, after 60 min, of 13.88% ± 4.62% (paired t‐test P < 0.005).
Figure 4.
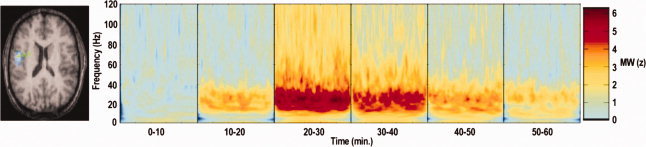
Time‐frequency representation of activity in the hand‐motor area in response to diazepam uptake. SAM analysis of beta power during isometric contraction of the right hand used to localize right primary motor cortex hand area (pseudo‐t = 2.9). The SAM peak (left) used to direct virtual electrode analysis using time frequency spectrograms (Morlett–Wavelet Mann–Whitney) to compare the activity in drug active phases with baseline in a representative individual (note a z‐value of 1.96 corresponds to P < 0.05).
Occipital Cortex
SAM analysis revealed an increase in alpha (7–14 Hz) activity bilaterally in occipital cortex in all participants (Fig. 2D). The spatial localization of the alpha peaks was consistent with those observed during eye closure (Supporting information Fig. 1), which can be seen as an increase in the alpha amplitude above the baseline throughout the duration of drug uptake (Fig. 3C). Occipital alpha power appears to increase gradually over time, reaching maximum typically at around 30 min, with a mean alpha power increase after 60 min of 15.83% ± 3.60% (paired t‐test P < 0.0005). SAM analysis revealed further large increases in occipital gamma (30–80 Hz) power bilaterally in the region of primary visual cortex of all participants (Fig. 2H). This increase was gradual, reaching maximum at ∼20–30 min and was sustained at increased amplitude for the full duration (Fig. 3G). These data show a mean gamma power increase after 60 min of 8.26% ± 3.61% (paired t‐test P < 0.001).
Temporal Cortex
SAM analysis reliably revealed that activity in the middle temporal cortex was restricted to the alpha frequency range (Fig. 2E), where power increases typically showed a gradual power increase, reaching maximum after ∼30 min (Fig. 3D). These data show a mean alpha power increase after 60 min of 9.60% ± 3.63% (paired t‐test P < 0.001).
Frontal Cortex
Frontal regions showed widespread reductions in oscillatory power in theta (4–7 Hz) band, which could be localized to a number of distinct regions. Prefrontal regions showed consistent bilateral reduction in oscillatory power (Fig. 2C), typically reaching maximal reduction after ∼20 min (Fig. 3B). Medial frontal regions showed power reduction (Fig. 2B), with a similar temporal profile to that observed in prefrontal regions (Fig. 3A). Finally, a unilateral reduction in theta power in the left middle/inferior frontal region (Fig. 2A), was evident in five of the eight participants (e.g. Fig. 1), although it appeared in the right hemisphere of one subject; possibly a reflection of the handedness of this participant. These data show a mean theta power decrease after 60 min of 12.53% ± 2.32% in the prefrontal cortex (paired t‐test P < 0.0005) and 15.30% ± 3.13% in the medial frontal cortex (paired t‐test P < 0.0005).
In addition to the reductions in theta power across the frontal cortex, there were observable increases in gamma power over the duration of the experiment. These increases were observed in all participants in bilateral prefrontal regions (Fig. 2K), typically reaching maximal amplitude after 30 min (Fig. 3H). Further frontal gamma frequency power increases were observed in medial frontal (Fig. 2J) and inferior frontal (Fig. 2I) regions. However, the location of these peaks was only reliably evident in five of the eight participants (e.g. Fig. 1). These data show a mean gamma power increase after 60 min of 8.00% ± 3.21% in the prefrontal cortex (paired t‐test P < 0.005).
Placebo–Control Data
SAM analysis of placebo–control data revealed no peaks of activity that were robust over the period of placebo uptake, either within of between participants. Reconstruction of beta activity in precentral cortex, alpha, and gamma activity in occipital cortex reveal that there are intrinsic fluctuations in power over the duration of the experiment. However, these display no robust or sustained profile over time and are variable between participants (Fig. 5).
DISCUSSION
It is a challenge for neuroimaging to reliably characterize neuronal activity change in response to neurochemical modulation. Specifically, whilst the spatial and temporal characteristics are readily discernible in isolation, the ability to observe these features in parallel is of fundamental importance to optimize application and translation of these methods. Here, we provide a preliminary solution in the form of pharmaco‐MEG, based upon an established beamforming‐based approach.
Oscillatory power changes following diazepam administration are observed across several cortical regions, varying in frequency, amplitude, and direction of power change. The spatial pattern of oscillatory change is consistent with observed GABAA receptor density in both animal [Pirker et al., 2000; Rudolph et al., 2001] and human [Fedi et al., 2006; Malizia et al., 1998; Zezula et al., 1988] studies. Whilst there are numerous levels of complexity between the receptor and networks of neurons, evidence suggests that MEG oscillations directly reflect receptor‐mediated changes in neuronal firing [Jensen et al., 2005], attributable to the regional cortical homogeneity, and spatial resolution of MEG, which affords a direct comparison with local network recordings [Hall et al., 2005].
It seems reasonable to assume that intrinsic oscillations, observable using MEG, can reflect information about receptor profiles in the cortex. However, given that oscillatory changes occur across the frequency range, with spatial overlap, in opposing directions and with possible involvement of deeper sources, a direct quantifiable relationship between receptor density and oscillatory power appears indeterminable with the current approach. However, the profile of synchronous power change in the prominent (β–γ) frequency range exhibits strong spatial similarities when compared with PET receptor occupancy studies (Fig. 6), where frontal, sensorimotor, temporal, and occipital cortices exhibit highest GABAA receptor binding [Chang et al., 2005; Fedi et al., 2006]. Here, we observe consistent changes across these regions, as discussed in further detail in the following sections.
Figure 6.
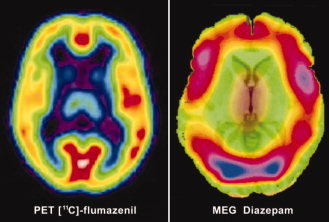
Comparison of pharmaco‐MEG and PET. Comparative image of PET receptor density mapping of benzodiazepine receptors using [11C]‐flumazenil and MEG activity in the β–γ (15–80 Hz) frequency range following diazepam administration. This image is partially composed (left panel) of a modified image from the study by Fedi et al. [ 2006].
Sensorimotor Cortex
Sensorimotor beta oscillations are believed to arise in the primary motor cortex (M1) [Baker et al., 1997; Murthy and Fetz, 1996], whilst the mu rhythm (10 Hz) is believed to originate in the somatosensory cortex [Salmelin and Hari, 1994]. Here, we observe that diazepam administration increases beta power, without an associated change in the mu rhythm, consistent with previous findings [Jensen et al., 2005]. This suggests that the neuronal population that mediates sensorimotor mu rhythm is a separate, non‐GABAergically mediated, network to the GABAergically driven network that generates the motor beta rhythm. The observation of elevated gamma power in somatosensory cortex is consistent with the findings that both somatosensory cortex gamma and motor cortex beta rhythms are mediated by GABAergic modulation of glutamatergic pyramidal cells [Roopun et al., 2006; Yamawaki et al., 2008], which contribute to the majority of the observed MEG signal [Murakami and Okada, 2006].
In response to limb movement, there is a reduction in power in both the beta and mu frequencies, believed to be related to neuronal activation [Pfurtscheller and Berghold, 1989]. Following movement termination, mu rhythm returns gradually to baseline, whilst beta power consistently exceeds premovement baseline levels [Pfurtscheller et al., 1996]. This increase in power is suggested to reflect transient inhibition of motor cortex [Salmelin and Makela, 1995] consistent with a recalibration of motor systems following movement [Kilner et al., 1999].
The persistently elevated beta power observed here is indicative of elevated local inhibitory network activity, consistent with the muscle relaxant properties of diazepam, and its use in the treatment of centrally mediated muscular tone disorders [Elkassabany et al., 2006]. This elevated inhibition is likely to mediate muscular tone via reduced beta coherence between muscle and motor cortex, as observed using EMG‐EEG measures [Baker and Baker, 2003].
The efficacy of benzodiazipine inhibitory action on motor‐cortical networks [Baker and Baker, 2003; Baker et al., 1997; Fingelkurts et al., 2004; Jensen et al., 2005] represents an attractive mechanism for the anticonvulsant properties of diazepam. The nature of epileptogenic activity, whereby high‐frequency neuronal activity results in seizure and in some cases convulsions, is consistent with a GABAA receptor‐mediated increase in neuronal firing refractory period resulting in reduced firing frequency of elevated amplitude in sensorimotor cortex. However, the ubiquitous CNS distribution of GABAA receptors, including other cortical regions and deeper structures, make it unclear whether the mechanism of anticonvulsant action mediated by the benzodiazepines is a focal or distributed process.
The spatial coexistence of beta and gamma activity in somatosensory cortex, whilst consistent with recent in vitro findings [Roopun et al., 2006], contradicts the suggestion that human beta rhythm measured using MEG is analogous to gamma rhythm observed in vitro [Jensen et al., 2005]; particularly as beta is observed in M1 in vitro and is elevated by GABAergic modulation [Yamawaki et al., 2008]. It is increasingly evident that beta and gamma oscillations are observed both interchangeably [Whittington et al., 2000] and in parallel [Roopun et al., 2006] in a single cortical region. Here, we observe that the hand area of the motor cortex, identified by isometric contraction [Taniguchi et al., 2000], is characterized by a progressive elevation in beta power, with an increase in gamma frequency only at maximal diazepam concentrations.
Visual Cortex
The predominant oscillation in the visual cortex is the alpha rhythm, a focus of electrophysiological research since reported by Berger in 1929. Although the gross dynamics of visual alpha are reasonably well defined, the physiological basis and functional significance are not entirely certain. Alpha is believed to be modulated by a thalamo‐cortico‐thalamic reentrant network, governed by GABAergic neurons of both the thalamus and cortex [Klimesch et al., 2007]. Alpha oscillations are directly influenced by input to the visual cortex; best demonstrated by an increase in alpha power during eye closure. Recent work by Ahveninen et al. [ 2007] demonstrated a reduction in occipital alpha power, particularly during eye‐closure, in participants administered the benzodiazepine lorezepam. Here, we describe a reduction in eye‐closure induced alpha power change during the diazepam condition (mean reduction 17.80%) compared to both baseline and placebo (Figs. 3 and 5; Supporting information Fig. 1). However, the observed increase in the intrinsic baseline alpha following diazepam administration is, surprisingly, in contrast to previous observations using lorazepam, [Ahveninen et al., 2007; Fingelkurts et al., 2004] or diazepam [Saletu et al., 1988] where reductions in alpha power were observed. These paradoxical observations are likely the consequence of dose‐dependent variations in specificity, particularly as the dose of diazepam used here is lower than those used in previous studies; this is of particular importance given the overall lack of specificity of these drugs for GABA‐A subunits. Nevertheless, what appears to be consistent is that visual alpha oscillations are under the direct influence of GABAergic modulation [Ahveninen et al., 2007; Klimesch et al., 2007].
Gamma oscillations in the primary visual cortex are observed in both animals [Gray and McCormick, 1996; Kreiter and Singer, 1996; Logothetis et al., 2001; Siegel and Konig, 2003] and humans [Adjamian et al., 2004b; Hadjipapas et al., 2007; Hall et al., 2005; Keil et al., 1999]. Visual gamma is implicated in visual feature integration [Gray and McCormick, 1996], object recognition [Tallon‐Baudry and Bertrand, 1999], and selective attention [Fell et al., 2003]. However, an increase in gamma power occurs in response to basic visual stimuli, such as gratings, and is directly dependant upon simple characteristics such as contrast [Hall et al., 2005] and spatial frequency [Adjamian et al., 2004b; Hadjipapas et al., 2007]. These data suggest that ongoing gamma oscillations in the primary visual cortex are GABAergically mediated, consistent with recent observations [Muthukumaraswamy et al., 2009].
Here, we show an increase in the intrinsic gamma power in primary visual cortex following diazepam, consistent with GABAergic interneuron‐modulated drive [Whittington and Traub, 2003], and with the model suggesting GABAergic modulation of visual gamma [Jensen et al., 2002]; whilst allowing for the possibility that modulation may also occur in response to more complex cognitively dependant modulations of the visual system [e.g. Tallon‐Baudry and Bertrand, 1999].
Temporal Cortex
The role of increased alpha oscillatory power, observed here, in the middle temporal cortex is unclear, although the role of this brain region in memory [Gonsalves et al., 2005] is of interest with regard to the amnestic properties of the benzodiazipines and other GABAA modulators. Alpha modulation is consistent with memory processing [Pesonen et al., 2007], known to be disrupted at various stages of encoding and retrieval by GABAergic modulators such as alcohol [Krause et al., 2002].
Increased temporal lobe alpha activity as a consequence of GABAergic modulation is consistent with the observed reduction in temporal lobe benzodiazepine binding in patients with generalized anxiety disorder [Tiihonen et al., 1997], for which benzodiazepines are a common therapeutic intervention. Similarly, changes in temporal lobe GABAA subunit expression in both temporal lobe epilepsy [Sperk et al., 2004] and absence seizures [Li et al., 2006], neither involving motor components, suggest that anticonvulsant effects via GABAergic modulation may be a globally mediated mechanism.
Frontal Cortex
EEG studies demonstrate that frontal midline theta reflects elevated anxiety levels, which are reduced following the administration of an anxiolytic medication [Suetsugi et al., 1998], with coincident physiological effects such as the galvanic skin response [Aftanas et al., 2002]. Reduced frontal theta power, observed here in response to diazepam is consistent with its anxiolytic properties as previously reported in the EEG following administration of the anxiolytic clomipramine [Suetsugi et al., 1998]. Sleep deprivation, known to reduce anxiety in patients suffering from anxiety disorders [Pflug, 1978], also modulates theta oscillatory power in frontal regions [Cajochen et al., 1999]. GABAA receptors have an established role in anxiety [Nutt and Malizia, 2001] with specific mediation through α2 receptors [Low et al., 2000; Rudolph et al., 2001]. These receptors are localized to the hippocampi, amygdalae, and basal forebrain [Fritschy and Mohler, 1995], whose connectivity with the cortex is important in the manifestation of anxiety. Observations in vivo of elevated hippocampal theta during “fear” in conditioned animals [Moita et al., 2003] is supported by human MEG studies demonstrating event related theta in response to unpleasant visual stimuli [Nishitani, 2003]. Connectivity between the hippocampi and prefrontal cortical regions is reflected by a phase relationship at theta frequency [Siapas et al., 2005]. Consequently, modulation of frontal theta, as observed here, may be an indication of hippocampal activity as a direct consequence of this phase relationship [Jensen and Lisman, 2005].
The observation of spatially coincident changes in theta and gamma power at several locations of the frontal cortex is suggestive of involvement of subcortical structures through an interaction of short‐ and long‐range connectivity, which appear as a function of scale [von Stein and Sarnthein, 2000]. Furthermore, spatially colocalized changes in theta and gamma are observed during memory paradigms [Burgess and Ali, 2002; Schack et al., 2002], consistent with the amnestic properties observed in the benzodiazepines at various doses [Bustos et al., 2006; Krause et al., 2002]. A recent study by Pesonen et al. [ 2007] using EEG, revealed increased theta and decreased alpha and beta power with increased memory load. Although these results were not presented in terms of spatial location, they are consistent with the decreased theta and increased alpha and beta following administration of an amnestic drug.
Placebo–Control Data
The placebo–control recordings here offer several useful insights into the efficacy of pharmaco‐MEG as a tool for further exploration of neuronal activity. First, the clear difference between diazepam‐induced changes in oscillatory power and placebo control suggest that typically low doses of a neuromodulator can be differentiated from intrinsic fluctuations in oscillatory power. Second, whilst placebo–control experiments do not show any characteristic sustained trend of oscillator modulation over the period of the experiment, there are clear fluctuations in intrinsic oscillations over the course of the experiment. This has potential implications for differentiating lower doses (e.g. microdoses) of a particular drug. Furthermore, over shorter durations, the “natural” fluctuations in oscillatory power may prove more difficult to disentangle from drug‐induced effects. Finally, there is a strong likelihood that these intrinsic fluctuations are attentionally modulated and therefore either shorter experiments or more sensitive measures of attention would be required to avoid or account for these effects.
CONCLUSION
The principal advantage of this approach is its ability to reconstruct the temporal profile of local neuronal activity at any point within the cortex. Therefore, we are provided with a representation of oscillatory power change in ROIs during drug uptake. This approach represents a more spatially refined and flexible method of reconstructing the neural pharmacodynamic profile, than those previously employed [Greenblatt et al., 2004]. The potential of this method could be fully realized through the observation of more specific therapeutic agents, such as those effective upon receptor subtypes. Whilst this pharmaco‐MEG approach is based upon the SAM beamforming method, it is noteworthy to mention that other approaches such as dynamic imaging of coherent sources are likely to offer similar outcomes for pharmacological investigation. Pharmaco‐MEG allows us to reconstruct the neuronal activity in response to pharmacological manipulation at a specific region of interest in the brain. This method is flexible, allowing a ROI to be determined in postprocessing, thereby offering the potential to characterize the specific pharmacodynamic profile of any cortical region. Furthermore, as a direct measure of neuronal activity, it is arguably less susceptible to the vascular, hemodynamic, and metabolic changes that pose potential difficulties for alternative pharmacological neuroimaging approaches such as pharmaco‐fMRI.
The continuous high‐temporal resolution approach used here affords the ability to generate accurate pharmacodynamic profiles of a range of similar drugs against which candidate agents can be compared (see Supporting information Fig. 2). Additionally, the time/frequency interrelationship affords us the opportunity to make more accurate inference about neuronal network activity and the comparison with animal in vitro/in vivo models. Furthermore, whilst this oral‐drug implementation focuses upon extended durations, IV administration typically elicits an immediate effect, for which high‐temporal resolution is an essential feature. Finally, whilst this proof‐of‐concept focuses on simple measures of oscillatory amplitude, the potential to extend the VE implementation to investigate the relationship between cortical regions involved have great potential.
As a proof‐of‐concept, this study highlights several potential limitations of the approach that are worthy of further consideration: First, as we observed with our placebo–control data, natural drifts in oscillatory power occur in all frequency bands. These occur unpredictably, without consistency either within or between participants and under conditions where attempts were made to maintain attention; possibly reflecting the need for more sensitive measures of attentional state. Second, spatial homogeneity is subject to greater variability than other MEG studies as spatial inhomogeneity exists in both the anatomical structure and the receptor distribution/density. This is potentially problematic when considered in the context of MEG source reconstruction and, in particular, group analysis where such variability can influence the robustness of localization. Finally, a consequence of the relatively high‐spatial resolution of the SAM beamforming approach, and indeed MEG generally, is insensitivity to deeper sources. The consequence in this case is that it is not possible to determine the inevitable effects of the drug on deep structures and, similarly, whether observations are the result of direct modulation of cortically located receptors or projections from distal structures; inevitably both are represented here. An integrative approach, using receptor density measures, such as PET to guide pharmaco‐MEG, has clear potential to circumvent many of the limitations discussed earlier. An attractive potential application of the approach presented here is in drug development. Whilst determination of the individual therapeutic characteristics for such a broad‐acting drug are not possible in isolation, a combined approach that determines the shared characteristics of a range of agents with a common therapeutic application offers a potentially invaluable tool (Supporting information Fig. 2). Such a tool could provide the information necessary to determine the characteristics of an effective substrate, whilst concurrently identifying potentially unwanted side effects. Furthermore, it is apparent that there is wide scope to apply more complex signal‐processing methods to investigate the profile of pharmacologically induced neuronal change and interactions between ROIs; an approach likely to offer more valuable information regarding the mechanistic bases of drug action.
This approach provides a method by which to determine the pharmacodynamic time course of drug action in a specific local cortical network, illustrated here by the mean uptake profiles of diazepam (Fig. 3). When combined with functional task localization, the pharmacodynamic profile can be determined for a functionally relevant ROI. The dose of diazepam used in this study was a low‐therapeutic dose that elicited no pronounced therapeutic effects in the participants tested, yet oscillatory power was increased by as much as 53% (precentral beta) in some participants. This raises the questions: what dose is detectable using this MEG method? And is it possible to detect changes at microdose levels? When combined with a therapeutic profile, the reduced risk factors associated with subtherapeutic observations have great potential value in the refinement and testing of potential drug candidates. Finally, this preliminary work demonstrates the potential of MEG‐based techniques to determine the pharmacodynamic profile of neuronal activity in the intact human brain as a consequence of drug modulation. This approach provides a direct comparator for animal electrophysiology measures and, consequently, has the potential to make a strong contribution to translational neuroscience research.
Supporting information
Additional Supporting Information may be found in the online version of this article.
Supplementary Figures
Re‐use of this article is permitted in accordance with the Terms and Conditions set out at http://wileyonlinelibrary.com/onlineopen#OnlineOpen_Terms
REFERENCES
- Adjamian P, Barnes GR, Hillebrand A, Holliday IE, Singh KD, Furlong PL, Harrington E, Barclay CW, Route PJG ( 2004a): Co‐registration of magnetoencephalography with magnetic resonance imaging using bite‐bar‐based fiducials and surface‐matching. Clin Neurophysiol 115: 691–698. [DOI] [PubMed] [Google Scholar]
- Adjamian P, Holliday IE, Barnes GR, Hillebrand A, Hadjipapas A, Singh KD ( 2004b): Induced visual illusions and gamma oscillations in human primary visual cortex. Eur J Neurosci 20: 587–592. [DOI] [PubMed] [Google Scholar]
- Aftanas LI, Varlamov AA, Pavlov SV, Makhnev VP, Reva NV ( 2002): Time‐dependent cortical asymmetries induced by emotional arousal: EEG analysis of event‐related synchronization and desynchronization in individually defined frequency bands. Int J Psychophysiol 44: 67–82. [DOI] [PubMed] [Google Scholar]
- Ahveninen J, Lin FH, Kivisaari R, Autti T, Hamalainen M, Stufflebeam S, Belliveau JW, Kahkonen S ( 2007): MRI‐constrained spectral imaging of benzodiazepine modulation of spontaneous neuromagnetic activity in human cortex. Neuroimage 35: 577–582. [DOI] [PubMed] [Google Scholar]
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, Gottwald MD, O'Neil N, Neuhaus JM, Segal MR, Lowenstein DH ( 2001): A comparison of lorazepam, diazepam, and placebo for the treatment of out‐of‐hospital status epilepticus. N Eng J Med 345: 631–637. [DOI] [PubMed] [Google Scholar]
- Baker MR, Baker SN ( 2003): The effect of diazepam on motor cortical oscillations and corticomuscular coherence studied in man. J Physiol Lond 546: 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SN, Olivier E, Lemon RN ( 1997): Coherent oscillations in monkey motor cortex and hand muscle EMG show task‐dependent modulation. J Physiol Lond 501: 225–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AP, Ali L ( 2002): Functional connectivity of gamma EEG activity is modulated at low frequency during conscious recollection. Int J Psychophysiol 46: 91–100. [DOI] [PubMed] [Google Scholar]
- Bustos SG, Maldonado H, Molina VA ( 2006): Midazolam disrupts fear memory reconsolidation. Neuroscience 139: 831–842. [DOI] [PubMed] [Google Scholar]
- Cajochen C, Khalsa SBS, Wyatt JK, Czeisler CA, Dijk DJ ( 1999): EEG and ocular correlates of circadian melatonin phase and human performance decrements during sleep loss. Am J Physiol Regulat Integr Comp Physiol 277: R640–R649. [DOI] [PubMed] [Google Scholar]
- Chang YS, Jeong JM, Yoon YH, Kang WJ, Lee SJ, Lee DS, Chung JK, Lee MC ( 2005): Biological properties of 2′‐[F‐18]fluoroflumazenil for central benzodiazepine receptor imaging. Nucl Med Biol 32: 263–268. [DOI] [PubMed] [Google Scholar]
- Cunningham MO, Davies CH, Buhl EH, Kopell N, Whittington MA ( 2003): Gamma oscillations induced by kainate receptor activation in the entorhinal cortex in vitro. J Neurosci 23: 9761–9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkassabany N, Tetzlaff JE, Argalious M ( 2006): Anesthetic management of a patient with stiff person syndrome. J Clin Anesth 18: 218–220. [DOI] [PubMed] [Google Scholar]
- Fedi M, Berkovic SF, Marini C, Mulligan R, Tochon‐Danguy H, Reutens DC ( 2006): A GABAA receptor mutation causing generalized epilepsy reduces benzodiazepine receptor binding. Neuroimage 32: 995–1000. [DOI] [PubMed] [Google Scholar]
- Fingelkurts AA, Fingelkurts AA, Kivisaari R, Pekkonen E, Ilmoniemi RJ, Kahkonen S ( 2004): The interplay of lorazepam‐induced brain oscillations: Microstructural electromagnetic study. Clin Neurophysiol 115: 674–690. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H ( 1995): GABAA‐receptor heterogeneity in the adult‐rat brain—Differential regional and cellular‐distribution of 7 major subunits. J Comp Neurol 359: 154–194. [DOI] [PubMed] [Google Scholar]
- Giusti P, Arban R ( 1993): Physiological and pharmacological bases for the diverse properties of benzodiazepines and their congeners. Pharmacol Res 27: 201–215. [DOI] [PubMed] [Google Scholar]
- Gonsalves BD, Kahn I, Curran T, Norman KA, Wagner AD ( 2005): Memory strength and repetition suppression: Multimodal imaging of medial temporal cortical contributions to recognition. Neuron 47: 751–761. [DOI] [PubMed] [Google Scholar]
- Gray CM, McCormick DA ( 1996): Chattering cells: Superficial pyramidal neurons contributing to the generation of synchronous oscillations in the visual cortex. Science 274: 109–113. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ ( 1992): Pharmacology of benzodiazepine hypnotics. J Clin Psychiatr 53: 7–13. [PubMed] [Google Scholar]
- Greenblatt DJ, Ehrenberg BL, Gunderman J, Locniskar A, Scavone JM, Harmatz JS, Shader RI ( 1989): Pharmacokinetic and electroencephalographic study of intravenous diazepam, midazolam, and placebo. Clin Pharmacol Therapeut 45: 356–365. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ, Ehrenberg BL, Culm KE, Scavone JM, Corbett KE, Friedman HL, Harmatz JS, Shader RI ( 2004): Kinetics and EEG effects of midazolam during and after 1‐minute, 1‐hour, and 3‐hour intravenous infusions. J Clin Pharmacol 44: 605–611. [DOI] [PubMed] [Google Scholar]
- Hadjipapas A, Adjamian P, Swettenham JB, Holliday IE, Barnes GR ( 2007): Stimuli of varying spatial scale induce gamma activity with distinct temporal characteristics in human visual cortex. Neuroimage 35: 518–530. [DOI] [PubMed] [Google Scholar]
- Hall SD, Barnes GR, Hillebrand A, Furlong PL, Singh KD, Holliday IE ( 2004): Spatio‐temporal imaging of cortical desynchronization in migraine visual aura: A magnetoencephalography case study. Headache 44: 204–208. [DOI] [PubMed] [Google Scholar]
- Hall SD, Holliday IE, Hillebrand A, Singh KD, Furlong PL, Hadjipapas A, Barnes GR ( 2005): The missing link: Analogous human and primate cortical gamma oscillations. Neuroimage 26: 13–17. [DOI] [PubMed] [Google Scholar]
- Haueisen J, Heuer T, Nowak H, Liepert J, Weiller C, Okada Y, Curio G ( 2000): The influence of lorazepam on somatosensory‐evoked fast frequency (600 Hz) activity in MEG. Brain Res 874: 10–14. [DOI] [PubMed] [Google Scholar]
- Hillebrand A, Singh KD, Holliday IE, Furlong PL, Barnes GR ( 2005): A new approach to neuroimaging with magnetoencephalography. Hum Brain Mapp 25: 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O, Lisman JE ( 2005): Hippocampal sequence‐encoding driven by a cortical multi‐item working memory buffer. Trends Neurosci 28: 67–72. [DOI] [PubMed] [Google Scholar]
- Jensen O, Hari R, Kaila K ( 2002): Visually evoked gamma responses in the human brain are enhanced during voluntary hyperventilation. Neuroimage 15: 575–586. [DOI] [PubMed] [Google Scholar]
- Jensen O, Goel P, Kopell N, Pohja M, Hari R, Ennentrout B ( 2005): On the human sensorimotor‐cortex beta rhythm: Sources and modeling. Neuroimage 26: 347–355. [DOI] [PubMed] [Google Scholar]
- Jurkiewicz MT, Gaetz WC, Bostan AC, Cheyne D ( 2006): Post‐movement beta rebound is generated in motor cortex: Evidence from neuromagnetic recordings. Neuroimage 32: 1281–1289. [DOI] [PubMed] [Google Scholar]
- Kasai K, Yamada H, Kamio S, Nakagome K, Iwanami A, Fukuda M, Yumoto M, Itoh K, Koshida I, Abe O, Kato N ( 2002): Do high or low doses of anxiolytics and hypnotics affect mismatch negativity in schizophrenic subjects? An EEG and MEG study. Clin Neurophysiol 113: 141–150. [DOI] [PubMed] [Google Scholar]
- Keil A, Mueller MM, Gruber T, Elbert T ( 1999): Oscillatory brain activity is modulated by affective properties of visual stimuli. Psychophysiology 36: S66. [Google Scholar]
- Kilner JM, Baker SN, Salenius S, Jousmaki V, Hari R, Lemon RN ( 1999): Task‐dependent modulation of 15‐30 Hz coherence between rectified EMGs from human hand and forearm muscles. J Physiol Lond 516: 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimesch W, Sauseng P, Hanslmayr S ( 2007): EEG alpha oscillations: The inhibition‐timing hypothesis. Brain Res Rev 53: 63–88. [DOI] [PubMed] [Google Scholar]
- Krause CM, Aromaki A, Sillanmaki L, Astrom T, Alanko K, Salonen E, Peltola O ( 2002): Alcohol‐induced alterations in ERD/ERS during an auditory memory task. Alcohol 26: 145–153. [DOI] [PubMed] [Google Scholar]
- Kreiter AK, Singer W ( 1996): Stimulus‐dependent synchronization of neuronal responses in the visual cortex of the awake macaque monkey. J Neurosci 16: 2381–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie AM, Tingey JJ, Harrison NL, Pritchett DB, Twyman RE ( 1997): Activation and deactivation rates of recombinant GABAA receptor channels are dependent on alpha‐subunit isoform. Biophys J 73: 2518–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HF, Kraus A, Wu J, Huguenard JR, Fisher RS ( 2006): Selective changes in thalamic and cortical GABAA receptor subunits in a model of acquired absence epilepsy in the rat. Neuropharmacology 51: 121–128. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A ( 2001): Neurophysiological investigation of the basis of the fMRI signal. Nature 412: 150–157. [DOI] [PubMed] [Google Scholar]
- Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, Fritschy JM, Rulicke T, Bluethmann H, Mohler H, Rudolph U ( 2000): Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290: 131–134. [DOI] [PubMed] [Google Scholar]
- Lucchesi LM, Braga NI, Pompeia S, Manzano GM, Tufik S ( 2003): Is the increase of the amplitude of the auditory N1‐P2 an specific effect of the hypnotic zolpidem? Sleep 26: A83–A84. [Google Scholar]
- Malizia AL, Cunningham VJ, Bell CJ, Liddle PF, Jones T, Nutt DJ ( 1998): Decreased brain GABAA‐benzodiazepine receptor binding in panic disorder—Preliminary results from a quantitative PET study. Arch Gen Psychiatr 55: 715–720. [DOI] [PubMed] [Google Scholar]
- Mamelak AN, Lopez N, Akhtari M, Sutherling WW ( 2002): Magnetoencephalography‐directed surgery in patients with neocortical epilepsy. J Neurosurg 97: 865–873. [DOI] [PubMed] [Google Scholar]
- Mehta MA, Richell RA ( 2005): Using neuroimaging to measure drug responses. Psychiatry 4: 10–13. [Google Scholar]
- Moita MAP, Rosis S, Zhou Y, Ledoux JE, Blair HT ( 2003): Hippocampal place cells acquire location‐specific responses to the conditioned stimulus during auditory fear conditioning. Neuron 37: 485–497. [DOI] [PubMed] [Google Scholar]
- Moolenaar F, Bakker S, Visser J, Huizinga T ( 1980): Biopharmaceutics of rectal administration of drugs in man. IX. Comparative biopharmaceutics of diazepam after single rectal, oral, intramuscular and intravenous administration in man. Int J Pharm 5: 127–137. [Google Scholar]
- Murakami S, Okada Y ( 2006): Contributions of principal neocortical neurons to magnetoencephalography and electroencephalography signals. J Physiol Lond 575: 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Fetz EE ( 1996): Oscillatory activity in sensorimotor cortex of awake monkeys: Synchronization of local field potentials and relation to behavior. J Neurophysiol 76: 3949–3967. [DOI] [PubMed] [Google Scholar]
- Muthukumaraswamy SD, Edden RA, Jones DK, Swettenham JB, Singh KD ( 2009): Resting GABA concentration predicts peak gamma frequency and fMRI amplitude in response to visual stimulation in humans. Proc Natl Acad Sci USA 106: 8356–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani N ( 2003): Dynamics of cognitive processing in the human hippocampus by neuromagnetic and neurochemical assessments. Neuroimage 20: 561–571. [DOI] [PubMed] [Google Scholar]
- Nutt DJ, Malizia AL ( 2001): New insights into the role of the GABAA‐benzodiazepine receptor in psychiatric disorder. Br J Psychiatr 179: 390–396. [DOI] [PubMed] [Google Scholar]
- Pesonen M, Hamalainen H, Krause CM ( 2007): Brain oscillatory 4‐30 Hz responses during a visual n‐back memory task with varying memory load. Brain Res 1138: 171–177. [DOI] [PubMed] [Google Scholar]
- Pflug B ( 1978): Influence of sleep‐deprivation on duration of endogenous depressive episodes. Arch Psychiatr Nervenkrankheiten 225: 173–177. [DOI] [PubMed] [Google Scholar]
- Pfurtscheller G, Berghold A ( 1989): Patterns of cortical activation during planning of voluntary movement. Electroencephalogr Clin Neurophysiol 72: 250–258. [DOI] [PubMed] [Google Scholar]
- Pfurtscheller G, Stancak A, Neuper C ( 1996): Post‐movement beta synchronization. A correlate of an idling motor area? Electroencephalogr Clin Neurophysiol 98: 281–293. [DOI] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G ( 2000): GABAA receptors: Immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101: 815–850. [DOI] [PubMed] [Google Scholar]
- Rangaswamy M, Porjesz B, Chorlian DB, Wang KM, Jones KA, Bauer LO, Rohrbaugh J, O'Connor SJ, Kuperman S, Reich T, Begleiter H ( 2002): Beta power in the EEG of alcoholics. Biol Psychiatr 52: 831–842. [DOI] [PubMed] [Google Scholar]
- Romano‐Torres M, Borja‐Lascurain E, Chao‐Rebolledo C, del‐Rio‐Portilla Y, Corsi‐Cabrera M ( 2002): Effect of diazepam on EEG power and coherent activity: Sex differences. Psychoneuroendocrinology 27: 821–833. [DOI] [PubMed] [Google Scholar]
- Roopun AK, Middleton SJ, Cunningham MO, Lebeau FEN, Bibbig A, Whittington MA, Traub RD ( 2006): A β2‐frequency (20‐30 Hz) oscillation in nonsynaptic networks of somatosensory cortex. Proc Natl Acad Sci USA 103: 15646–15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Mohler H ( 2001): GABAA receptor subtypes: Dissecting their pharmacological functions. Trends Pharmacol Sci 22: 188–194. [DOI] [PubMed] [Google Scholar]
- Saletu B, Anderer P, Kinsperger K, Grunberger J, Sieghart W ( 1988): Comparative bioavailability studies with a new mixed‐micelles solution of diazepam utilizing radioreceptor assay, psychometry and EEG brain mapping. Int Clin Psychopharmacol 3: 287–323. [DOI] [PubMed] [Google Scholar]
- Salmelin R, Hari R ( 1994): Spatiotemporal characteristics of sensorimotor neuromagnetic rhythms related to thumb movement. Neuroscience 60: 537–550. [DOI] [PubMed] [Google Scholar]
- Salmelin R, Makela J ( 1995): Magnetic signals in the study of human brain dynamics. Rivista Neuroradiol 8: 329–344. [Google Scholar]
- Schack B, Vath N, Petsche H, Geissler HG, Moller E ( 2002): Phase‐coupling of δ‐γ EEG rhythms during short‐term memory processing. Int J Psychophysiol 44: 143–163. [DOI] [PubMed] [Google Scholar]
- Siapas AG, Lubenov EV, Wilson MA ( 2005): Prefrontal phase locking to hippocampal theta oscillations. Neuron 46: 141–151. [DOI] [PubMed] [Google Scholar]
- Siegel M, Konig P ( 2003): A functional gamma‐band defined by stimulus‐dependent synchronization in area 18 of awake behaving cats. J Neurosci 23: 4251–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KD, Barnes GR, Hillebrand A, Forde EME, Williams AL ( 2002): Task‐related changes in cortical synchronization are spatially coincident with the hemodynamic response. Neuroimage 16: 103–114. [DOI] [PubMed] [Google Scholar]
- Sperk G, Furtinger S, Schwarzer C, Pirker S ( 2004): GABA and its receptors in epilepsy. Recent Adv Epilepsy Res 548: 92–103. [DOI] [PubMed] [Google Scholar]
- Suetsugi M, Mizuki Y, Ushijima I, Yamada M, Imaizumi J ( 1998): Anxiolytic effects of low‐dose clomipramine in highly anxious healthy volunteers assessed by frontal midline theta activity. Prog Neuropsychopharmacol Biol Psychiatr 22: 97–112. [DOI] [PubMed] [Google Scholar]
- Takita M, Kuramochi M, Izaki Y, Ohtomi M ( 2007): In vivo temporal property of GABAergic neural transmission in collateral feed‐forward inhibition system of hippocampal‐prefrontal pathway. Brain Res 1150: 69–73. [DOI] [PubMed] [Google Scholar]
- Tallon‐Baudry C, Bertrand O ( 1999): Oscillatory gamma activity in humans and its role in object representation. Trends Cogn Sci 3: 151–162. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Kato A, Fujita N, Hirata M, Tanaka H, Kihara T, Ninomiya H, Hirabuki N, Nakamura H, Robinson SE, Cheyne D, Yoshimine T ( 2000): Movement‐related desynchronization of the cerebral cortex studied with spatially filtered magnetoencephalography. Neuroimage 12: 298–306. [DOI] [PubMed] [Google Scholar]
- Tiihonen J, Kuikka J, Rasanen P, Lepola U, Koponen H, Liuska A, Lehmusvaara A, Vainio P, Kononen M, Bergstrom K, Yu M, Kinnunen I, Akerman K, Karhu J ( 1997): Cerebral benzodiazepine receptor binding and distribution in generalized anxiety disorder: A fractal analysis. Mol Psychiatr 2: 463–471. [DOI] [PubMed] [Google Scholar]
- Van Veen BD, van DW, Yuchtman M, Suzuki A ( 1997): Localization of brain electrical activity via linearly constrained minimum variance spatial filtering. IEEE Trans Biomed Eng 44: 867–880. [DOI] [PubMed] [Google Scholar]
- von Stein A, Sarnthein J ( 2000): Different frequencies for different scales of cortical integration: From local gamma to long range alpha/theta synchronization. Int J Psychophysiol 38: 301–313. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Ebert B ( 2006): Gaboxadol—A new awakening in sleep. Curr Opin Pharmacol 6: 30–36. [DOI] [PubMed] [Google Scholar]
- Whiting PJ ( 2006): GABAA receptors: A viable target for novel anxiolytics? Curr Opin Pharmacol 6: 24–29. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Traub RD ( 2003): Interneuron diversity series: Inhibitory interneurons and network oscillations in vitro. Trends Neurosci 26: 676–682. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Lebeau FEN, Buhl EH ( 2000): On the relationship between inhibition‐based gamma oscillations and field potentials in the hippocampal slice. J Physiol Lond 525: 60P. [Google Scholar]
- Yamazaki A, Kumagai Y, Fujita T, Hasunuma T, Yokota S, Maeda M, Otani Y, Majima M ( 2007): Different effects of light food on pharmacokinetics and pharmacodynamics of three benzodiazepines, quazepam, nitrazepam and diazepam. J Clin Pharm Ther 32: 31–39. [DOI] [PubMed] [Google Scholar]
- Yamawaki N, Stanford IM, Hall SD, Woodhall GL ( 2008): Pharmacologically induced and stimulus evoked rhythmic neuronal oscillatory activity in the primary motor cortex in vitro. Neuroscience 151: 386–395. [DOI] [PubMed] [Google Scholar]
- Zezula J, Cortes R, Probst A, Palacios JM ( 1988): Benzodiazepine receptor‐sites in the human‐brain—Autoradiographic mapping. Neuroscience 25: 771–795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
Supplementary Figures