

Abstract
We report measurements of atmospheric composition over a tropical rainforest and over a nearby oil palm plantation in Sabah, Borneo. The primary vegetation in each of the two landscapes emits very different amounts and kinds of volatile organic compounds (VOCs), resulting in distinctive VOC fingerprints in the atmospheric boundary layer for both landscapes. VOCs over the Borneo rainforest are dominated by isoprene and its oxidation products, with a significant additional contribution from monoterpenes. Rather than consuming the main atmospheric oxidant, OH, these high concentrations of VOCs appear to maintain OH, as has been observed previously over Amazonia. The boundary-layer characteristics and mixing ratios of VOCs observed over the Borneo rainforest are different to those measured previously over Amazonia. Compared with the Bornean rainforest, air over the oil palm plantation contains much more isoprene, monoterpenes are relatively less important, and the flower scent, estragole, is prominent. Concentrations of nitrogen oxides are greater above the agro-industrial oil palm landscape than over the rainforest, and this leads to changes in some secondary pollutant mixing ratios (but not, currently, differences in ozone). Secondary organic aerosol over both landscapes shows a significant contribution from isoprene. Primary biological aerosol dominates the super-micrometre aerosol over the rainforest and is likely to be sensitive to land-use change, since the fungal source of the bioaerosol is closely linked to above-ground biodiversity.
Keywords: biogenic volatile organic compounds, tropospheric ozone, hydroxyl radical, atmospheric aerosol, rainforest, oil palm
1. Introduction
Land-system change is one of the nine ‘planetary boundaries’, by which Rockström et al. [1] assessed the state of the ‘‘‘planetary playing field’ for humanity”, and links to another planetary boundary: climate change. It does so via biogeophysical changes (albedo, aerodynamic roughness and the ratio of energy flux to the atmosphere via sensible and latent heat, or Bowen ratio; see also Fowler et al. [2]) and biogeochemical changes to atmospheric composition [3,4].
The present paper addresses the latter link, between land-system change and atmospheric composition, particularly with respect to reactive trace gases and particles. Volatile organic compound (VOC) [5] and nitrogen oxide emissions can change, and gas and particle deposition can change [6] as land-use changes over time, affecting both air quality and climate.
Setting aside the wholesale changes brought about by urbanization, rural land-system change in the tropics can produce significant changes in atmospheric state and composition. For example, in Amazonia, it has been shown that conversion of forest to savannah can, inter alia, increase dry season mixing-layer heights by 50 per cent [7], decrease rainfall, increase regional aerosol loading as a result of biomass burning, increase the frequency and intensity of lightning and alter regional photochemistry (see below). In South East Asia, the severe impact of using fire to convert peaty rainforest landscapes to agricultural land is well attested [8–10] and the biogenic VOC emissions from a traditional agricultural crop (rubber) have been measured [11,12].
Land-system change in the tropics is proceeding at an unprecedented rate. The precise pressures on rural land use vary across the tropics, but always include conversion to agricultural land for food, fuel and fibre. Some of the effects of tropical land-use change are becoming apparent, but there is a steep west–east ‘knowledge gradient’ in our understanding, particularly of the effects of land-system change on biogenic emissions and atmospheric photochemistry. We know much more about Amazonia [13–18, and references therein, 19,20] than we do about the African wet tropics [21–24], and we know more about tropical Africa than we do about South [25,26] and South East Asia [27,28]. The paucity of direct measurements of atmospheric composition in South East Asia was a primary driver for the measurements reported below.
Photochemistry in the troposphere largely depends on the VOC/NOx concentration ratio (e.g.[29–31]); biogenic emissions of NOx and VOCs are especially important in the remote tropics, distant from urban sources of air pollutants. It is becoming apparent that both gas phase and particle phase chemistry are affected by the [VOC]/[NOx] ratio, and hence the prevailing chemical regime and concentrations of photochemical products in the atmosphere will change as the ratios of the precursor concentrations change. It is also becoming clear that major gaps remain in our knowledge of what was, until recently, believed to be relatively simple chemistry. The oxidation of trace gases in the atmosphere is largely initiated by reaction with the hydroxyl radical, OH, whose concentration is controlled by local VOC and NOx concentrations [32]. The biogenic VOC isoprene reacts rapidly with the hydroxyl radical (OH), but recent field observations indicate that our understanding of isoprene chemistry in low-NOx conditions remains very incomplete [19,33–36]. Reactions of isoprene may also lead to the formation of secondary organic aerosol (SOA) particles (e.g.[37–39]) that are active radiatively and act as cloud condensation nuclei (CCN).
In the tropics, there is a rapid production rate of OH from the reaction of O(1D) atoms with water vapour (the O(1D) coming from near-UV photolysis of ozone) owing to the presence of intense sunlight and high humidities. OH is closely coupled with peroxy radicals (HO2 and organic peroxy radicals (ΣiRiO2)), these species being key intermediates and chain carriers in the photochemical cycling of ozone in polluted air [40]. Peroxy radicals are formed primarily via the OH-initiated oxidation of anthropogenic and biogenic species in the atmosphere. Ozone is produced via the peroxy radical-catalysed oxidation of NO to NO2 and subsequent photolysis of NO2, while ozone can also be destroyed through reaction with HO2 [40]. Owing to the short lifetime of OH (less than 1 s) and peroxy radicals (HO2 has a lifetime of the order of a minute in clean air, much less than a minute in polluted air [40]), these species are not strongly influenced by transport processes. In addition, the self- and cross-reactions of peroxy radicals to form peroxides (e.g. H2O2) are a major sink for HO2 and OH [41].
Measurements of OH, HO2 and RO2 concentrations can be compared with model calculations in order to probe the understanding of the photochemistry of this region, and to validate chemical schemes that are used in climate models to predict future changes in composition, for example, of greenhouse gases and secondary pollutants. There have been relatively few measurements of OH and HO2 radicals in the tropical troposphere, for example from aircraft above the Pacific region [42–47] and from a ship in the Atlantic Ocean [48], and also aircraft measurements over West Africa and in Mexico City [49]. However, measurements of OH and HO2 within or above a tropical rainforest are particularly sparse, with only one previous study (GABRIEL), in which aircraft measurements were made over Suriname [19,50]. Prior to the GABRIEL study, it was expected that, owing to the elevated concentrations of isoprene and other biogenic VOCs within the rainforest boundary layer, OH concentrations would be low, and hence the degree of chemical processing relatively modest. Model calculations of the global distribution of OH showed suppressed OH in tropical rainforests, with concentrations much lower than surrounding oceanic areas. The GABRIEL study, however, gave the surprising result that OH and HO2 concentrations measured in the boundary layer above the forest were up to a factor of 5 or more higher than the calculations of a box model constrained by measurements of trace gases and radiation parameters measured simultaneously on the aircraft. It was hypothesized that the reaction of HO2 with isoprene peroxy radicals was able to recycle OH radicals, with good agreement for OH achieved with the model by approximately two to three OH molecules recycled [19]. However, there is no evidence in the laboratory for this reaction generating a high yield of OH. An alternative explanation is that air containing high concentrations of isoprene does not mix well with the surrounding troposphere, and that the effective rate constant for removal of isoprene by OH is lower than that measured in the laboratory (see §7).
Until the measurements discussed below, peroxy radical measurements in forested regions had been made exclusively in Boreal forested regions [35,36,51–53]. No previous ground-based rainforest measurements have been obtained of OH, HO2 or peroxy radicals (ΣiRiO2), and an overview of such measurements, obtained during the OP3/ACES campaign in Borneo, is presented below.
We investigate issues related to atmospheric photochemistry and land-system change using measurements of atmospheric composition obtained in Sabah, Malaysia, as part of the ‘Oxidant and particle photochemical processes above a South East Asian tropical rainforest/Aerosol Coupling in the Earth System’ (OP3/ACES) campaign. A detailed description of the rationale behind the OP3/ACES campaign, a description of the measurement sites and an initial survey of results are provided by Hewitt et al. [54], and only a very brief summary is presented here. The OP3/ACES campaign was carried out by a consortium consisting of eight research groups from the UK, along with Malaysian, Italian, USA and other collaborators.
2. The op3/aces campaign
Ground-based measurements were obtained at the Bukit Atur Global Atmosphere Watch (GAW) station in the Danum Valley forest conservation area in Sabah, Malaysia (4°58′49.33′ N, 117°50′39.05′ E, 426 m above mean sea level (a.s.l.)) and at the Sabahmas oil palm plantation, also in Sabah (5°14′52.67 N, 118°27′14.96 E), owned by Wilmar International Ltd (figure 1). Campaigns focused on the rainforest took place between 7 April and 4 May 2008 (OP3-I) and between 23 June and 23 July 2008 (OP3-III). A sub-set of instruments were deployed in the oil palm plantation during the period 11 May–20 June 2008 (OP3-II). Measurements using the FAAM BAe146 research aircraft were obtained over northern Borneo (Sabah) at typical altitudes of approximately 150, 1500, 3000 and 6000 m above ground, over rainforest and extensive areas of agro-industrialized oil palm landscape [31]. Measurements of fluxes of trace gases and particles to and from the forest canopies were made, coupled with ground, tower and aircraft-based atmospheric composition measurements and complementary modelling activities.
Figure 1.

The geographical region (northern Borneo) studied in OP3/ACES. Shading shows land use; white areas are predominantly oil palm plantation. The coloured dots are isoprene mixing ratios measured by the FAAM BAe146 research aircraft flying when in the boundary layer. The strong black line shows the path usually taken by the BAe146 to reach Bukit Atur at low altitude, and is the section along which the measurements shown in figure 4 were made. The location of the oil palm site and the nearest urban area are shown. Adapted from fig. 1 of Hewitt et al. [31].
Bukit Atur, the ground-based rainforest measurement site, is a small hill approximately 260 m above the nearby valley floor. On the top of the hill is a small grassy clearing approximately 150 × 50 m, surrounded by secondary rainforest, rising to approximately 10 m on three sides. The surrounding rainforest is either virgin or has not been logged since 1988 [55]. Measurements at Bukit Atur were obtained at a range of heights from 5 to 75 m above ground level, using a 100 m steel-lattice measurement tower. The Danum Valley region is hilly, having peaks above 500 m a.s.l. and steep-sided valleys with valley floors at about 200 m a.s.l. [56,57] (figure 2). The ground-based oil palm plantation measurements were obtained in a flat 33 ha section of the much larger Sabahmas oil palm plantation, situated on the low-lying plain to the north and east of Danum Valley. The palms where we measured were all 12 year-old Elaeis guineensis × Elaeis oleifera hybrids of the progeny ‘Gutherie’, with an average height of 12 m and planted at the standard plantation density of 150 trees per hectare. The plantation site comprised a 15 m hydraulic tower and 8 m canopy access platform; instruments were housed in a hut at the base of the platform. Aircraft and ground-based measurements show good agreement at both sites [31], suggesting that the ground-based measurements are representative of the well-mixed boundary layer during daytime.
Figure 2.

Time-sequence of photographs from the GAW tower at Bukit Atur through a typical day.
The emissions measurements obtained during OP3/ACES, and composition measurements obtained within the rainforest canopy, are reported in one companion paper [2]; implications of the measurements for regional- and global-scale atmospheric composition, chemistry and climate are discussed in another [58]. Below, we first give the meteorological context for the measurements and report the chemical regimes (i.e. typical ozone, nitrogen oxide and the total VOC mixing ratios) present over the rainforest and oil palm plantation landscapes. We then discuss our measurements, in both landscapes, of fast photochemistry and radicals and of aerosol size distributions and particle composition. Finally, we draw the measurements together to assess the level of our understanding of the impacts of South East Asian land-system change on atmospheric composition.
3. Boundary layer characteristics
Climatologically, the OP3 sites in northern Borneo are in the superwet tropics [59]. Such a climate is ideal for lowland evergreen broadleaf rainforest, which grades into montane rainforest at higher elevations [60]. The local atmospheric boundary-layer structure is dominated by the interaction of strong insolation with this topography and land cover: solar radiation is efficiently captured at the top of the forest canopy and partitioned into latent and sensible heating [2,61]. The boundary layer is well-mixed, several hundred metres deep and capped by shallow cumulus during the middle of the day, while at night the hill tops are de-coupled from the valleys, in which fog forms (figure 2). In the afternoon, convection is often strong enough to produce thunderstorms, giving an afternoon peak in rainfall [54,62].
During OP3, boundary-layer characteristics were monitored using in situ instruments on the GAW tower [63] and remotely using Doppler lidar [56,57]. At the oil palm plantation, eddy covariance measurements at the top of the canopy provide boundary-layer characterization [2]. Aircraft measurements of atmospheric state parameters also provide information on the boundary layer. Figure 3 shows potential temperature profiles from aircraft data above the rainforest and plantation. The depth of well-mixed boundary layer (constant potential temperature) is approximately constant at about 800 m over the two landscapes.
Figure 3.

Mean vertical profiles of dry-bulb potential temperature over the rainforest (squares) and oil palm plantation (circles) landscapes from observations using the FAAM BAe146 research aircraft during OP3-III. Data are binned in 100 m bins plotted at the mid-point of the bin, and are plotted against height above ground. Horizontal bars are ± 1 s.d.
The rainforest lidar data give no information on nocturnal boundary-layer structure because the instrument was sited in a valley and so was usually in fog during the night (figure 2). The depth of the daytime mixed layer can be calculated in a variety of ways from lidar data: using aerosol backscatter or metrics based on the vertical wind. There is no consensus on which measure of mixing-layer depth should be adopted; the choice of metric depends on the issue being studied. Measures that emphasize the ‘high-tide line’ reached by turbulent eddies give a mixing-layer depth of up to 1300 m; measures that emphasize gradients in tracer (i.e. aerosol) abundance give a mixing depth of no more than 500 m [57]. Mixing depth estimated by the standard deviation of the vertical wind—which emphasizes the depth of atmosphere through which turbulent eddy mixing is efficient and which maximizes at about 800 m—is perhaps the most useful definition for budget calculations and discussions of atmospheric composition. It is used in the box-modelling studies described below (see [64] and the associated online discussion for further details). A mixing-layer depth of 800 m is substantially shallower than those found in Amazonian studies, in which the mixing layer is found typically to be about 1500 m deep.
The differences in eddy momentum, sensible heat and latent heat fluxes between the rainforest and the plantation are described by Fowler et al. [2]. Both sites had daytime momentum fluxes between 0.1 and 0.2 N m−2. More cloudiness and rainfall were experienced at the rainforest site during the measurement period, and this led to lower available energy in the rainforest boundary layer. How typical this difference is, and what the difference implies for atmospheric composition at the two sites, remains to be determined.
Sabah is known as ‘the land below the wind’, and this colloquial description held true during OP3/ACES. Mean horizontal winds near the surface were 1.8 ± 1 m s−1, with a maximum of approximately 3.5 m s−1 recorded as gust fronts passed [57]. Mean winds remained below 4 m s−1 for heights up to 1000 m above ground level. These low winds speeds suggest, for example, a transit time of ca 17 h for a near-surface air parcel to traverse the region depicted in figure 1. Twenty-four hour backward air-mass trajectories during the OP3-I and OP3-III rainforest intensives are predominantly from the south (see [60], figure 3). Air arriving at Bukit Atur or the plantation site from the southeast would be expected to show maritime influence, whereas air arriving from the southwest passed over the Borneo hinterland and would be expected to show the greatest influence of terrestrial emissions and deposition. This variation in air mass history is most evident in aerosol composition (see below).
4. Chemical regimes
(a). Aircraft measurements of composition
The differences in composition between the rainforest and the oil palm plantations can be seen in aircraft measurements throughout the boundary layer. Aircraft measurements at low altitude require descent over flat, low-lying terrain. Because plantations, predominantly of oil palm, cover the lowlands of northern Borneo, and because we wished to fly as low as possible over the rainforest station, the sorties of the BAe146 FAAM research aircraft included low-level runs across the plantation–rainforest boundary (figure 1). Figure 4 shows aircraft level runs across the plantation—rainforest boundary, which the aircraft made in order to reach Bukit Atur at low altitude. The statistics of atmospheric composition for all measurements made by the aircraft over the landscapes are discussed in Hewitt et al. [31] and summarized here. Over the rainforest, median boundary-layer mixing ratios of NO, NO2 and isoprene are 35, 161 and 767 pptv, respectively, and over the plantation the median mixing ratios are 67, 288 and 3870 pptv, respectively. The changed VOC and NOx conditions clearly have an impact on the atmospheric chemistry: mixing ratios of the secondary pollutant peroxy acetyl nitrate are greatly enhanced (median mixing ratios are 6.0 pptv over the rainforest and 11.5 pptv over the plantation landscape). Ozone mixing ratios are little changed, however (rainforest: 10.8 ppbv; oil palm plantation: 12.1 ppbv), because the NOx enhancement is proportionately much less than the VOC enhancement, and the chemistry remains severely NOx-limited over both landscapes. Ozone deposition may also change between the two landscapes [2], with deposition to the plantation landscape being smaller than to the rainforest. However, our modelling shows that chemical fluxes are comparable, and usually larger in magnitude, than deposition fluxes (see §7). Independent, longer-term, ozone measurements at the Bukit Atur GAW station confirm that our measured mixing ratios are typical.
Figure 4.

The influence of land use on atmospheric composition, as shown by aircraft data in flight segments traversing plantation and rainforest at low altitude. From top to bottom panels, the quantities plotted are: isoprene mixing ratio (pptv), NOx (pptv), CO (ppbv), acetone (pptv), peroxy acetyl nitrate (PAN, pptv), O3 (ppbv), sulphate aerosol mass concentration (g m−3) and organic aerosol mass concentration (g m−3).
Measurements of OH and HO2 were made aboard the BAe146 during the July OP3 campaign between 500 and 6500 m. OH concentrations were highest in the free-troposphere (up to approx. 107 molecule cm−3). Using the Master Chemical Mechanism [65] to model the aircraft measurements significantly under-predicts OH, with the discrepancy between modelled and measured OH correlating with isoprene. Introducing the mechanism of Peeters et al. [66], which recycles OH, leads to a significant improvement in modelled OH (n.b.: this study of the OP3 aircraft data is not so constrained by measurements as that described below and in Pyle et al. [58], because the ground-based measurements were more comprehensive than the airborne measurements).
(b). Variations in total reactive carbon and volatile organic compound speciation between landscapes
Figure 5 shows average mass spectra from the proton transfer reaction mass spectrometers (PTR-MS) operated at Bukit Atur and Sabahmas. PTR is a soft ionization method that protonates chemicals with a higher proton affinity than water, making it very suitable for detection of trace levels of gas-phase organic compounds [67,68]. Although the library of PTR-MS spectra is not as complete as that for more established ionization methods (e.g. the NIST/EPA/NIH Mass Spectral Library, http://webbook.nist.gov/chemistry/name-ser.html), and although any compound identification based solely on mass-spectrometry is limited by the single dimension (i.e. mass/charge ratio) of the chemical information, we have assigned the main features of the mass spectra in figure 5. This is made easier because (i) the proton transfer reactions between 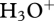 and VOCs tend not to produce excess energy that would cause the protonated VOC to fragment (that is, the reactions are not highly exoergic), (ii) a dual-channel gas chromatograph with flame ionization detectors (DC-GC-FID [69]) provided speciated hydrocarbon measurements at the rainforest site with 1 hour resolution, and (iii) adsorbent-tube samples of ambient air and of leaf emissions were collected for later analysis by a gas chromatograph with a mass-spectrometry detector (GC-MS).
and VOCs tend not to produce excess energy that would cause the protonated VOC to fragment (that is, the reactions are not highly exoergic), (ii) a dual-channel gas chromatograph with flame ionization detectors (DC-GC-FID [69]) provided speciated hydrocarbon measurements at the rainforest site with 1 hour resolution, and (iii) adsorbent-tube samples of ambient air and of leaf emissions were collected for later analysis by a gas chromatograph with a mass-spectrometry detector (GC-MS).
Figure 5.

Average proton-transfer reaction mass spectrometer (PTR-MS) fingerprints from (a) the rainforest at Danum and the (b) nearby Sabahmas oil palm plantation. Peaks are assigned, as discussed in the main text. Tentative assignments are given in grey. Note the different y-axes on the plots.
The total VOC spectrum and mass loading are very different between rainforest and plantation (figure 5). In the average mass spectrum for the rainforest, only isoprene is present at mixing ratios greater than 1 ppbv; for the plantation spectrum, nine compounds are present at mixing ratios greater than 1 ppbv. Summing each spectrum to estimate the volatile carbon, our tentative compound identification suggests that five times as much volatile reactive organic carbon is present in the plantation air than in the rainforest air (see also Fowler et al. [2]). This is perhaps surprising, since the plantation exists to convert atmospheric carbon dioxide into organic compounds for use as food and fuel and so one might expect plants bred for maximum oil yield to minimize their ‘waste’ carbon and hence emit less as VOCs. Overall, however, the carbon loss owing to VOC emission is a small fraction of carbon uptake, and the relative ‘wastefulness’ of the oil palm plants does not scale with the relative change in VOC abundance across the landscapes: the sum of the VOC fluxes measured represents a 0.4 per cent loss of daytime assimilated carbon by the rainforest canopy [70], and a loss of 0.8 per cent by the plantation canopy [71].
The PTR-MS was also used to target specific compounds to allow for higher resolution measurements of mixing ratios and flux measurements. Some of these compounds are shown in figure 6, which compares the diurnal pattern in mixing ratios measured above each landscape. Table 1 shows the correlation coefficients between compounds measured in one landscape, and between compounds across landscapes. Methanol, acetone and acetaldehyde all share broadly similar trends above each landscape and are of a similar magnitude. Similarly, emission rates of these compounds were either small (acetone and acetaldehyde) or negative (methanol) over both landscapes, which, considering their relatively high ambient mixing ratios, suggests that photochemistry may be an important source for these compounds. In contrast, flux measurements of isoprene showed significant emissions from both landscapes [2] and, of the compounds shown in figure 6, isoprene emissions represented approximately 80 per cent of the reactive carbon emitted from both rainforest and oil palm.
Figure 6.

Average diurnal cycles of VOCs targeted in the PTR-MS observations, measured at 75 m above ground at the Bukit Atur rainforest site. Grey shading and error bars show the variability (±1 s.d.) about the mean. Solid line, rainforest; circles with solid line, oil palm.
Table 1.
Pearson's coefficient of determination (r2) for the average diurnal patterns of the compounds detected in rainforest (a) and oil palm landscapes (b). Correlations of the average diurnal patterns across landscapes are shown in (c). Note that a high r2 value is not sufficient in itself to imply a causal link between the diurnal patterns observed. All correlations, except the two italicized, are significant at p = 0.05.
| methanol | acetaldehyde | acetone | isoprene | MVK + MACR | hexanal | Σmonoterpenes | toluene | estragole | |
|---|---|---|---|---|---|---|---|---|---|
| rainforest | |||||||||
| (a) rainforest | |||||||||
| methanol | 1 | 0.33 | 0.77 | 0.55 | 0.83 | 0.87 | 0.13 | — | — |
| acetaldehyde | 1 | 0.71 | 0.89 | 0.47 | 0.60 | 0.83 | — | — | |
| acetone | 1 | 0.89 | 0.83 | 0.92 | 0.49 | — | — | ||
| isoprene | 1 | 0.64 | 0.80 | 0.76 | — | — | |||
| MVK + MACR | 1 | 0.94 | 0.17 | — | — | ||||
| hexanal | 1 | 0.33 | — | — | |||||
| Σmonoterpenes | 1 | — | — | ||||||
| toluene | — | — | |||||||
| estragole | — | ||||||||
| oil palm | |||||||||
| (b) oil palm | |||||||||
| methanol | 1 | 0.01 | 0.26 | 0.10 | 0.09 | 0.27 | 0.20 | 0.27 | 0.22 |
| acetaldehyde | 1 | 0.19 | 0.15 | 0.15 | 0.12 | 0.14 | 0.06 | 0.16 | |
| acetone | 1 | 0.66 | 0.72 | 0.81 | 0.63 | 0.45 | 0.34 | ||
| isoprene | 1 | 0.95 | 0.76 | 0.40 | 0.14 | 0.08 | |||
| MVK + MACR | 1 | 0.79 | 0.47 | 0.14 | 0.07 | ||||
| hexanal | 1 | 0.66 | 0.39 | 0.26 | |||||
| Σmonoterpenes | 1 | 0.36 | 0.45 | ||||||
| toluene | 1 | 0.81 | |||||||
| estragole | 1 | ||||||||
| rainforest | |||||||||
| (c) oil palm | |||||||||
| methanol | 0.17 | 0.18 | 0.23 | 0.25 | 0.23 | 0.27 | 0.10 | — | — |
| acetaldehyde | 0.04 | 0.19 | 0.11 | 0.19 | 0.11 | 0.15 | 0.16 | — | — |
| acetone | 0.21 | 0.83 | 0.62 | 0.80 | 0.43 | 0.53 | 0.71 | — | — |
| isoprene | 0.09 | 0.73 | 0.43 | 0.67 | 0.11 | 0.26 | 0.92 | — | — |
| MVK + MACR | 0.06 | 0.71 | 0.41 | 0.64 | 0.11 | 0.24 | 0.90 | — | — |
| hexanal | 0.22 | 0.73 | 0.60 | 0.73 | 0.34 | 0.48 | 0.67 | — | — |
| Σmonoterpenes | 0.28 | 0.48 | 0.52 | 0.56 | 0.42 | 0.52 | 0.37 | — | — |
| toluene | 0.79 | 0.50 | 0.80 | 0.65 | 0.93 | 0.89 | 0.15 | — | — |
| estragole | 0.77 | 0.40 | 0.66 | 0.55 | 0.89 | 0.86 | 0.09 | — | — |
Although not currently producing a significant effect on ozone, the much increased reactive VOC emissions from the oil palm plantation make that landscape vulnerable to episodic high ground-level ozone should nitrogen oxide mixing ratios increase as a result of regional development [31,58].
Figure 6e shows the mixing ratios of methyl vinyl ketone (MVK) and methacrolein (MACR). MVK and MACR share the same molecular mass so that their sum is measured by the PTR-MS. MVK and MACR are unique first-generation reaction products of isoprene oxidation, and consequently analysis of the ratio of their sum to isoprene can be informative. This ratio was similar above both landscapes, with the smallest ratio of around 0.15 occurring around mid-day when the production rate of isoprene was exceeding the photochemical turnover; ratios as high as 0.35 could be observed during both the morning and afternoon. The similarity of the ratios between the two sites is surprising when the height at which the measurements were obtained above the canopy is considered—1 m for the oil palm and between 100 and 150 m at the rainforest—demonstrating that the convective mixing timescale was much quicker than the chemical lifetime of isoprene.
In addition to MVK + MACR, second-generation reaction products were also detected above both landscapes. Hydroxyacetone (HA) is a breakdown product of MACR and is thought to relate to the signal observed at m/z 75 (figure 5). Although previous studies have reliably measured HA at m/z 75 [72], contributions from other compounds such as propionic acid and/or biogenic esters (mainly propionates), some of which are known kairomones (i.e. signalling chemicals that produce responses between species) of the oil palm weevil [73], cannot be ruled out. The signal observed at m/z 75 was not specifically targeted, i.e. with a long dwell-time during the PTR-MS measurement cycle, but a strong signal was detected at this m/z at the oil palm plantation using the full-mass-range scans. The signal was far stronger above the oil palm plantation than the rainforest, and its ratio to isoprene was 0.1 in the morning, 0.2 during mid-day, increasing sharply in the late afternoon to 1, when isoprene emissions were decreasing rapidly. HA has a relatively long atmospheric lifetime of 4 days with respect to OH [74], so can potentially accumulate in the shallow boundary layer after its collapse in the evening, which may explain the sharp increase at this time.
Figure 6 shows the diurnal variability of total monoterpene signal, as derived by the PTR-MS technique. Since the rate constants for the reactions of monoterpene species with both ozone and OH are highly dependent on precise molecular structure, it is important to also understand the diurnal variability in the individual monoterpenes. Above the rainforest in Danum, monoterpene composition was principally composed of α-pinene, camphene, limonene and γ-terpinene, as identified by supporting GC-FID analysis [75]. These species comprised around 14 per cent of ambient reactive carbon (when accounting for C2–C8 non-methane hydrocarbons, carbonyls measured by GC and PTR-MS and speciated monoterpenes), or 18 per cent of the directly emitted reactive carbon from the canopy as estimated by eddy covariance of PTR-MS measurements. Figure 7 shows the average individual diurnal cycles of three speciated monoterpenes, with all species showing a daytime maximum and night-time minimum. However, the exact shapes of monoterpene diurnal profiles do differ between species. For example, terpinene mixing ratios are notably elevated in the early evening compared with other species. In contrast to isoprene, night-time monoterpene mixing ratios, measured at 5 m above ground, are non-zero. This behaviour is consistent with monoterpene emissions that are partially dependent on temperature only rather than sunlight and temperature. Monoterpenes present in the very shallow and unmixed nocturnal boundary layer may continue to undergo dark reactions with ozone. Such reactions maintain the formation of carbonyl species of relevance to SOA partitioning, and may also help sustain non-zero night-time OH mixing ratios close to the ground. Monoterpenes are known to have a high-SOA-forming potential, with mass yields of 30–50% [76,77]. We can produce a steady-state, box-type, budget for SOA, based on the observed monoterpene emission rates, E (ca 0.2 mgC m−2 h−1, [2]), the average oxidized organic aerosol (OOA) fraction, C (0.48 µg m−3), observed by aerosol mass spectrometry and a well-mixed boundary-layer height, h, of 800 m. At steady state, the ratio Ch/E defines the product of the loss timescale, τ, and the yield factor for conversion of emitted monoterpene into SOA, χ. The observations at the OP3 rainforest site produce χτ ≈ 7000 s, suggesting that there is ample monoterpene to supply the OOA aerosol fraction for yields greater than 5 per cent or (wet plus dry) deposition lifetimes greater than 2 days.
Figure 7.

Diurnal average mixing ratios of the monoterpenes camphene, limonene and γ-terpinene and the isoprene breakdown product methacrolein, as measured by gas chromatography at 5 m above ground at the Bukit Atur rainforest site.
In stark contrast to observations at the rainforest, ambient levels of monoterpenes at the oil palm plantation were small and leaf-level emission measurements confirmed oil palm to be a very low monoterpene emitter [2]. The absence of significant monoterpene emissions might suggest the oil palm landscape to have a lower SOA-forming potential, especially given the recent findings of Kiendler-Scharr et al. [78], which suggest that isoprene emissions may actually suppress aerosol yields by scavenging OH. Nonetheless, significant fluxes and mixing ratio of estragole, a known precursor for SOA [77], were observed above the plantation [79]. Mixing ratio of estragole, shown in figure 6i, averaged 3 ppbv in the middle of the day, but gradually increased throughout the afternoon (3.8 ppbv) and evening (4.5 ppbv). Unlike isoprene, which was emitted directly from the oil palm fronds as a response to the ambient light and temperature, estragole emissions originate from the oil palm flowers (of which there are thousands on one inflorescence) and lag the ambient light and temperature by several hours. The emissions are, therefore, not well correlated with isoprene and monoterpenes (table 1). Estragole acts as an attractant for the pollinating weevil, which in turn enhances the plant yield. This ecological function makes estragole emissions hugely important for the oil palm industry. Once in the atmosphere, estragole reacts with the hydroxyl radical (estimated lifetime against OH attack approx. 55 min) and ozone (estimated lifetime against O3 attack approx.18 h) [80], also making it important in regional photochemistry and SOA formation. Using the same box-budget approach as for monoterpenes, above, the estragole observations at the OP3 plantation site produce χτ ≈ 2880 s. In this calculation, we use the observed estragole emission rates, E (ca 0.5 mg m−2 h−1 [79]), the average observed OOA fraction, C (0.5 µg m−3), and a well-mixed boundary-layer height, h, of 800 m. Such a value of χτ suggests that there is ample estragole to supply the OOA aerosol fraction for yields greater than 1 per cent or (wet plus dry) deposition lifetimes greater than 2 days. There is also potentially a direct contribution to particle loading from estragole, which has a relatively high octanol–air partition coefficient of 5.194. Comparison of PTR- and aerosol mass spectra suggest that the direct contribution of estragole to particle mass is 1/1000–1/10 000 of the gas-phase concentration [79], i.e. up to a few tens of ng m−3. For estragole, then, the potential for secondary aerosol is much greater than the potential for primary aerosol.
Figure 6h, shows the diurnal profile of m/z 93, which had a very strong signal (approx. 1.5 ppbv) above the oil palm plantation, but was not detected above the rainforest. We attribute this signal to toluene [71] as contributions at this m/z from other compounds such as p-cymene, hydrated HA and chloroacetone were each tested and ruled out. Large fluxes of m/z 93 confirm the source of the emission to be below the canopy and thus rule out its possible advection from local anthropogenic sources, and the diurnal pattern is highly suggestive of a biogenic source. Biogenic toluene emissions have previously been reported by Heiden et al. [81] from sunflowers and more recently by White et al. [82] from alfalfa fields, although the absolute emission rates were smaller. Enclosure studies suggest that, like estragole, the source of biogenic toluene in the oil palm is the palm inflorescences.
5. Fast photochemistry
Ground-based measurements of OH, HO2, ΣiRiO2, OH reactivity and other parameters relevant for the interpretation of radical measurements (CO, biogenic VOCs, ozone, NOx, spectrally resolved actinic flux) were measured at the Bukit Atur site in the rainforest; OH, HO2 and ΣiRiO2 were also measured on the aircraft. OH and HO2 radicals were measured using laser-induced fluorescence spectroscopy at low pressure, using the fluorescence assay by gas expansion (FAGE) technique. With FAGE, OH is measured directly, whereas HO2 is converted first to OH by titration with added nitric oxide, NO. The sum of HO2 and ΣiRiO2 were measured using a peroxy radical chemical amplifier (PERCA; [83]). OH reactivity is a relatively new measurement that enables the total rate of removal of OH by reaction with its sinks to be established [84–86]. As OH is such a short-lived molecule (less than 1 s), its concentration is in steady state, being a balance of the rate of production and removal. By generating OH radicals artificially in a flowtube and measuring their rate of decay following reaction with sampled ambient air, the OH reactivity (inverse OH lifetime) can be measured.
Despite the concentration of isoprene being elevated in the rainforest (figures 5 and 6), the OH concentrations in the first campaign were relatively high, the average diurnal profile of OH showing a maximum of approximately 2.5 × 106 molecule cm−3. The corresponding maximum value of the OH reactivity was approximately 30 s−1 (equivalent to a lifetime of approx. 30 ms.). In order to investigate the source able to maintain this concentration of OH despite its rapid rate of removal, the OH concentration was calculated using a selection of the measured source strengths. Previously identified sources of OH included the photolysis of ozone followed by the reaction of O(1D) atoms with water vapour, the photolysis of other trace gases (e.g. HONO), the reaction of ozone with alkenes and the reaction of HO2 with NO. However, including these sources of OH resulted in a calculated OH peak concentration that was only 10 per cent of the measured value in the rainforest, demonstrating that a major source of OH was missing in the calculations. Recently, two theoretical studies have suggested sources of OH and HO2 following the reaction of OH with isoprene [66,87]. Peeters et al. [66] calculated the barrier heights on the potential energy surface for this reaction and the lifetime of the isoprene peroxy radical intermediates against isomerization (namely a hydrogen atoms shift) to form OH, HO2 and photolabile products. Under the low-NOx conditions observed at Bukit Atur, a major channel for the OH reaction with isoprene is calculated by Peeters et al. to be direct recycling of OH and HO2, and also the formation of multi-functional-oxygenated intermediates, which may photolyse readily to form further radical species. If unity yield of OH from this reaction is assumed, the calculated OH is still much less than that measured, and unless the yield is much higher, this reaction cannot explain the levels of OH observed (and then the HO2 concentration is over-predicted). In a study in the Pearl River delta region of China, Hofzumanhaus et al. [33] also measured OH levels significantly higher than could be explained by known sources, and suggested that RO2 is converted to HO2 and HO2 to OH by an unknown species, X. In the rainforest it would appear that the same, as yet unknown, process is necessary to convert HO2 into OH in order for the model and measured OH and HO2 to agree with each other. In a chamber study, Paulot et al. [37] observed the formation of epoxide species following the oxidation of isoprene by OH, which are likely to react further or photolyse to form OH and HO2, but, as these species were not a focus of measurements at Bukit Atur, it is difficult to evaluate this mechanism against the measured radical data.
The only previous measurements of OH reactivity in a tropical rainforest were obtained in Suriname [88]. The measured reactivity was found to be about a factor of 3 higher than that calculated using the co-located sinks of OH measured at the same time. During OP-3, the measured OH reactivity (average diurnal peak approx. 30 s−1) was also found to be considerably higher than that calculated from the observed sinks of OH (approx. 13 s−1). Of the measured OH sinks, the removal was dominated by isoprene. A model was used to calculate the reactivity towards OH of intermediates in the oxidation chain of isoprene and other biogenic VOCS, but including these additional sinks still left approximately 50 per cent of the OH reactivity unaccounted for.
Clearly, in this environment there are both sources and sinks of OH that are yet to be identified. Although measurements of radicals were not made at ground-level in the oil palm region, levels of isoprene were considerably higher than in the rainforest (figure 6), suggesting that similar poorly understood chemistry may operate there, and this is certainly implied by the modelling studies discussed below.
Figure 8 shows the mean hourly averaged HO2 + ΣRO2 diurnal cycles measured at Bukit Atur. The mid-day average concentration (with the 95% confidence interval of the mean) of the peroxy radicals was 33 ± 3.5 pptv in OP3-I and 35.6 ± 3.5 pptv in OP3-II. The mid-day averages of the j(O1D) were 2.67 ± 0.45 × 10−5 s−1 in OP3-I and 2.97 ± 0.49 × 10−5 s−1 in OP3-II. The diurnal cycles show a classical bell shape with a slight asymmetry ascribed in previous low-NOx environments to the persistence of RO2 radicals [89] into the night, owing to the relative rates of radical cross- and self-reactions. Using the OP3 measurements and the methods described in [90], net ozone production rates were calculated (figure 9). These show that overall the conditions encountered during this work at the rainforest were net ozone productive. This is a reflection of the small, but significant NO levels (ca 40 pptv) present in the rainforest. The derived ozone production rate is strongly dependent on the isoprene concentration, having a linear sensitivity (dln(P(O3))/dln(isoprene) = 1.08) averaged from the two different campaign periods. The results imply that the P(O3) is strongly sensitive in the rainforest boundary layer to changes in isoprene concentration.
Figure 8.

Mean hourly averaged HO2 + ΣRO2 diurnal cycles taken at Bukit Atur during OP3-I (Apr/May 2008) and OP3-III (June/July 2008). Diamonds with solid line, HO2+RO2/OP3-I; triangles with solid line, HO2+RO2/OP3-III.
Figure 9.

Mean day-time cycle for net rate of ozone change, N[O3] (asterisks with solid line), ozone production rate, P[O3] (plus symbols with solid line), and ozone loss rate, L[O3] (triangles with solid line), from OP3-I.
6. Aerosol composition
The composition of sub-micrometre aerosol in regions of oil palm and rainforest was compared using measurements obtained aboard the FAAM aircraft. These were performed using an Aerodyne compact time-of-flight aerosol mass spectrometer (C-ToF-AMS) [91] that measures the bulk composition of sub-micrometre non-refractory aerosol particles with a high time resolution. Though 70 eV electron impact ionization is employed, key fragments in the mass spectrum have been shown to provide information on organic composition [92,93]. In particular, the organic signal m/z 44 (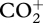 ) has been shown to be high when the organic aerosol is very oxidized and implies significant ageing, whereas that at m/z 43 is either owing to an alkyl ion (
) has been shown to be high when the organic aerosol is very oxidized and implies significant ageing, whereas that at m/z 43 is either owing to an alkyl ion (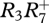 ) or to an oxygenated peak (
) or to an oxygenated peak (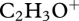 ), the latter of which has been demonstrated to arise in rather less-processed SOA [93–95], hence the m/z 44 to m/z 43 ratio gives a good indication of the level of oxidation of the particulate organic matter (OM). The mass fragment m/z 60 is a typical fragment of anhydrous sugars [96,97] and is associated with fresh biomass burning [98,99].
), the latter of which has been demonstrated to arise in rather less-processed SOA [93–95], hence the m/z 44 to m/z 43 ratio gives a good indication of the level of oxidation of the particulate organic matter (OM). The mass fragment m/z 60 is a typical fragment of anhydrous sugars [96,97] and is associated with fresh biomass burning [98,99].
The particle mass concentrations presented here use more robust averaging methods and are slightly revised compared with the preliminary data in Hewitt et al. [54]. Considering only boundary-layer data, average mass concentrations of organic and sulphate aerosol were elevated over the oil palm plantations when compared with the rainforest, by 17 per cent and 20 per cent, respectively—modest compared with the increases in isoprene concentrations observed. The organic particulate over the oil palm is also more oxidized than the rainforest, with ratios of mean organic contributions at m/z 44 and 43 of 1.35 and 1.26, respectively.
There is a little correlation of either OM or O : C with tracers of anthropogenic emission, suggesting that primary anthropogenic OM is not dominant, although processed anthropogenic OM may be significant. However, NOx, BC and CO were all enhanced over the oil palms when compared with the rainforest, suggesting there is some increase in anthropogenic influence. The less OOA measured over the rainforest is likely to be locally produced biological SOA from rainforest VOC emissions. Air mass analysis [100] shows that the main source of sulphate aerosol is external to the island. The elevated sulphate levels over the oil palms are consistent with this, being closer to the coast.
Palm oil processing plants are also major local sources of particulate matter, although the regional impact remains un-assessed. Sampling of a processing plant chimney plume near-source showed organic particle loadings of more than 100 µg m−3, with a mass spectrum typical of flaming biomass burning aerosol [96,97]. Our data suggest that, despite large changes in VOC, there appear to be only small changes to SOA and sulphate burden over the palm oil compared with the rainforest.
A substantial fraction (up to 15% by mass) of atmospheric sub-micrometre organic aerosol measured during OP3 was detected as methylfuran (MF), identified by a strong peak at m/z = 82 by AMS and two-dimensional gas chromatography with detection by time-of-flight mass spectrometry (GC × GC/ToF-MS) analysis [101,102]. This MF signal correlated strongly with MVK and MACR. MF can be produced in the gas phase by isoprene oxidation, but has thermophysical properties that make it unlikely to partition significantly into the particle phase. Instead, we hypothesize that lower volatility isoprene oxidation products partition into the particle phase, and these are converted into MF during the thermal volatilization of particles prior to detection (by AMS or GC × GC/ToF-MS). The m/z 82 peak is greater over regions of oil palm, which is consistent with higher levels of isoprene emission from the plantation landscape.
Super-micrometre particles, measured at the rainforest site, exhibited a primary biological aerosol (PBA) mode [103] between 2 and 20 µm (optical equivalent diameter) and so it is expected that the relative PBA contribution to giant CCN and ice nuclei (IN) is larger in the rainforest than elsewhere. Bioaerosol data are not available for all sites, but it is likely that the quantity and morphology of bioaerosol over plantations would be different to those in the rainforest, because of the removal of a microclimate suitable for fungi. This may have implications for precipitation over the two landscapes.
7. Modelling local atmospheric composition
(a). Rainforest
Box-modelling studies of the rainforest [64,104] have found that NOx and O3 chemistry can be represented well during the daytime, but not VOC concentrations and those of the principal oxidant, OH. Specifically, in default model runs, the concentration of OH during the day was substantially underestimated, and the concentrations of MVK and MACR were overestimated [64]. Overestimation of these oxidation products could be corrected by implementing wet deposition in the model, and also including a substantial dry deposition velocity (approx. 1.5 cm s−1) for MVK and MACR.
For this particular location, implementing an isoprene oxidation scheme with OH recycling [19,66] could correct the modelled (OH) underestimation, but caused the model fit to isoprene in particular to be significantly degraded [64]. The only way in which to yield simultaneously a good model fit to both isoprene and OH and remain within the constraints provided by the measured isoprene fluxes was to invoke a 50 per cent reduction in the rate of reaction between isoprene and OH, following Butler et al. [34]. One can justify a reduction in reaction rate by envisaging the segregation of isoprene and OH within the PBL. That is, parcels of air which have been in contact with the isoprene source (the forest canopy) have high isoprene concentrations. Let us call these ‘source-impacted’ air parcels. Source-impacted air parcels also have low OH concentrations owing to the rapid reaction between isoprene and OH. Other air parcels, distant from the source but still within the local planetary boundary layer (PBL), contain lower isoprene concentrations, because the isoprene has reacted away, and higher OH. Let us call these ‘aged’ air parcels. Turbulent flow in the PBL stirs and folds source-impacted and aged air parcels, mixing them together relatively slowly, so that instruments measure high-frequency variations as air parcels of different kinds move past the instrument. Since it is not possible to include segregation directly in a box model, which, by definition, assumes instantaneous mixing throughout the box, segregation must be parametrized by reducing the rate coefficient for the isoprene OH reaction. The 50 per cent reduction in rate constant used by Butler et al. [34] and Pugh et al. [64] is at the high end of estimates using large-eddy simulations [105], and larger than the segregation measured in the single high-frequency field measurement [106]. Pugh et al. [107] have subsequently developed a more direct method than box-model optimization to calculate the intensity of segregation of isoprene and OH. This method uses the high-frequency measurements of isoprene concentration [70] made in the PBL above the rainforest during OP3. Direct measurements of segregation intensity require co-located, high-frequency time series for both species. High-frequency OH measurements were not available during OP3, so the steady-state expression for OH, constrained to the observed isoprene (and other) concentrations, was used instead to generate a suitable time series. A test of this method against the only measured dataset of isoprene and OH segregation in the PBL [106] yielded very satisfactory results. Using the method for the OP3 data showed that segregation of isoprene and OH above the rainforest was equivalent to a reaction rate reduction between these two compounds of no more than 15 per cent [107]. With this measure of segregation, modelled isoprene and OH concentrations cannot be reconciled with measurements.
In common with the Amazonian studies of Ganzeveld et al. [15], OP3 box-modelling studies substantially underestimated night-time NO and O3 concentrations [64]. Incorrect model concentrations of NOx and O3 at the end of the night can adversely influence the chemistry of the following day, so some care is needed when using box models over several days. Comparison of a one-dimensional model with the OP3 data suggests that the nocturnal measurements are representative of a very shallow (ca 5 m) layer. It is then more informative to compare box-model results with boundary-layer-average mixing ratios from a one-dimensional model tuned to fit the observations of NO, NO2 and O3. When NOx mixing ratios are high (above ca 1 ppbv), chemistry and transport interact such that there appears to be no general strategy or parametrization that can produce box-model results at the end of the night in reasonable agreement with boundary-layer averages from one-dimensional simulations. However, under low-NOx conditions (i.e. mixing ratios less than ca 1 ppbv), the use of a lower, effective, deposition velocity for O3 (only) during night time in a box model can produce NO2 and O3 mixing ratios in agreement with one-dimensional simulations at the end of the night. A parametrization has been developed to derive the appropriate effective deposition velocity for O3, based on friction velocity and the Monin–Obukhov length [108]. When the parametrization is employed, more ozone remains available in the box to convert nocturnal NO emissions to NO2, as is the case in a stable boundary layer in which the ozone aloft cannot mix down to the surface efficiently and NO emissions percolate upwards, converting to NO2 as they do so.
(b). Oil palm plantation
The 24 h ozone budgets for rainforest and oil palm landscapes are shown in table 2. The large magnitudes of chemical production and loss show the importance of chemistry in determining the atmospheric composition at these sites. The model study, from which this budget is derived [31], treats both landscapes as having the same deposition velocities for ozone and other species. If, as Fowler et al. [2] now suggest, the deposition velocity to oil palm is lower, then the significance of chemistry to the budget will increase.
Table 2.
Twenty four hour average boundary-layer ozone budgets for rainforest and oil palm plantation, estimated using the CiTTyCAT model [31]. Tendencies are reported in pptv h−1.
| rainforest | oil palm plantation | |
|---|---|---|
| chemical production | 508 | 957 |
| chemical loss | −178 | −721 |
| due to isoprene | −37 | −398 |
| due to monoterpenes | −15 | −5 |
| dry deposition | −379 | −333 |
The difficulties in modelling surface-layer atmospheric chemistry are not any less over the plantation landscape than over the rainforest landscape. The ambient NOx mixing ratios (figure 4) are higher over the plantation landscape owing, we believe, to emissions from the palm-oil processing plants and to fertilizer application [2]. This should tend to make the atmospheric chemistry modelling easier, because the isoprene–OH ‘problem’ is prominent under low-NOx conditions, but isoprene mixing ratios are also much increased relative to the rainforest (figures 5 and 6) so that, overall, the plantation atmosphere has a [VOC]/[NOx] at which atmospheric composition is poorly modelled by current chemical schemes. Where plantations are sited next to highly developed urban areas and highways, the modelling in Hewitt et al. [31,58] suggests that ozone production will be efficient, resulting in air-quality problems on occasion.
8. Discussion and conclusions
The OP3 project was initially designed to focus exclusively on the atmospheric chemistry of the remote tropical rainforest in South East Asia. Additional funding (the NERC ACES project), substantial help in kind from an oil palm plantation operator and the particular logistics of flying low-altitude research sorties in Borneo have resulted in an extensive dataset over contrasting land systems in South East Asia. The ground-based and airborne measurements show that the atmospheric composition above each landscape is dominated by local emissions [2], because horizontal wind speeds are typically low. Air above the rainforest is rich in isoprene (with a typical mixing ratio of approx. 1 ppbv) and other biogenic VOCs emitted from the forest. Air above the rainforest is low in NOx (typically approx. 200 pptv), this NOx provided by soil emissions. Even greater amounts of isoprene are present in air above the oil palm plantation (typically approx. 4 ppbv), a result of the very strong isoprene emissions from oil palm. NOx mixing ratios over the plantation are also elevated (typically 350 pptv) compared with the rainforest, as a result both of nitrogen fertilizer application and on-site palm-oil processing. Although isoprene is the dominant VOC over both landscapes, and there is a mass-spectrometric signal indicating that isoprene is a substantial source of SOA, other VOCs—monoterpenes over the rainforest and estragole over the plantation—are also present in quantities that could account for the oxidized organic fraction of the observed atmospheric aerosol, given reasonable estimates of yields and loss rates.
Our ground-based studies of the fast photochemistry taking place, and of boundary-layer structure, were restricted for logistic and funding reasons to the rainforest location. Here, we found concentrations of OH (maximum daily values typically greater than 2 × 106 molecule cm−3) that were high considering the abundance of reactive biogenic VOCs, particularly isoprene. These observations are consistent with those of Lelieveld et al. [19] from Amazonia, suggesting that the sustenance of atmospheric oxidizing capacity is a generic property of tropical rainforests. We expect the oil palm plantation landscape to exhibit similar chemistry, except where the plantation is close to highly developed urban areas and highways, in which case the additional anthropogenic NOx will make the chemistry more straightforward and ozone production efficient.
Sub-micrometre aerosol composition, measured at the rainforest and at the plantation, shows a strong contribution from isoprene. Since isoprene is the major component of reactive VOCs emitted into the atmosphere, this finding could have significant implications for the global aerosol budget. Larger, super-micrometre, aerosol particles at the rainforest were dominated by PBA, particularly fungal spores. Although we were not able to make measurements of PBA at the plantation, it is very likely that the quantity and morphology of primary bioaerosol from the plantation landscape will differ significantly from that of the rainforest, since fungal communities exist in closely synergistic relations with the above-ground biomass. Changing the primary bioaerosol emission from a landscape may have impacts on rainfall, since cloud-ice formation, a key process in rain-making, even in the tropics, may be sensitive to the concentration of bioaerosol present. As land is converted from natural rainforest to oil palm agriculture, the contribution of isoprene SOA from atmospheric oxidation of isoprene emissions, and aerosol from oil palm processing, will both increase, though our data suggest that overall, there appear to be only small changes to SOA and sulphate burden over the two regions measured.
Although OP3/ACES was probably the largest field deployment of the UK atmospheric chemistry community to date, and had substantial help from Malaysian and other international partners, the consequences of land-use change on atmospheric composition in South East Asia cannot be said to be adequately characterized. Measurements at the oil palm plantation did not include those relevant to fast photochemistry and boundary-layer dynamics. Longer term measurements are needed to ensure that the differences between oil palm and rainforest atmospheric composition, and the differences between South East Asian and Amazonian rainforest are robust. Further night-time measurements at rainforest (South East Asian and Amazonian) and oil palm plantation sites would be valuable in order to improve our understanding of the impact of nocturnal chemistry and deposition on the atmospheric composition of subsequent days. Our hypothesis that ozone production from oil palm plantation emissions will greatly increase as socio-economic development drives up vehicle use remains to be tested. There are regions of South East Asia, such as Kuala Lumpur and surroundings, in which this chemistry could already be tested.
Acknowledgements
We thank the Malaysian and Sabah Governments for their permission to conduct research in Malaysia; the Malaysian Meteorological Department (MMD, now Met Malaysia) for access to the Bukit Atur Global Atmosphere Watch station and their long-term ozone record; Leong Chow Peng (formerly of MMD) for her support in the early stages of the project; and Wilmar International Ltd (particularly Simon Siburat and his staff) for access to and considerable logistical support at their PPB Oil Palms Bhd Sabahmas Estate. For all their assistance with the fieldwork, we gratefully acknowledge: Dr Waidi Sinun of Yayasan Sabah and his staff; Dr Glen Reynolds and his staff at the Royal Society's Danum Valley Research Centre; Dr Nick Chappell and Dr Brian Davison of Lancaster University; the ground staff, engineers, scientists and pilots of the UK Natural Environment Research Council/UK Meteorological Office's BAe 146-301 large atmospheric research aircraft; and the rest of the OP3 project team for their individual and collective efforts. This research was funded by NERC under the OP3 (grant ref: NE/D002117/1) and ACES (grant ref: NE/E011233/1) projects. This paper is dedicated to the memory of Kate Furneaux, a core member of the OP3 project team, who was killed in a traffic accident while cycling in Leeds on 28 July 2009. This paper constitutes Publication Number A/572 of the Royal Society South East Asia Rainforest Research Programme.
References
- 1.Rockström J., et al. 2009. A safe operating space for humanity. Nature 461, 472–475 10.1038/461472a (doi:10.1038/461472a) [DOI] [PubMed] [Google Scholar]
- 2.Fowler, et al. 2011. Effects of land use on surface–atmosphere exchanges of trace gases and energy in Borneo: comparing fluxes over oil palm plantations with a rainforest. Phil Trans. R. Soc. B 366, 3196–3209 10.1098/rstb.2011.0055 (doi:10.1098/rstb.2011.0055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forster P., et al. 2007. Changes in atmospheric constituents and in radiative forcing. In Climate change 2007: the physical science basis. Contribution of Working Group I To The Fourth Assessment Report Of The Intergovernmental Panel On Climate Change (eds Solomon S., Qin D., Manning M., Chen Z., Marquis M., Averyt K. B., Tignor M., Miller H. L.), Cambridge, UK and New York, NY: Cambridge University Press [Google Scholar]
- 4.Feddema J. J., Oleson K. W., Bonan G. B., Mearns L. O., Buja L. E., Meehl G. A., Washington W. M. 2005. The importance of land-cover change in simulating future climates. Science 310, 1674–1678 10.1126/science.1118160 (doi:10.1126/science.1118160) [DOI] [PubMed] [Google Scholar]
- 5.Lathiere J., Hauglustaine D. A., Friend A. D., De Noblet-Ducoudre N., Viovy N., Folberth G. A. 2006. Impact of climate variability and land use changes on global biogenic volatile organic compound emissions. Atmos. Chem. Phys. 6, 2129–2146 10.5194/acp-6-2129-2006 (doi:10.5194/acp-6-2129-2006) [DOI] [Google Scholar]
- 6.Fowler D., et al. 2009. Atmospheric composition change: ecosystems–atmosphere interactions. Atmos. Environ. 43, 5193–5267 10.1016/j.atmosenv.2009.07.068 (doi:10.1016/j.atmosenv.2009.07.068) [DOI] [Google Scholar]
- 7.Fisch G., Tota J., Machado L. A. T., Dias M., Lyra R. F. D., Nobre C. A., Dolman A. J., Gash J. H. C. 2004. The convective boundary layer over pasture and forest in Amazonia. Theor. Appl. Climatol. 78, 47–59 10.1007/s00704-004-0043-x (doi:10.1007/s00704-004-0043-x) [DOI] [Google Scholar]
- 8.Duncan B. N., Martin R. V., Staudt A. C., Yevich R., Logan J. A. 2003. Interannual and seasonal variability of biomass burning emissions constrained by satellite observations. J. Geophys. Res. Atmos. 108, 4100. (doi:10.1029/2002jd002378) [Google Scholar]
- 9.Duncan B. N., et al. 2003. Indonesian wildfires of 1997: impact on tropospheric chemistry. J. Geophys. Res. Atmos. 108, 4458. (doi:10.1029/2002jd003195) [Google Scholar]
- 10.Thompson A. M., Witte J. C., Hudson R. D., Guo H., Herman J. R., Fujiwara M. 2001. Tropical tropospheric ozone and biomass burning. Science 291, 2128–2132 10.1126/science.291.5511.2128 (doi:10.1126/science.291.5511.2128) [DOI] [PubMed] [Google Scholar]
- 11.Baker B., et al. 2005. Wet and dry season ecosystem level fluxes of isoprene and monoterpenes from a southeast Asian secondary forest and rubber tree plantation. Atmos. Environ. 39, 381–390 10.1016/j.atmosenv.2004.07.033 (doi:10.1016/j.atmosenv.2004.07.033) [DOI] [Google Scholar]
- 12.Wang Y. F., Owen S. M., Li Q. J., Penuelas J. 2007. Monoterpene emissions from rubber trees (Hevea brasiliensis) in a changing landscape and climate: chemical speciation and environmental control. Global Change Biol. 13, 2270–2282 10.1111/j.1365-2486.2007.01441.x (doi:10.1111/j.1365-2486.2007.01441.x) [DOI] [Google Scholar]
- 13.Kesselmeier J., et al. 2002. Concentrations and species composition of atmospheric volatile organic compounds (vocs) as observed during the wet and dry season in Rondonia (Amazonia). J. Geophys. Res. Atmos. 107, 8053. 10.1029/2000jd000267 (doi:10.1029/2000jd000267) [DOI] [Google Scholar]
- 14.Geron C., Guenther A., Greenberg J., Loescher H. W., Clark D., Baker B. 2002. Biogenic volatile organic compound emissions from a lowland tropical wet forest in Costa Rica. Atmos. Environ. 36, 3793–3802 10.1016/S1352-2310(02)00301-1 (doi:10.1016/S1352-2310(02)00301-1) [DOI] [Google Scholar]
- 15.Ganzeveld L., et al. 2008. Surface and boundary layer exchanges of volatile organic compounds, nitrogen oxides and ozone during the GABRIEL campaign. Atmos. Chem. Phys. 8, 6223–6243 10.5194/acp-8-6223-2008 (doi:10.5194/acp-8-6223-2008) [DOI] [Google Scholar]
- 16.Greenberg J. P., Guenther A. B., Petron G., Wiedinmyer C., Vega O., Gatti L. V., Tota J., Fisch G. 2004. Biogenic VOC emissions from forested Amazonian landscapes. Global Change Biol. 10, 651–662 10.1111/j.1365-2486.2004.00758.x (doi:10.1111/j.1365-2486.2004.00758.x) [DOI] [Google Scholar]
- 17.Karl T., Guenther A., Yokelson R. J., Greenberg J., Potosnak M., Blake D. R., Artaxo P. 2007. The tropical forest and fire emissions experiment: emission, chemistry, and transport of biogenic volatile organic compounds in the lower atmosphere over Amazonia. J. Geophys. Res. Atmos. 112, D18302. 10.1029/2007jd008539 (doi:10.1029/2007jd008539) [DOI] [Google Scholar]
- 18.Guenther A., et al. 1995. A global model of natural volatile organic compound emissions. J. Geophys. Res. Atmos. 100, 8873–8892 10.1029/94JD02950 (doi:10.1029/94JD02950) [DOI] [Google Scholar]
- 19.Lelieveld J., et al. 2008. Atmospheric oxidation capacity sustained by a tropical forest. Nature 452, 737–740 10.1038/nature06870 (doi:10.1038/nature06870) [DOI] [PubMed] [Google Scholar]
- 20.Kubistin D., et al. 2008. Hydroxyl radicals in the tropical troposphere over the suriname rainforest: comparison of measurements with the box model mecca. Atmos. Chem. Phys. Discuss. 8, 15 239–15 289 10.5194/acpd-8-15239-2008 (doi:10.5194/acpd-8-15239-2008) [DOI] [Google Scholar]
- 21.Klinger L. F., Greenberg J., Guenther A., Tyndall G., Zimmerman P., M'Bangui M., Kenfack D. 1998. Patterns in volatile organic compound emissions along a savanna–rainforest gradient in central Africa. J. Geophys. Res. Atmos. 103, 1443–1454 10.1029/97JD02928 (doi:10.1029/97JD02928) [DOI] [Google Scholar]
- 22.Saxton J. E., Lewis A. C., Kettlewell J. H., Ozel M. Z., Gogus F., Boni Y., Korogone S. O. U., Sera D. 2007. Isoprene and monoterpene measurements in a secondary forest in northern Benin. Atmos. Chem. Phys. 7, 4095–4106 10.5194/acp-7-4095-2007 (doi:10.5194/acp-7-4095-2007) [DOI] [Google Scholar]
- 23.Capes G., Murphy J. G., Reeves C. E., McQuaid J. B., Hamilton J. F., Hopkins J. R., Crosier J., Williams P. I., Coe H. 2009. Secondary organic aerosol from biogenic VOCs over West Africa during Amma. Atmos. Chem. Phys. 9, 3841–3850 10.5194/acp-9-3841-2009 (doi:10.5194/acp-9-3841-2009) [DOI] [Google Scholar]
- 24.Saunois M., Reeves C. E., Mari C. H., Murphy J. G., Stewart D. J., Mills G. P., Oram D. E., Purvis R. M. 2009. Factors controlling the distribution of ozone in the West African lower troposphere during the Amma (African monsoon multidisciplinary analysis) wet season campaign. Atmos. Chem. Phys. 9, 6135–6155 10.5194/acp-9-6135-2009 (doi:10.5194/acp-9-6135-2009) [DOI] [Google Scholar]
- 25.Padhy P. K., Varshney C. K. 2005. Isoprene emission from tropical tree species. Environ. Pollut. 135, 101–109 10.1016/j.envpol.2004.10.003 (doi:10.1016/j.envpol.2004.10.003) [DOI] [PubMed] [Google Scholar]
- 26.Padhy P. K., Varshney C. K. 2005. Emission of volatile organic compounds (VOC) from tropical plant species in India. Chemosphere 59, 1643–1653 10.1016/j.chemosphere.2005.01.046 (doi:10.1016/j.chemosphere.2005.01.046) [DOI] [PubMed] [Google Scholar]
- 27.Wang Y.-F., Li J. 2005. BVOCs emitted from plants of terrestrial ecosystems and their ecological functions. Zhiwu Shengtai Xuebao 29, 487–496 [Google Scholar]
- 28.Geron C., Owen S., Guenther A., Greenberg J., Rasmussen R., Bai J. H., Li Q., Baker B. 2006. Volatile organic compounds from vegetation in southern Yunnan province, China: emission rates and some potential regional implications. Atmos. Environ. 40, 1759–1773 10.1016/j.atmosenv.2005.11.022 (doi:10.1016/j.atmosenv.2005.11.022) [DOI] [Google Scholar]
- 29.Sillman S. 1999. The relation between ozone, NOx and hydrocarbons in urban and polluted rural environments. Atmos. Environ. 33, 1821–1845 10.1016/S1352-2310(98)00345-8 (doi:10.1016/S1352-2310(98)00345-8) [DOI] [Google Scholar]
- 30.Colbeck I., MacKenzie A. R. 1994. Air pollution by photochemical oxidants, 1st edn. Amsterdam, The Netherlands: Elsevier [Google Scholar]
- 31.Hewitt C. N., et al. 2009. Nitrogen management is essential to prevent tropical oil palm plantations from causing ground-level ozone pollution. Proc. Natl Acad. Sci. USA 106, 18 447–18 451 10.1073/pnas.0907541106 (doi:10.1073/pnas.0907541106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heard D. E., Pilling M. J. 2003. Measurement of OH and HO2 in the troposphere. Chem. Rev. 103, 5163–5198 10.1021/cr020522s (doi:10.1021/cr020522s) [DOI] [PubMed] [Google Scholar]
- 33.Hofzumahaus A., et al. 2009. Amplified trace gas removal in the troposphere. Science 324, 1702–1704 10.1126/science.1164566 (doi:10.1126/science.1164566) [DOI] [PubMed] [Google Scholar]
- 34.Butler T. M., Taraborrelli D., Fischer C. B. H., Harder H., Martinez M., Williams J., Williams J., Lawrence M. G., Lelieveld J. 2008. Improved simulation of isoprene oxidation chemistry with the ECHAM5/MESSy chemistry-climate model: lessons from the GABRIEL airborne field campaign. Atmos. Chem. Phys. 8, 4529–4546 10.5194/acp-8-4529-2008 (doi:10.5194/acp-8-4529-2008) [DOI] [Google Scholar]
- 35.Tan D., et al. 2001. HOx budgets in a deciduous forest: results from the prophet summer 1998 campaign. J. Geophys. Res. Atmos. 106, 24 407–24 427 10.1029/2001JD900016 (doi:10.1029/2001JD900016) [DOI] [Google Scholar]
- 36.Carslaw N., et al. 2001. OH and HO2 radical chemistry in a forested region of north-western Greece. Atmos. Environ. 35, 4725–4737 10.1016/S1352-2310(01)00089-9 (doi:10.1016/S1352-2310(01)00089-9) [DOI] [Google Scholar]
- 37.Paulot F., Crounse J. D., Kjaergaard H. G., Kurten A., St Clair J. M., Seinfeld J. H., Wennberg P. O. 2009. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science 325, 730–733 10.1126/science.1172910 (doi:10.1126/science.1172910) [DOI] [PubMed] [Google Scholar]
- 38.Claeys M., et al. 2004. Formation of secondary organic aerosols through photooxidation of isoprene. Science 303, 1173–1176 10.1126/science.1092805 (doi:10.1126/science.1092805) [DOI] [PubMed] [Google Scholar]
- 39.Kroll J. H., Seinfeld J. H. 2008. Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 42, 3593–3624 10.1016/j.atmosenv.2008.01.003 (doi:10.1016/j.atmosenv.2008.01.003) [DOI] [Google Scholar]
- 40.Monks P. S. 2005. Gas-phase radical chemistry in the troposphere. Chem. Soc. Rev. 34, 376–395 10.1039/b307982c (doi:10.1039/b307982c) [DOI] [PubMed] [Google Scholar]
- 41.Reeves C. E., Penkett S. A. 2003. Measurements of peroxides and what they tell us. Chem. Rev. 103, 5199–5218 10.1021/cr0205053 (doi:10.1021/cr0205053) [DOI] [PubMed] [Google Scholar]
- 42.Hoell J. M., Davis D., Liu S., Newell R. E., Shipman M., Akimoto H., McNeal R. J., Bendura R. J., Drewry J. W. 1996. Pacific Exploratory Mission-West A (PEM_West A) September–October 1991. J. Geophys. Res. Atmos. 101, 1641–1653 10.1029/95JD00622 (doi:10.1029/95JD00622) [DOI] [Google Scholar]
- 43.Hoell J. M., Davis D., Liu S., Newell R. E., Akimoto H., McNeal R. J., Bendura R. J. 1997. The Pacific Exploratory Mission-West phase B: February–March 1994. J. Geophys. Res. Atmos. 102, 28 223–28 239 10.1029/97JD02581 (doi:10.1029/97JD02581) [DOI] [Google Scholar]
- 44.Hoell J. M., Davis D., Jacob D. J., Rodgers M. O., Newell R. E., Fuelberg H. E., McNeal R. J., Raper J. L., Bendura R. J. 1999. Pacific Exploratory Mission in the tropical Pacific: PEM-tropics A, August–September 1996. J. Geophys. Res. Atmos. 104, 5567–5583 10.1029/1998JD100074 (doi:10.1029/1998JD100074) [DOI] [Google Scholar]
- 45.Raper J. L., Kleb M. M., Jacob D. J., Davis D., Newell R. E., Fuelberg H. E., Bendura R. J., Hoell J. M., McNeal R. J. 2001. Pacific Exploratory Mission in the tropical Pacific: PEM-tropics B, March–April 1999. J. Geophys. Res. Atmos. 106, 32 401–32 425 10.1029/2000JD900833 (doi:10.1029/2000JD900833) [DOI] [Google Scholar]
- 46.Mauldin R. L., III, Tanner D., Eisele F. 1999. Measurements of OH during PEM-tropics A. J. Geophys. Res. Atmos. 104, 5817–5827 10.1029/98JD02305 (doi:10.1029/98JD02305) [DOI] [Google Scholar]
- 47.Mauldin R. L., III, et al. 2001. Measurements of OH aboard the NASA p-3 during PEM-tropics B. J. Geophys. Res. Atmos. 106, 32 657–32 666 10.1029/2000JD900832 (doi:10.1029/2000JD900832) [DOI] [Google Scholar]
- 48.Brauers T., Hausmann M., Bister A., Kraus A., Dorn H. P. 2001. OH radicals in the boundary layer of the Atlantic ocean 1. Measurements by long-path laser absorption spectroscopy. J. Geophys. Res. Atmos. 106, 7399–7414 10.1029/2000JD900679 (doi:10.1029/2000JD900679) [DOI] [Google Scholar]
- 49.Shirley T. R., et al. 2006. Atmospheric oxidation in the Mexico city metropolitan area (MCMA) during April 2003. Atmos. Chem. Phys. 6, 2753–2765 10.5194/acp-6-2753-2006 (doi:10.5194/acp-6-2753-2006) [DOI] [Google Scholar]
- 50.Martinez M., et al. 2010. Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: Airborne measurements. Atmos. Chem. Phys. 10, 3759–3773 10.5194/acp-10-3759-2010 (doi:10.5194/acp-10-3759-2010) [DOI] [Google Scholar]
- 51.Handisides G. M., Plass-Dulmer C., Gilge S., Bingemer H., Berresheim H. 2003. Hohenpeissenberg photochemical experiment (HOPE 2000): measurements and photostationary state calculations of OH and peroxy radicals. Atmos. Chem. Phys. 3, 1565–1588 10.5194/acp-3-1565-2003 (doi:10.5194/acp-3-1565-2003) [DOI] [Google Scholar]
- 52.Mihele C. M., Hastie D. R. 2003. Radical chemistry at a forested continental site: results from the PROPHET 1997 campaign. J. Geophys. Res. Atmos. 108, 4450. 10.1029/2002JD002888 (doi:10.1029/2002JD002888) [DOI] [Google Scholar]
- 53.Ren X. R., Brune W. H., Cantrell C. A., Edwards G. D., Shirley T., Metcalf A. R., Lesher R. L. 2005. Hydroxyl and peroxy radical chemistry in a rural area of central Pennsylvania: observations and model comparisons. J. Atmos. Chem. 52, 231–257 10.1007/s10874-005-3651-7 (doi:10.1007/s10874-005-3651-7) [DOI] [Google Scholar]
- 54.Hewitt C. N., et al. 2010. Overview: oxidant and particle photochemical processes above a south-east Asian tropical rainforest (the OP3 project): introduction, rationale, location characteristics and tools. Atmos. Chem. Phys. 10, 169–199 10.5194/acp-10-169-2010 (doi:10.5194/acp-10-169-2010) [DOI] [Google Scholar]
- 55.Tangki H., Chappell N. A. 2008. Biomass variation across selectively logged forest within a 225-km(2) region of Borneo and its prediction by Landsat TM. For. Ecol. Manag. 256, 1960–1970 10.1016/j.foreco.2008.07.018 (doi:10.1016/j.foreco.2008.07.018) [DOI] [Google Scholar]
- 56.Collier C. G., Davies F., Pearson G. N. 2010. The land below the wind: doppler lidar observations from the tropical rain forest of Sabah, Borneo, Malaysia. Weather 65, 45–50 10.1002/wea.429 (doi:10.1002/wea.429) [DOI] [Google Scholar]
- 57.Pearson G., Davies F., Collier C. 2010. Remote sensing of the tropical rain forest boundary layer using pulsed doppler lidar. Atmos. Chem. Phys. Discuss. 10, 5021–5049 10.5194/acpd-10-5021-2010 (doi:10.5194/acpd-10-5021-2010) [DOI] [Google Scholar]
- 58.Pyle J. A., et al. 2011. The impact of local surface changes in Borneo on atmospheric composition at wider spatial scales: coastal processes, land-use change and air quality. Phil. Trans. R. Soc. B 366, 3210–3224 10.1098/rstb.2011.0060 (doi:10.1098/rstb.2011.0060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh R. P. D. 1996. Climate. In The tropical rain forest (ed. Richards P. W.), pp. 159–205 Cambridge, UK: Cambridge University Press [Google Scholar]
- 60.Hewitt C. N., et al. 2010. Overview: oxidant and particle photochemical processes above a south-east Asian tropical rainforest (the OP3 project): introduction, rationale, location characteristics and tools. Atmos. Chem. Phys. 10, 563–563 10.5194/acp-10-563-2010 (doi:10.5194/acp-10-563-2010) [DOI] [Google Scholar]
- 61.Whitehead J. D., et al. 2010. Aerosol fluxes and dynamics within and above a tropical rainforest in south-east Asia. Atmos. Chem. Phys. Discuss. 10, 12 023–12 061 10.5194/acpd-10-12023-2010 (doi:10.5194/acpd-10-12023-2010) [DOI] [Google Scholar]
- 62.Chappell N. A., Discenza A. R., Tych W., Whittaker J., Bidin K. 2009. Simulating hourly rainfall occurrence within an equatorial rainforest, Borneo island. Hydrol. Sci. J. 54, 571–581 10.1623/hysj.54.3.571 (doi:10.1623/hysj.54.3.571) [DOI] [Google Scholar]
- 63.Helfter C., Phillips G. J., Coyle M., Di Marco C. F., Langford B., Whitehead J., Dorsey J. R., Gallagher W. M., Nemitz E. Momentum and heat exchange above a South East Asian rainforest in complex terrain. In preparation. [Google Scholar]
- 64.Pugh T. A. M., et al. 2010. Simulating atmospheric composition over a South-East Asian tropical rainforest: performance of a chemistry box model. Atmos. Chem. Phys. 10, 279–298 10.5194/acp-10-279-2010 (doi:10.5194/acp-10-279-2010) [DOI] [Google Scholar]
- 65.Bloss C., et al. 2005. Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons. Atmos. Chem. Phys. 5, 641–664 10.5194/acp-5-641-2005 (doi:10.5194/acp-5-641-2005) [DOI] [Google Scholar]
- 66.Peeters J., Nguyen T. L., Vereecken L. 2009. HOx radical regeneration in the oxidation of isoprene. Phys. Chem. Chem. Phys. 11, 5935–5939 10.1039/b908511d (doi:10.1039/b908511d) [DOI] [PubMed] [Google Scholar]
- 67.Lindinger W., Hansel A., Jordan A. 1998. Proton-transfer-reaction mass spectrometry (PTR-MS): on-line monitoring of volatile organic compounds at pptv levels. Chem. Soc. Rev. 27, 347–354 10.1039/a827347z (doi:10.1039/a827347z) [DOI] [Google Scholar]
- 68.Hewitt C. N., Hayward S., Tani A. 2003. The application of proton transfer reaction-mass spectrometry (PTR-MS) to the monitoring and analysis of volatile organic compounds in the atmosphere. J. Environ. Monit. 5, 1–7 10.1039/b204712h (doi:10.1039/b204712h) [DOI] [PubMed] [Google Scholar]
- 69.Lewis A. C., et al. 2007. Chemical composition observed over the mid-Atlantic and the detection of pollution signatures far from source regions. J. Geophys. Res. Atmos. 112, D10S03. 10.1029/2006jd007584 (doi:10.1029/2006jd007584) [DOI] [Google Scholar]
- 70.Langford B., Misztal P. K., Nemitz E., Davison B., Helfter C., Pugh T. A. M., MacKenzie A. R., Lim S. F., Hewitt C. N. 2010. Fluxes and concentrations of volatile organic compounds from a south-east Asian tropical rainforest. Atmos. Chem. Phys. Discuss. 10, 11 975–12 021 10.5194/acpd-10-11975-2010 (doi:10.5194/acpd-10-11975-2010) [DOI] [Google Scholar]
- 71.Oke T. R. 1982. The energetic basis of the urban heat-island. Q. J. R. Meteorol. Soc. 108, 1–24 [Google Scholar]
- 72.Karl T., Guenther A., Turnipseed A., Tyndall G., Artaxo P., Martin S. 2009. Rapid formation of isoprene photo-oxidation products observed in Amazonia. Atmos. Chem. Phys. 9, 7753–7767 10.5194/acp-9-7753-2009 (doi:10.5194/acp-9-7753-2009) [DOI] [Google Scholar]
- 73.Gries G., Gries R., Perez A. L., Gonzales L. M., Pierce H. D., Oehlschlager A. C., Rhainds M., Zebeyou M., Kouame B. 1994. Ethyl propionate—synergistic kairomone for African palm weevil, Rhynchophorus phoenicis L (Coleoptera, Curculionidae). J. Chem. Ecol. 20, 889–897 10.1007/BF02059585 (doi:10.1007/BF02059585) [DOI] [PubMed] [Google Scholar]
- 74.Orlando J. J., Tyndall G. S., Fracheboud J. M., Estupinan E. G., Haberkorn S., Zimmer A. 1999. The rate and mechanism of the gas-phase oxidation of hydroxyacetone. Atmos. Environ. 33, 1621–1629 10.1016/S1352-2310(98)00386-0 (doi:10.1016/S1352-2310(98)00386-0) [DOI] [Google Scholar]
- 75.Jones C. E., Hopkins J. R., Lewis A. C. 2011. In situ measurements of isoprene and monoterpenes within a South-East Asian tropical rainforest. Atmos. Chem. Phys. Discuss. 11, 1189–1218 10.5194/acpd-11-1189-2011 (doi:10.5194/acpd-11-1189-2011) [DOI] [Google Scholar]
- 76.Hallquist M., et al. 2009. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 9, 5155–5236 10.5194/acp-9-5155-2009 (doi:10.5194/acp-9-5155-2009) [DOI] [Google Scholar]
- 77.Lee A., Goldstein A. H., Kroll J. H., Ng N. L., Varutbangkul V., Flagan R. C., Seinfeld J. H. 2006. Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes. J. Geophys. Res. Atmos. 111, D17305. 10.1029/2006jd007050 (doi:10.1029/2006jd007050) [DOI] [Google Scholar]
- 78.Kiendler-Scharr A., et al. 2009. New particle formation in forests inhibited by isoprene emissions. Nature 461, 381–384 10.1038/nature08292 (doi:10.1038/nature08292) [DOI] [PubMed] [Google Scholar]
- 79.Misztal P. K., et al. 2010. Large estragole fluxes from oil palms in Borneo. Atmos. Chem. Phys. 10, 4343–4358 10.5194/acp-10-4343-2010 (doi:10.5194/acp-10-4343-2010) [DOI] [Google Scholar]
- 80.Bouvier-Brown N. C., et al. 2009. Methyl chavicol: characterization of its biogenic emission rate, abundance, and oxidation products in the atmosphere. Atmos. Chem. Phys. 9, 2061–2074 10.5194/acp-9-2061-2009 (doi:10.5194/acp-9-2061-2009) [DOI] [Google Scholar]
- 81.Oke T. R. 1973. City size and urban heat island. Atmos. Environ. 7, 769–779 10.1016/0004-6981(73)90140-6 (doi:10.1016/0004-6981(73) 90140-6) [DOI] [Google Scholar]
- 82.White M. L., et al. 2009. Are biogenic emissions a significant source of summertime atmospheric toluene in the rural northeastern United States? Atmos. Chem. Phys. 9, 81–92 10.5194/acp-9-81-2009 (doi:10.5194/acp-9-81-2009) [DOI] [Google Scholar]
- 83.Green T. J., Reeves C. E., Fleming Z. L., Brough N., Rickard A. R., Bandy B. J., Monks P. S., Penkett S. A. 2006. An improved dual channel PERCA instrument for atmospheric measurements of peroxy radicals. J. Environ. Monit. 8, 530–536 10.1039/b514630e (doi:10.1039/b514630e) [DOI] [PubMed] [Google Scholar]
- 84.Ingham T., et al. 2009. A flow-tube based laser-induced fluorescence instrument to measure OH reactivity in the troposphere. Atmos. Meas. Tech. 2, 465–477 10.5194/amt-2-465-2009 (doi:10.5194/amt-2-465-2009) [DOI] [Google Scholar]
- 85.Ren X. R., et al. 2006. OH, HO2, and OH reactivity during the PMTACS–NY whiteface mountain 2002 campaign: observations and model comparison. J. Geophys. Res. Atmos. 111, D10S03. 10.1029/2005jd006126 (doi:10.1029/2005jd006126) [DOI] [Google Scholar]
- 86.Ren X. R., Harder H., Martinez M., Lesher R. L., Oliger A., Shirley T., Adams J., Simpas J. B., Brune W. H. 2003. HOx concentrations and OH reactivity observations in New York city during PMTACS–NY2001. Atmos. Environ. 37, 3627–3637 10.1016/s1352-2310(03)00460-6 (doi:10.1016/s1352-2310(03)00460-6) [DOI] [Google Scholar]
- 87.Da Silva G., Graham C., Wang F. 2010. Unimolecular beta-hydroxyperoxy radical decomposition with OH recycling in the photochemical oxidation of isoprene. Environ. Sci. Technol. 44, 250–256 10.1021/es900924d (doi:10.1021/es900924d) [DOI] [PubMed] [Google Scholar]
- 88.Sinha V., Williams J., Crowley J. N., Lelieveld J. 2008. The comparative reactivity method—a new tool to measure total OH reactivity in ambient air. Atmos. Chem. Phys. 8, 2213–2227 10.5194/acp-8-2213-2008 (doi:10.5194/acp-8-2213-2008) [DOI] [Google Scholar]
- 89.Monks P. S., Carpenter L. J., Penkett S. A., Ayers G. P. 1996. Night-time peroxy radical chemistry in the remote marine boundary layer over the Southern Ocean. Geophys. Res. Lett. 23, 535–538 10.1029/96GL00306 (doi:10.1029/96GL00306) [DOI] [Google Scholar]
- 90.Salisbury G., Monks P. S., Bauguitte S., Bandy B. J., Penkett S. A. 2002. A seasonal comparison of the ozone photochemistry in clean and polluted air masses at Mace Head, Ireland. J. Atmos. Chem. 41, 163–187 10.1023/A:1014202229304 (doi:10.1023/A:1014202229304) [DOI] [Google Scholar]
- 91.Drewnick F., et al. 2005. A new time-of-flight aerosol mass spectrometer (TOF-AMS)—instrument description and first field deployment. Aerosol Sci. Technol. 39, 637–658 10.1080/02786820500182040 (doi:10.1080/02786820500182040) [DOI] [Google Scholar]
- 92.Zhang Q., Alfarra M. R., Worsnop D. R., Allan J. D., Coe H., Canagaratna M. R., Jimenez J. L. 2005. Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry. Environ. Sci. Technol. 39, 4938–4952 10.1021/es048568l (doi:10.1021/es048568l) [DOI] [PubMed] [Google Scholar]
- 93.Ng N. L., et al. 2010. Organic aerosol components observed in northern hemispheric datasets from aerosol mass spectrometry. Atmos. Chem. Phys. 10, 4625–4641 10.5194/acp-10-4625-2010 (doi:10.5194/acp-10-4625-2010) [DOI] [Google Scholar]
- 94.Jimenez J. L., et al. 2009. Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 10.1126/science.1180353 (doi:10.1126/science.1180353) [DOI] [PubMed] [Google Scholar]
- 95.Morgan W. T., Allan J. D., Bower K. N., Capes G., Crosier J., Williams P. I., Coe H. 2009. Vertical distribution of sub-micron aerosol chemical composition from north-western Europe and the north-east Atlantic. Atmos. Chem. Phys. 9, 5389–5401 10.5194/acp-9-5389-2009 (doi:10.5194/acp-9-5389-2009) [DOI] [Google Scholar]
- 96.Alfarra M. R., Prevot A. S. H., Szidat S., Sandradewi J., Weimer S., Lanz V. A., Schreiber D., Mohr M., Baltensperger U. 2007. Identification of the mass spectral signature of organic aerosols from wood burning emissions. Environ. Sci. Technol. 41, 5770–5777 10.1021/es062289b (doi:10.1021/es062289b) [DOI] [PubMed] [Google Scholar]
- 97.Capes G., Johnson B., McFiggans G., Williams P. I., Haywood J., Coe H. 2008. Aging of biomass burning aerosols over West Africa: aircraft measurements of chemical composition, microphysical properties, and emission ratios. J. Geophys. Res. Atmos. 113, D00C15. 10.1029/2008jd009845 (doi:10.1029/2008jd009845) [DOI] [Google Scholar]
- 98.Simoneit B. R. T. 1999. A review of biomarker compounds as source indicators and tracers for air pollution. Environ. Sci. Pollut. Res. 6, 159–169 10.1007/BF02987621 (doi:10.1007/BF02987621) [DOI] [PubMed] [Google Scholar]
- 99.Jordan T. B., Seen A. J., Jacobsen G. E., Gras J. L. 2006. Radiocarbon determination of woodsmoke contribution to air particulate matter in Launceston, Tasmania. Atmos. Environ. 40, 2575–2582 10.1016/j.atmosenv.2005.12.024 (doi:10.1016/j.atmosenv.2005.12.024) [DOI] [Google Scholar]
- 100.Robinson N. H., et al. 2011. Source attribution of Bornean air masses by back trajectory analysis during the OP3 project. Atmos. Chem. Phys. Discuss. 11, 15 157–15 226 10.5194/acpd-11-15157-2011 (doi:10.5194/acpd-11-15157-2011). [DOI] [Google Scholar]
- 101.Hamilton J. F., Webb P. J., Lewis A. C., Hopkins J. R., Smith S., Davy P. 2004. Partially oxidised organic components in urban aerosol using GCXGC-TOF/MS. Atmos. Chem. Phys. 4, 1279–1290 10.5194/acp-4-1279-2004 (doi:10.5194/acp-4-1279-2004) [DOI] [Google Scholar]
- 102.Kautzman K. E., et al. 2010. Chemical composition of gas- and aerosol-phase products from the photooxidation of naphthalene. J. Phys. Chem. A. 114, 913–934 10.1021/jp908530s (doi:10.1021/jp908530s) [DOI] [PubMed] [Google Scholar]
- 103.Gabey A. M., Gallagher M. W., Whitehead J., Dorsey J. R., Kaye P. H., Stanley W. R. 2010. Measurements and comparison of primary biological aerosol above and below a tropical forest canopy using a dual channel fluorescence spectrometer. Atmos. Chem. Phys. 10, 4453–4466 10.5194/acp-10-4453-2010 (doi:10.5194/acp-10-4453-2010) [DOI] [Google Scholar]
- 104.Pike R. C., et al. 2009. Can a global model chemical mechanism reproduce NO, NO2, and O3 measurements above a tropical rainforest? Atmos. Chem. Phys. Discuss. 9, 27 611–27 648 10.5194/acpd-9-27611-2009 (doi:10.5194/acpd-9-27611-2009) [DOI] [Google Scholar]
- 105.Surratt J. D., et al. 2010. Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc. Natl Acad. Sci. USA 107, 6640–6645 10.1073/pnas.0911114107 (doi:10.1073/pnas.0911114107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dlugi R., et al. 2009. Turbulent exchange and segregation of HOx radicals and volatile organic compounds above a deciduous forest. Atmos. Chem. Phys. Discuss. 9, 24 423–24 476 10.5194/acpd-9-24423-2009 (doi:10.5194/acpd-9-24423-2009) [DOI] [Google Scholar]
- 107.Pugh T. A. M., MacKenzie A. R., Langford B., Nemitz E., Misztal P. K., Hewitt C. N. 2010. The influence of small-scale variations in isoprene concentrations on atmospheric chemistry over a tropical rainforest. Atmos. Chem. Phys. Discuss. 10, 18 197–18 234 10.5194/acpd-10-18197-2010 (doi:10.5194/acpd-10-18197-2010) [DOI] [Google Scholar]
- 108.Pugh T. A. M., et al. In preparation. A Lagrangian model of air-mass photochemistry and mixing using a trajectory ensemble: the Cambridge tropospheric trajectory model of chemistry and transport (CiTTyCAT). Geosci. Model Dev. Discuss. [Google Scholar]