

Abstract
The metalloid arsenic is a natural environmental contaminant to which humans are routinely exposed in food, water, air, and soil. Arsenic has a long history of use as a homicidal agent, but in the past 100 years arsenic, has been used as a pesticide, a chemotherapeutic agent and a constituent of consumer products. In some areas of the world, high levels of arsenic are naturally present in drinking water and are a toxicological concern. There are several structural forms and oxidation states of arsenic because it forms alloys with metals and covalent bonds with hydrogen, oxygen, carbon, and other elements. Environmentally relevant forms of arsenic are inorganic and organic existing in the trivalent or pentavalent state. Metabolism of arsenic, catalyzed by arsenic (+3 oxidation state) methyltransferase, is a sequential process of reduction from pentavalency to trivalency followed by oxidative methylation back to pentavalency. Trivalent arsenic is generally more toxicologically potent than pentavalent arsenic. Acute effects of arsenic range from gastrointestinal distress to death. Depending on the dose, chronic arsenic exposure may affect several major organ systems. A major concern of ingested arsenic is cancer, primarily of skin, bladder, and lung. The mode of action of arsenic for its disease endpoints is currently under study. Two key areas are the interaction of trivalent arsenicals with sulfur in proteins and the ability of arsenic to generate oxidative stress. With advances in technology and the recent development of animal models for arsenic carcinogenicity, understanding of the toxicology of arsenic will continue to improve.
Keywords: arsenic, cancer, exposure
The word “arsenic” elicits a fearful response in most people. This is because arsenic has a long history of being a poison, both intentional and unintentional, to humans. However, most laymen do not know or understand that we are constantly exposed to arsenic because it is naturally present in the environment, is used in commercial products, and has medical applications. Although most typical environmental exposures to arsenic do not pose a health risk, several areas of the world contain arsenic from natural or anthropogenic sources at levels that create a toxicological concern. Many of these areas have been identified, and efforts are being made to either remediate these areas or limit access to them.
Arsenic is the number one substance in the most recent (ATSDR, 2007a) Comprehensive, Environmental, Response, Compensation and Liability Act (CERCLA) Priority List of Hazardous Substances published by the Agency for Toxic Substances and Disease Registry (ATSDR). This list is comprised of substances found at hazardous waste sites on the National Priorities List. The substances are ranked on frequency or occurrence, toxicity, and potential for human exposure.
An understanding of the chemistry of arsenic is needed to appreciate the toxicology of this metalloid, which shares properties of metals and nonmetals. (A metal has luster, conducts heat and electricity, and is malleable and ductile. Elemental arsenic tends to be nonductile.) In the environment, arsenic is found in inorganic and organic forms and in different valence or oxidation states. The valence states of arsenic of environmental interest are the trivalent (III) and pentavalent (V) states. Elemental arsenic has a valence state of (0). Arsine and arsenides have a valence of (–III). In this review, we will be focused on the arsenicals in the trivalent and pentavalent states that are found in the environment and to which humans are exposed. A list of relevant environmental arsenicals is shown in Table 1. The structure of some of these arsenicals is shown in Figure 1.
TABLE 1.
Arsenic Compounds of Environmental and Human Relevance
| Trivalent oxidation state | Pentavalent oxidation state |
| Arsenite | Arsenate |
| Arsenic trioxide | Arsenic pentoxide |
| Monomethylarsonous acid | Monomethylarsonic acid |
| Dimethylarsinous acid | Dimethylarsinic acid |
| Trimethylarsine oxide | |
| Arsanilic acid | |
| Arsenobetaine |
FIG. 1.

Chemical structure of some relevant arsenic compounds.
The most toxicologically potent arsenic compounds are in the trivalent oxidation state. This has to do with their reactivity with sulfur containing compounds and generation of reactive oxygen species (ROS). However, humans are exposed to both trivalent and pentavalent arsenicals. In this review, we will discuss in a historical context the exposure of these compounds, how we have learned that the metabolism of arsenic is a critical determinant of its toxic effects, and potential modes of action (MOA), animal carcinogenicity, and the epidemiology of this metalloid. Table 2 highlights some of the historical aspects of arsenic over the past 250 years.
TABLE 2.
Timeline of Some Historic Events in the Toxicology of Arsenic
| 1786 | Fowler’s solution (1% potassium arsenite) |
| 1836 | Marsh Test for detection of arsenic developed |
| 1842 | Dimethylarsinic acid detected in the environment |
| 1867 | Paris green (copper acetoarsenite) used as insecticide in United States |
| 1887 | Hutchison proposes arsenic is a human skin carcinogen |
| 1910 | Salvarsan used as a chemotherapeutic agent |
| 1940s | British antilewisite is developed |
| 1942 | U.S. arsenic drinking water interim standard set at 50 μg/l |
| 1968 | Tseng et al. publish findings on prevalence of skin cancer in an arsenic-exposed Taiwanese population |
| 1975 | EPA adopts drinking water interim standard at 50 μg/l |
| 1993 | WHO recommends drinking water standard of 10 μg/l |
| 1995 | Dimethylarsinic acid a tumor promoter in four rat organs |
| 1998 | Dimethylarsinic acid a complete carcinogen in rat urinary bladder |
| 2000 | FDA approves arsenic trioxide for leukemia chemotherapy |
| 2001 | EPA lowers U.S. arsenic drinking water standard to 10 μg/l (ruling delayed to 2002) |
| 2001 | Inorganic arsenic a complete carcinogen in adult mice after transplacental exposure |
| 2002 | Arsenic (+3 oxidation state) methyltransferase isolated in rat liver cytosol |
| 2010 | Inorganic arsenic a complete carcinogen in adult mice after whole life exposure |
Arsenic as an Intentional Homicidal and Suicidal Poison
Arsenic is a naturally occurring element that an individual typically encounters every day in food, water, soil, and air. While understanding how environmental exposures may affect human health, especially at low levels, is currently an active area of research, humans have known on some level about the toxicity of arsenic for centuries.
In the Middle Ages, arsenic gained notoriety as an effective homicidal and suicidal agent, both because of the frequency of its use and because of its involvement in many high-profile murders. In fact, arsenic is often referred to as the “king of poisons” and the “poison of kings” because of its potency and the discreetness, by which it could be administered, particularly with the intent of removing members of the ruling class during the Middle Ages and Renaissance (Vahidnia et al., 2007). For example, it is well documented that arsenic was among the poisons used by the Medici and Borgia families to eradicate rivals (Cullen, 2008). Arsenic continued to enjoy its reputation as a high-profile poison and was implicated in several other prominent murder cases, most famously in the death of Napoleon Bonaparte in 1851, which some conspiracy theorists claim was a political assassination (Cullen, 2008).
Up until the mid-1850s, arsenic remained a popular poison for several reasons. Arsenic was readily available and because it is odorless and tasteless, it was undetectable in food or beverages (Bartrip, 1992). The most visible symptoms of acute arsenic poisoning—nausea, vomiting, diarrhea, and abdominal pain—could be easily confused with other common diseases at the time (e.g., cholera and pneumonia) (ATSDR, 2007b). Also, importantly, for a long time, there was no reliable analytical method for detecting, much less measuring, arsenic in tissue or other media, although early tests for arsenic were introduced in the mid-1700s. Interestingly, in the first trial ever recorded to present forensic evidence, a woman was sentenced to death because a white power recovered by a servant was “proven” to be arsenic, based on appearance, texture, behavior in water, and garlic-like odor when burned (Caudill, 2009; Cullen, 2008). The detection of arsenic took a leap forward in 1832 when James Marsh decided to investigate analytical methods to provide juries with more reliable evidence of “visible arsenic” (Cullen, 2008). His test method was first used in the trial of Marie LaFarge in France in 1840, in which Mme. LaFarge was charged with poisoning her husband with arsenic-laden cakes (Cullen, 2008). Generally, the tests involved mixing the sample of interest with zinc and acid and heating the vessel with a flame, which would cause a silvery substance to accumulate on the glass vessel; this was considered diagnostic for arsenic in amounts as low as 0.02 mg (Marsh, 1837; Newton, 2007). Although this method would be considered primitive by today's standards, the Marsh test represented a turning point in arsenic analytics and the beginning of the end of undetected arsenic poisonings.
Although stories of murder by arsenic appeal to the morbid interests of the public, these murders provided important insights that advanced the knowledge of arsenic toxicology. For example, information on the acute effects of arsenic and the target organs involved was obtained by studying poisonings. Importantly, these cases also precipitated the development of analytical methods for different media, including biological samples, which eventually led to an increased understanding of metabolism of arsenic. Due to improved understanding of arsenic measurement, one cannot readily “get away with murder” by using arsenic anymore. Nonetheless, incidents do still occur. As recently as 2003, arsenic poisoning made headlines when arsenic was detected in coffee served at a church meeting in Maine (Maine Rural Health, 2008; Zernike, 2003).
Medicinal Uses of Arsenic
Despite its toxicity—or perhaps because of it—arsenic has been used beneficially to treat certain ailments. Documented cases of arsenic as a therapeutic agent date back to before 2000 BCE (Antman, 2001; Hyson, 2007). The Father of Medicine, Hippocrates, is thought to have used an arsenic paste to treat ulcers and abscesses (Riethmiller, 2005; Waxman and Anderson, 2001). Other pioneering physicians (e.g., Aristotle and Paracelsus) are also reported to have used arsenic medicinally (Cullen, 2008; Jolliffe, 1993).
Although arsenic has been used throughout history, more detailed documentation of its use began in the late 18th century. Fowler’s solution, which was discovered in 1786, is a 1% solution of potassium arsenite that was used in the treatment of various diseases, including malaria, syphilis, asthma, chorea, eczema, and psoriasis (Rohe, 1896; Scheindlin, 2005). In 1910, Paul Ehrlich introduced a new arsenic-based drug called Salvarsan, which became known as the “magic bullet” for treating syphilis and was used until the use of penicillin became more prevalent in the 1940s (Riethmiller, 2005; Yarnell, 2005).
Arsenic also has a rich history as a cancer chemotherapeutic. As reported by Antman (2001), pharmacology texts from the 1880s described the use of arsenical pastes for the treatment of skin and breast cancer. In 1878, it was found that Fowler’s solution could be effective in lowering the white blood cell count in leukemia patients (Antman, 2001, 210-9361). Although the use of Fowler's solution eventually declined over time due to its overt toxicity, a more detailed understanding of arsenic mechanism of action has allowed arsenic trioxide to emerge as an effective chemotherapeutic drug for treating acute promyelocytic leukemia (Rust and Soignet, 2001; Zhang et al., 2001). With the success of this drug, the treatment of other cancers with arsenic trioxide is also being investigated (Murgo, 2001; Sekeres, 2007).
Arsenic as a Pigment and Use in Other Products
Arsenic’s use as a pigment (e.g., Paris Green or copper acetoarsenite) in the 1800s was suspected as a major source of unintentional arsenic poisonings. Although the arsenic-based pigment was used in many consumer products (e.g., toys, candles, and fabric), its use in wallpaper was particularly linked to widespread sickness and death during this period (Scheindlin, 2005; Wood, 1885). Concerns associated with the use of wallpaper containing arsenic-based pigment were reported as early as 1839, and the theory was eventually proposed that illnesses from wallpaper were related to the biotransformation of the arsenic compounds by mold to a toxic arsenic gas (Gosio gas) (Cullen and Bentley, 2005). This theory gained momentum, and in 1893, Bartolomeo Gosio, an Italian physician, demonstrated that arsenic could be volatilized from arsenic-containing compounds, including Paris Green (Buck and Stedman, 1904; Cullen and Bentley, 2005). Although it became widely accepted at the time that arsenic gas from the wallpaper was responsible for the deaths and illnesses, this notion has been challenged recently by scientists who believe that there were insufficient quantities of the gas generated (now known to be trimethylarsine) to cause the reported effects; and possibly the mold, itself, was the responsible agent (Cullen and Bentley, 2005). Regardless of the toxicity of the wallpaper, the work conducted by Gosio and later by Frederick Challenger (in the late 1930s), laid the groundwork for today’s understanding of arsenic metabolism, namely that the metabolism of arsenic involves sequential reduction and oxidative methylation steps (Cullen and Bentley, 2005).
Although arsenic use has been phased out of pigment products, it is still used in the production of glass and semiconductors (ATSDR, 2007b).
Arsenic as a Pesticide
Although it was recognized that the arsenic used in pigments could be toxic to humans, Paris Green was used as an insecticide from 1867 to 1900; it was effective in controlling Colorado potato beetles and mosquitoes (Cullen, 2008; Peryea, 1998). In the late 1800s, another arsenic-based pesticide, lead arsenate, became extensively used; it was an effective pesticide but was less toxic to plants than Paris Green (Peryea, 1998). Lead arsenate was widely used as a pesticide for apple and cherry orchards through the early 1900s. By 1960, most uses of lead arsenate were phased out after it was recognized that its use was associated with health effects in orchard workers and an increasing concern that arsenic residues on fruits were a public health concern (Frisbie, 1936; Nelson et al., 1973). The official United States ban in lead arsenate use did not take place until 1988 (Peryea, 1998).
Up to the early 1900s, concerns about arsenic toxicity had focused on the acute effects of arsenic; the use of arsenic in insecticides furthered the understanding of how low levels of exposure over longer periods might affect public health. Studies in orchard workers were some of the first to propose a link between arsenic exposure and cancer (lung cancer in this case) (Mabuchi et al., 1980; Nelson et al., 1973; Tollestrup et al., 1995). Also, because arsenic was released to the environment in concentrated amounts, the use of arsenic-based pesticides helped develop knowledge of arsenic’s fate and transport (Peryea, 1998).
The use of lead arsenate pesticides has been effectively eliminated for over 50 years. However, because of the pesticide’s environmental persistence, it is estimated that millions of acres of land are still contaminated with lead arsenate residues. This presents a potentially significant public health concern in some areas of the United States (e.g., New Jersey, Washington, and Wisconsin), where large areas of land used historically as orchards have been converted into residential developments (Hood, 2006).
Some modern uses of arsenic-based pesticides still exist. Chromated copper arsenate (CCA) has been registered for use in the United States since the 1940s as a wood preservative, protecting wood from insects and microbial agents. In 2003, CCA manufacturers instituted a voluntary recall of residential uses of CCA-treated wood. CCA is still approved for use in nonresidential applications, such as in marine facilities (pilings and structures), utility poles, and sand highway structures (U.S. EPA, 2008; WPSC, 2008).
The use of organic arsenical pesticides began in the 1950s and has continued into the present day. Overall, organic arsenicals in the pentavalent oxidation state are much less toxic than inorganic arsenicals because, unlike inorganic arsenic, these ingested organic arsenicals are not readily taken up into cells and undergo limited metabolism (Cohen et al., 2006). Key pesticides based on organic arsenicals include monosodium methanearsonate (MSMAsV) and dimethylarsinic acid (DMAsV), also known as cacodylic acid. The use of these compounds as pesticides has given scientists the opportunity to comparatively study organic and inorganic arsenic toxicity and carcinogenicity, including investigations on metabolites of organic and inorganic arsenic compounds (e.g., the trivalent methylated arsenic compounds). In particular, the study of cacodylic acid has helped elucidate a potential mode of carcinogenic action for arsenic. In light of this, the U.S. Environmental Protection Agency (EPA) (2006) chose not evaluate DMAsV as carcinogenic to humans in the reregistration decision for this pesticide (U.S. EPA, 2006). Similarly, in 2007, a U.S. EPA Science Advisory Board (SAB) determined that DMAsV had the potential to cause cancer in humans only when doses are high enough to cause bladder cell cytotoxicity (U.S. EPA, 2007). Some uses of these compounds as herbicides were discontinued in 2009 (U.S. EPA, 2009). At that time, EPA reregistered MSMAsV for use on cotton but decided to stop the use of it on golf courses, sod farms, and right-of-ways in 2013. However, EPA agreed to reevaluate new information on the carcinogenicity of inorganic arsenic, the environmental degradation product of the organic arsenical products in 2012. EPA will consider the results of the reevaluation in make decisions regarding new applications for registration of MSMAsV.
Copper Smelting
Studies of orchard workers have provided a basis for understanding some of the long-term effects of occupational exposure to arsenic. However, occupational exposure studies in the copper smelting industry are much more extensive and have established definitive links between arsenic, a by-product of copper smelting, and lung cancer via inhalation (e.g., Enterline et al., 1995; Jarup et al., 1989; Pinto et al., 1977; Welch et al., 1982). Dermal and neurological effects were also increased in some of these studies (Dunlap, 1921; Feldman et al., 1979; Lagerkvist et al., 1986). Although exposures to arsenic changed over time (i.e., as time went on, occupational controls became more stringent and workers were exposed to reduced arsenic concentrations), the arsenic exposures measured from these studies ranged from about 0.05 to 0.3 mg/m3 and are significantly higher than airborne environmental exposures to arsenic (which range from 0 to 0.000003 mg/m3) (ATSDR, 2007b; European Commission, 2000). The pathway of exposure for these workers was mainly via inhalation of arsenic dusts but could also be from arsenic trioxide vapors (Yager et al., 1997).
Communities near smelters have also been studied (Marsh et al., 1997; Tollestrup et al., 2003). Exposure in these studies was often not well characterized but might have been via inhalation or from the ingestion of arsenic-contaminated soil, depending on the activity of the smelter. Overall, no clear association has been established between environmental exposure to arsenic and adverse health effects in communities residing near smelters (as reviewed in European Commission, 2000).
Arsenic in the Environment
Historical uses of arsenic in products, pharmaceuticals, and industry have led to important insights on arsenic’s toxicity. In the modern era, most interest in arsenic toxicology comes from naturally occurring background exposures in food, water, and soil. Understanding the environmental levels that can cause a public health concern is a key area of research.
Arsenic in Drinking Water
Arsenic found in water is almost entirely in the inorganic form and can be stable as both arsenite and arsenate, trivalent and pentavalent inorganic arsenicals, respectively (Saxe et al., 2006). The U.S. Geological Survey estimates that the median groundwater concentration is 1 μg/l or less, although some groundwater aquifers, particularly in the western United States, can contain much higher levels (Focazio et al., 1999). For example, median levels in Nevada were about 8 μg/l (Focazio et al., 1999) but levels of naturally occurring arsenic as high as 1000 μg/l have been measured in the United States in drinking water (Lewis et al., 1999; Steinmaus et al., 2003).
Several regions outside the United States also have high levels of arsenic in groundwater, averaging several hundred μg/l, resulting in significant human exposures via drinking water (for review, see Nordstrom, 2002). An extensive body of research studying the health effects associated with arsenic in drinking water both within and outside the United States has been published. It is through these studies that ingestion of arsenic has been definitively linked to increased incidence of cancer in lung, bladder, skin, kidney, liver, and potentially prostate. A number of noncancer effects also are linked to exposure in drinking water, including skin lesions, cardiovascular disease, neurological effects, and diabetes (ATSDR, 2007b; NRC, 1999, 2001). A historical perspective on the important contributions that drinking water studies have made to our understanding of oral chronic toxicity from arsenic is described in more detail later in this paper.
Arsenic in the Diet
Although inorganic arsenic was added to food as a preservative in the late 1800s and early 1900s, today, inorganic arsenic is not intentionally added to food (Buck and Stedman, 1904; Taylor, 1873). Nonetheless, because arsenic is ubiquitous in the environment, diet is the largest source of both inorganic and organic arsenic for typical individuals. Estimates of dietary inorganic arsenic intakes vary. In the United States, Schoof et al. (1999) estimated an average adult intake of 3.2 μg/day, with a range of 1–20 μg/day. Estimates for children were similar (Yost et al., 2004). Recently, the European Food Safety Authority (EFSA) estimated a higher intake level, although estimates depended on simplifying assumptions regarding the ratio of inorganic arsenic to total arsenic in food. The analysis estimated a typical intake of 0.13–0.56 μg/kg/day for average consumers (9.1–39.2 μg/day for a 70 kg adult) (EFSA, 2009).
The possibility that arsenic is an essential nutrient has received some research attention, especially in the 1970s and 1980s, although some interest extends into the present day (Uthus, 1992, 2003). In 1988, the U.S. EPA convened a scientific panel to specifically evaluate the potential essentiality of arsenic (U.S. EPA, 1988). Based on an extensive review of the literature, this panel concluded, “information from experimental studies with rats, chicks, minipigs, and goats demonstrates the plausibility that arsenic, at least in inorganic form, is an essential nutrient.” Since 1988, very little new research on the essentiality of arsenic has been conducted. In 1999, a National Research Council report stated that essentiality of arsenic in humans had not been tested to date and there was no known biochemical process for which arsenic was essential (NRC, 1999). Later, an EPA arsenic science advisory panel cited similar arsenic essentiality evidence as the 1988 panel and determined that arsenic essentiality is in “need of further research” (U.S. EPA, 2007).
Food also contains many organic arsenic compounds, which are generally considered to have low toxicity, although toxicity does vary among the individual compounds. Developing analytical methods to identify these compounds has been important for distinguishing these compounds from the more toxic inorganic forms. The key organic arsenic compounds that can be routinely found in food (depending on food type) include monomethylarsonic acid (MMAsV), DMAsV, arsenobetaine, arsenocholine, arsenosugars, and arsenolipids. DMAsV or MMAsV can be found in various types of fin fish, crabs, and mollusks, but often at very low levels (Borak and Hosgood, 2007). Arsenobetaine is the major form of arsenic in marine animals, and, by all accounts, it is considered a compound that is nontoxic under conditions of human consumption (ATSDR, 2007b; EFSA, 2009). Although arsenobetaine is little studied, available information indicates it is not mutagenic, immunotoxic, or embryotoxic (Borak and Hosgood, 2007). Arsenocholine, which is mainly found in shrimp, is chemically similar to arsenobetaine, and is considered to be “essentially nontoxic” (ATSDR, 2007b).
Arsenosugars and arsenolipids have recently been identified. Exposure to these compounds and toxicological implications are currently being studied. Arsenosugars are detected mainly in seaweed but are also found to a lesser extent in marine mollusks (EFSA, 2009). Concerns about the potential toxicity of arsenosugars have been raised because there is evidence that arsenosugars are metabolized to DMAsV (Andrewes et al., 2004). Studies addressing arsenosugar toxicity, however, have largely been limited to in vitro studies, which show that arsenosugars are significantly less toxic than both inorganic arsenic and trivalent methylated arsenic metabolites (Kaise et al., 1996). Arsenolipids, which are a component of fish oil, have only been recently characterized; their toxicity has not been studied (Schmeisser et al., 2006).
Arsenic in Soil
The natural content of arsenic in soils globally ranges from 0.01 to over 600 mg/kg, with an average of about 2–20 mg/kg (Yan-Chu, 1994). In the United States, a nationwide survey conducted in areas that were judged not to have anthropogenic sources of arsenic reported that natural background concentrations in soil ranged from less than 1 to 97 mg/kg (Shacklette and Boerngen, 1984). Arsenic in soil is almost entirely in the inorganic form, except in areas with intentional organic arsenic application, where higher levels of organic compounds can be found (Saxe et al., 2006). In soils, pentavalent arsenic predominates due to oxidation of trivalent arsenicals (Gong et al., 2001).
Exposure to arsenic in soil can occur through multiple pathways. Incidental ingestion is typically the most significant exposure pathway for soil. Compared with the intake of naturally occurring arsenic from water and the diet, soil arsenic constitutes only a small fraction of intake (Petito Boyce et al., 2008); this is a reflection of the relatively small amounts of inorganic arsenic in soil that is typically ingested on a daily basis as well as the reduced bioavailability of arsenic in soil compared with water. Overall, a large number of studies have shown that the relative oral bioavailability of arsenic in soils to be less than 50% (Roberts et al., 2002).
Other potential exposure pathways for soil arsenic include dermal absorption and inhalation of wind-blown soil particles (i.e., fugitive dust). However, arsenic is not readily absorbed through the skin from soil (U.S. EPA, 2001), and, as discussed below, the amount of arsenic measured in ambient air is low.
Arsenic in Air
Compared with arsenic exposure from food and water, exposure to arsenic in air, which is almost entirely as inorganic arsenic, is generally very low. The European Commission (2000) reports that levels of arsenic in air range 0–1 ng/m3 in remote areas, 0.2–1.5 ng/m3 in rural areas, 0.5–3 ng/m3 in urban areas, and up to about 50 ng/m3 in the vicinity of industrial sites. Based on these data, the European Commission (2000) estimated that in relation to food, cigarette smoking, water, and soil, air contributes less than 1% of total arsenic exposure, even when assuming an arsenic air exposure that is significantly above typical background (i.e., 20 ng/m3).
Arsenic Metabolism
Study of the metabolism of inorganic arsenic in mammals is rooted in 19th century studies of arsenic metabolism in microorganisms (Cullen, 2008). As discussed above, identification of Gosio gas as trimethylarsine, a volatile product emitted by microorganisms exposed to inorganic arsenic, presaged work on microbial conversion of inorganic arsenic into methylated metabolites. Based on these studies, Challenger (1947, 1951) provided a chemically plausible scheme (I) for the methylation of inorganic arsenic.
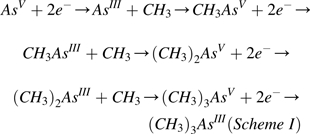 |
(AsV, arsenate; AsIII, arsenite; CH3AsV, monomethylarsonic acid; CH3AsIII, monomethylarsonous acid; (CH3)2AsV, dimethylarsinic acid; (CH3)2AsIII, dimethylarsinous acid; (CH3)3AsV, trimethylarsine oxide; (CH3)3AsIII, trimethylarsine).
For Challenger’s scheme of arsenic biotransformation (i.e., alternate steps involving reduction of arsenic from pentavalency to trivalency and subsequent oxidative methylation of trivalent arsenicals) to move from a plausible theory to a well-accepted biological principle, a variety of chemical and biochemical approaches were explored. Over the past half century, research has revealed a remarkable unity in patterns of arsenic metabolism in organisms as diverse as simple prokaryotes and members of the deuterostomal superphylum, including humans. Elucidating the pathway for arsenic metabolism has provided new insights into production of metabolites of inorganic arsenic. Advancements in our understanding suggest that these metabolites may mediate some of the toxic and carcinogenic effects associated with exposure to inorganic arsenic. From these efforts has emerged a fuller picture of the complex relations between the metabolism and toxicity of arsenic and the role that interindividual variation in capacity to metabolize arsenic may play as a determinant of risk. Continuing exploration of the metabolic pathway will likely provide more insights into the linkage of metabolism and toxicity. Here, we review our evolving understanding of arsenic metabolism over the past 5 decades and describe some of the challenges for future research.
The possibility that inorganic arsenic undergoes chemical conversion in higher organisms was noted at least 60 years ago in a study that found “discrepancies in analytical data” in mice and rats treated with arsenic-containing water and sludge samples (Clements et al., 1951). Because analysis of arsenic in tissues and excreta by then-standard colorimetric assays did not account for all the administered arsenic, the authors conjectured that some of the missing mass of arsenic might be present in tissues and excreta in previously unknown and undetected forms. Improvement in analytical techniques over the next 2 decades made possible detection of methylated arsenicals in environmental samples and human urine. Braman and Foreback (1973) used hydride generation–cryotrapping-column chromatography-atomic absorption spectrometry to demonstrate that natural waters and human urine contained both inorganic and organic (methylated) arsenicals. In this study of four individuals, inorganic arsenic accounted for about 25%, methyl arsenic accounted for about 8%, and dimethyl arsenic for about 66% of the total arsenic in urine. Remarkably, despite the many refinements and improvements in analytical techniques for arsenic speciation over intervening years, these estimates of fractional distribution of arsenic metabolites in urine are consistent with currently reported values. Using a single volunteer, Crecelius (1977) explored the metabolism of arsenic after ingestion of wine or water that contained inorganic arsenic. After ingestion of wine or water, the concentration of inorganic arsenic in urine peaked within 24 h; peak concentrations of methyl and dimethyl arsenic in urine occurred at later time points.
The temporal pattern for the appearance of inorganic and methylated arsenicals in urine reported by Crecelius was broadly compatible with the pattern predicted by the Challenger scheme, suggesting that internal dosimetry of inorganic arsenic and its metabolites would follow a similar pattern. These findings were the impetus for a large body of work on the distribution, retention, and clearance of arsenicals in humans and other species. These dosimetric data have supported development of several pharmacokinetic models for humans and other species (El-Masri and Kenyon, 2008; Evans et al., 2008; Gentry et al., 2004; Mann et al., 1996a, 1996b; Yu, 1999). Such models are useful for estimating tissue exposures and can be used in risk assessment (Liao et al., 2008).
Three decades ago, the biochemical reactions underlying arsenic methylation were unknown. Braman and Foreback (1973) suggested that methylation of arsenic was due to “methylcobalamin-methionene (sic) reactions in the body.” This hypothesis reflected then-current ideas about methylation of metals and metalloids as exemplified by studies of microbial methylation of inorganic mercury (Landner, 1972). Wood (1974) formally stated this chemical scheme for arsenic methylation in a review of toxic element methylation processes.
Early work on formation of methylated arsenicals in mammals used tissue homogenates and subcellular fractions to explore the requirements for in vitro methylation. Buchet and Lauwerys (1985) showed addition of S-adenosylmethionine (AdoMet) and glutathione (GSH) to reaction mixtures containing rat liver cytosol supported conversion of inorganic arsenic to methylated products. Focus on AdoMet as a methyl group donor for arsenic methylation was consistent with the then-contemporary finding that enzymatically catalyzed methylation of selenium used AdoMet for methyl group donation (Mozier et al., 1988). Thus, by the late 1980’s, it was possible to define the operational characteristics of the enzyme that catalyzed the conversion of inorganic arsenic to methylated metabolites. It was likely to be a cytosolic AdoMet-dependent methyltransferase that required the presence of a reductant. Although it was unclear whether the reductant protected the enzyme against oxidation or functioned as a cofactor in enzyme catalysis, an absolute requirement for a reductant imposed a number of limitations on any strategy used to isolate and characterize the protein.
Purification of a mammalian arsenic methyltransferase was the object of intensive research in the 1990s. In a series of studies summarized by Aposhian (1997), a ∼60 kDa protein purified from rabbit liver cytosol catalyzed AdoMet-dependent methylation of arsenite and monomethylarsonous acid (MMAsIII). The presence of a monothiol (e.g., cysteine, GSH) or a dithiol (e.g., dithiothreitol) was required for these activities (Zakharyan et al., 1999). In related work, Lin et al., (2002) purified a ∼43 kDa protein from mouse liver cytosol that catalyzed AdoMet-dependent methylation of arsenite and MMAsIII. A partial amino acid sequence of this protein showed that it was the product of the cyt19 gene previously annotated as a methyltransferase of unknown function. Subsequently, this mouse gene was annotated as arsenic (+3 oxidation state) methyltransferase (As3mt) and cloned (Walton et al., 2003). Mouse As3mt proved to be a prototype for arsenic methyltransferases in many genomes. Genes encoding proteins closely related to mouse As3mt occur in genomes of organisms ranging from sea squirts to humans (Thomas et al., 2007). These proteins share conserved sequences commonly found in nonnucleic acid methyltransferases and cysteinyl residues required for catalytic function as an arsenic methyltransferase (Fomenko et al., 2007). Given differences in molecular masses of proteins isolated from rabbit liver and mouse liver, they are unlikely to be identical. In the absence of full sequencing of the rabbit liver protein, it has proven difficult to pursue the characteristics of this putative arsenic methyltransferase. Indeed, subsequent research has focused on the role of As3mt and its protein products in the metabolism of arsenicals.
Although the presence of GSH in buffers preserved enzymatic activity during purification, dithiol-containing reductants (thioredoxin, Tx; glutaredoxin, Gx; reduced lipoic acid, LA) were found to support high rates of catalysis in vitro by recombinant rat As3mt (Waters et al., 2004b). Although GSH modulated production of mono-, di-, and trimethylated arsenicals from inorganic arsenic, it could be replaced by Tx, Gx, or LA, indicating that its presence was not required for catalysis (Waters et al., 2004a). These findings suggested that redox cycling of physiological dithiol-containing reductants was required for catalytic activity of As3mt. Because all intermediates predicted by the Challenger scheme, including methylated arsenicals containing trivalent or pentavalent arsenic, can be found in reaction mixtures with recombinant rat As3mt, a Tx/Tx reductase/NADPH cycling system, and AdoMet, As3mt may catalyze both oxidative methylation of trivalent arsenic-containing substrates and reduction of pentavalent arsenic in methylated products. The fusing of two intimately related catalytic functions in one enzyme would assure that metabolic transformation proceeded without accumulation of reactive and toxic intermediates. Notably, there is evidence for separate pathways for reduction of pentavalent arsenic to trivalent arsenic in mammalian cells. For example, GSH transferase omega (GSTO) catalyzes in vitro reduction of monomethylarsonic acid (MMAsV) to MMAsIII (Zakharyan et al., 2001). Whether GSTO contributes significantly to arsenic reduction in intact organisms is unclear; studies in GSTO knockout mice failed to detect differences in patterns of arsenic methylation and excretion (Chowdhury et al., 2006). Phosphorolytic-arsenolytic enzymes such as purine nucleoside phosphorylase (PNP) can function as catalysts in reaction schemes in which an activated arsenate ester is reduced by thiols to arsenite (Gregus et al., 2009; Nemeti et al., 2010). Existence of several mechanisms for reduction of pentavalent arsenic could be an example of functional redundancy in which independent processes mediate a single important biochemical reaction (Wang and Zhang, 2009). Understanding interrelations between catalysis of methylation and of reduction in the cell will be critical to more fully comprehend the molecular basis for arsenic metabolism.
Studies in which altered As3mt expression leads to changes in capacity for arsenic methylation confirm its critical role. Heterologous expression of rat As3mt in human uroepithelial cells that do not methylate arsenic confers capacity to methylate arsenic (Drobna et al., 2005). Silencing of AS3MT expression in human hepatoma cells by RNA interference diminishes, but does not eliminate, capacity to methylate arsenic (Drobna et al., 2006). The fate of arsenic differs radically in As3mt knockout mice and wild-type mice. Following single or repeated oral doses of arsenate, tissues from As3mt knockout mice attain higher concentrations of inorganic arsenic than do those from wild-type mice and the rate of whole body clearance of arsenic is much slower in knockout than in wild-type mice (Drobna et al., 2009; Hughes et al., 2010). A lower rate of whole body clearance of inorganic arsenic and its methylated metabolites in As3mt knockout mice is consistent with earlier findings that methylated and dimethylated arsenic clear faster than inorganic arsenic (Buchet et al., 1981; Marafante et al., 1987). These phenotypic changes in As3mt knockout mice are also associated with increased susceptibility to damage to the uroepithelial cells following exposure to inorganic arsenic (Yokohira et al., 2010).
Capacity to produce methylated metabolites of inorganic arsenic can depend on environmental or genetic factors. For example, environmental factors such as selenium nutriture or protein and lipotrope deficiency affect patterns of urinary metabolites in mice treated with inorganic arsenic (Kenyon et al., 1997; Vahter and Marafante, 1987). Similarly, there is evidence that genotypic variation in the AS3MT gene in humans alters the capacity to methylate inorganic arsenic. Changes in the capacity to methylate inorganic arsenic can be referred to as the arsenic methylation phenotype. Thus, it is possible to look for connections between AS3MT genotype and arsenic methylation phenotype.
A linkage between AS3MT genotype and arsenic methylation phenotype was first noted in a study of arsenite metabolism by cultured primary human hepatocytes (Drobna et al., 2004). Sequencing of cDNA from one of eight hepatocyte donors detected a single missense allelic mutation in exon 9 that would change amino acid 287 in wild-type AS3MT from methionine to threonine (M287T), indicating that this donor was a heterozygote expressing both wild-type AS3MT and M287T AS3MT. This genotypic difference was associated with an altered arsenic methylation phenotype. Cells from the affected donor showed a higher capacity for production of monomethylarsenic at medium concentrations of arsenite compared with cells from donors homozygous for wild-type AS3MT (which showed reduced mono- and dimethylarsenic production). In populations worldwide, the nonsynonymous single nucleotide polymorphism (SNP) for the M287T mutation occurs at an allele frequency of 0.01 to 0.1 (Fujihara et al., 2007; Wood et al., 2006). Hence, under Hardy-Weinberg conditions, the frequency of AS3MT heterozygotes (wild-type/M287T) would range from 0.02 to 0.18 and the frequency of M287T AS3MT homozygotes from 0.0001 to 0.01. COS-1 cells expressing M287T AS3MT had 350% higher levels of arsenic methylation activity and 190% higher levels of immunoreactive AS3MT than did cells expressing wild-type AS3MT (Wood et al., 2006). This finding suggested that regulation of AS3MT expression or turnover of AS3MT was affected by the M287T mutation.
Population-based studies have shown that M287T genotype and genotype differences at other exonic and intronic sites affect arsenic methylation phenotype. Compared with the wild-type genotype, M287T genotype is associated with a significant increase in the odds ratio for percentage of urinary arsenic present as % monomethylarsenic (%MMAs) in men working in a copper smelter (Hernandez et al., 2007, 2008). A similar increase in %MMAs was associated with the M287T genotype in men, but not women, who were exposed to inorganic arsenic from drinking water and food (Lindberg et al., 2007). An altered sequence in exon 1 encoding part of the 5′ untranslated region of variable number tandem repeats is associated with a significant increase in the odds ratio for percentage of urinary arsenic present as monomethylarsenic in men working in a copper smelter (Hernandez et al., 2007). In contrast to the effects of exonic changes, several sequence changes in introns 6, 7, and 9 of AS3MT result in decreased %MMAs and increased % dimethylarsenic in urine compared with those found in individuals with wild-type AS3MT (Agusa et al., 2009; Chung et al., 2009; Schläwicke Engström et al., 2007).
The concept of genotype-phenotype correlations can be extended to determine if changes in AS3MT genotype can be linked to evidence of altered susceptibility to adverse health effects associated with chronic exposure to inorganic arsenic. In a Mexican population that chronically uses drinking water contaminated with inorganic arsenic, M287T AS3MT genotype was associated with significantly increased %MMAs (Valenzuela et al., 2009). A marginal difference (p < 0.055) was found in frequencies of the M287T genotype and occurrence of premalignant skin lesions, suggesting that a genotype change associated with a change in the arsenic methylation profile could be linked to increased susceptibility to a type of skin lesion commonly associated with chronic exposure to arsenic. Another study in this Mexican population found that the percentage of DNA damage in peripheral blood leukocytes (PBLs) of children (measured by the comet assay) and the level of arsenic exposure was significantly influenced (p < 0.034) by M287T genotype (Sampayo-Reyes et al., 2010). A follow-up study in a Taiwanese population that examined the relation between urinary profiles of arsenicals and cancer risk over a 15 year period during which exposure to inorganic arsenic decreased found associations between earlier cancer incidence and higher baseline urinary %MMAs or smaller temporal change in urinary %MMAs (Chung et al., 2009). This finding suggests that persistently high levels of monomethylarsenic in urine related to the M287T genotype are associated with increased cancer risk. Taken together, these studies show that the M287T genotype that affects the arsenic methylation phenotype can also be associated with increased risk of either noncancer or cancer health effects. Additional studies will show whether other AS3MT polymorphisms produce these or other phenotypic effects.
The effects of polymorphisms in AS3MT should be evaluated in the context of polymorphisms in other genes that may affect the arsenic methylation phenotype or influence susceptibility to the adverse health effects associated with chronic exposure to inorganic arsenic. Polymorphisms in two genes encoding enzymes used in AdoMet production, methylenetetrahydrofolate reductase, and cystathione β synthase, affect the profile of arsenicals found in urine (Porter et al., 2010; Steinmaus et al., 2007). An exon 3 polymorphism in PNP, a putative arsenate reductase, increases the risk of skin cancer in individuals exposed to inorganic arsenic in drinking water (De Chaudhuri et al., 2008). Associations have been identified between polymorphisms in several genes encoding members of the GSH transferase (GST) family and urinary metabolite profiles or risk for arsenic-induced cancers (Lin et al., 2007; McCarty et al., 2007a, 2007b; Paiva et al., 2010; Wang et al., 2009). Although the single-gene strategy for identifying modifiers is effective, a more heuristic approach to understanding the role of genes in control of arsenic metabolism and toxicity has looked for haplotypes associated with altered metabolism or response. For example, three SNPs in AS3MT are in linkage disequilibrium (LD) and associated with an altered arsenic methylation phenotype (Meza et al., 2005). A LD cluster on human chromosome 10 near the AS3MT locus indicates that several genes may be acting to modify the arsenic methylation phenotype or to alter susceptibility (Gomez-Rubio et al., 2010). Further studies of gene associations and of the role of members of the LD cluster will be needed to identify the modifiers.
Although this review has focused on metabolism of arsenicals mediated by the host organism, particularly the central role of AS3MT as a determinant of metabolic capacity and susceptibility, the organisms that compose the microbiome of the gastrointestinal tract also have the capacity to convert inorganic arsenic to methylated species. The microbiome of the human gastrointestinal tract consists of nearly 10-times as many cells as does the body of the host and contains about 3-times as many genes as does the human genome (Zhu et al., 2010). Given the size and genetic diversity of organisms of the gut microbiome, it is probably not surprising that a complex interaction has evolved between these cells and cells that constitute the host component of the gastrointestinal system (Spor et al., 2011). Evidence for a role of the microbiota of the gastrointestinal tract in the metabolism of arsenicals comes primarily from in vitro studies in which the microbiota of the mouse cecum is incubated under strictly anaerobic conditions with arsenicals. Studies with arsenate and DMAsV found that these arsenicals were rapidly converted to methylated metabolites (Kubachka et al., 2009; Pinyayev et al., 2011). In addition to oxyarsenical metabolites, the products of these reactions included thioarsenicals in which O is replaced with S. The prevalence of thioarsenicals among the products of metabolism of anaerobic microorganisms presumably reflects low O tension and the abundance of H2S in cultures. In terms of risk, the significance of metabolism of arsenic by the microbiota of the gastrointestinal tract (preabsorptive metabolism) depends on whether the bioavailabilities of the metabolites are materially different from those of the parent compounds. Additional studies are needed to resolve this issue.
Over the past half century, the study of arsenic metabolism has progressed from descriptive studies of kinetic behavior of inorganic arsenic and its metabolites to an effort to understand the molecular basis of metabolism and the interactions among genes that affect the capacity for metabolism and modify the biological response to this arsenic. It has recently been suggested that arsenic is a toxicant that could profitably be studied by newly available techniques in genomics, metabolomics, and proteomics (Vlaanderen et al., 2010). It will be interesting to see what new insights these technologies will bring to our understanding of arsenic metabolism and toxicity.
Arsenic Mode of Action
The precise MOA for the many disease endpoints following acute and chronic arsenic exposure are not known, although research in this area has been ongoing for many years. A clear understanding of the MOA for arsenic, indeed for any toxic chemical, will facilitate selection of the appropriate human risk assessment model (e.g., linear vs. nonlinear). The most appropriate dose-response model for low environmental exposures to arsenic is the subject of debate. It is likely that several MOAs account for the adverse effects of arsenic; indeed, these MOAs may be interdependent. Although inorganic arsenic’s MOA for all disease endpoints is not fully elucidated, it is apparent that there are clear differences in the toxicities of arsenicals, largely due to valence state. Trivalent arsenicals (e.g., arsenite, monomethylarsonous acid, dimethylarsinous acid) are more potent toxicants than pentavalent arsenicals (e.g., arsenate, monomethylarsonic acid, dimethylarsinic acid). Several review articles have been published recently on the MOA for arsenic (Druwe and Vaillancourt, 2010; Kitchin and Conolly, 2010; Kitchin and Wallace, 2008; Kumagi and Sumi, 2007; Platanias, 2009; Rossman, 2003; Schoen et al., 2004; Schumacher-Wolz et al., 2009; Tapio and Grosche, 2006). Several proposed MOA for arsenic and examples of the biochemical effects that occur are shown in Figure 2.
FIG. 2.

Proposed MOA for arsenic and examples of biochemical effects that result from this action.
Interactions with Sulfur
The chemistry of arsenic is an important aspect of its MOA. Observations in the late 19th century noted that arsenic exists in the body in pentavalent and trivalent oxidation states and that it interacts with sulfur (Parascandola, 1977). One of the first proposed MOAs for arsenic, suggested by Binz and Schulz in 1879 (Parascandola, 1977), was the interference of cellular oxidation from the cycling of oxygen during the interconversion of arsenate and arsenite. This would suggest that both arsenicals are equally potent, but as it became apparent, arsenite is more potent than arsenate, so this proposal was soon disfavored.
Early mechanistic studies centered on an aromatic arsenic compound used to treat syphilis and trypanosomiasis. This compound, Salvarsan, was synthesized by Paul Ehrlich’s laboratory in the early 1900s and was the first man-made antibiotic. (See Riethmiller, 2005 for a review of the discovery of Salvarsan and Lloyd et al., 2005 for determination of its structure.) Ehrlich proposed that the chemotherapeutic (toxic) effect of Salvarsan involved its binding to a chemoreceptor in the microorganism and that this receptor might contain a hydroxyl or sulfhydryl group (Parascandola, 1977). Carl Voegtlin and colleagues conducted a series of studies investigating the MOA of Salvarsan and similarly structured arsenicals (reviewed in Voegtlin (1925)). They determined that the most potent arsenicals were in the trivalent arsenious oxide form (R-As=O, 3-amino-4-hydroxyphenyl arsenious oxide, “arsenoxide”) and that Salvarsan must be converted to this in the body to be therapeutically effective. Voegtlin et al. (1923), knowing that arsenic reacts chemically with sulfur, investigated the effect of a recently found protein-like substance with a sulfhydryl group, “glutathione,” on the trypanocidal effect of arsenoxide (note at this time, the glutathione used was a dipeptide containing glutamic acid and cysteine). In vitro and in vivo studies showed that glutathione and other sulfyhydryl compounds such as cysteine and thioglycollate antagonized the trypanocidal effect of arsenoxide. Chemicals without a sulfhydryl group, such as glucose, were ineffective. It was later determined that trivalent arsenic, not pentavalent arsenic, binds to “fixed” sulfhydryl groups of tissue proteins (Peters, 1949; Rosenthal, 1932; Voegtlin et al., 1923). Voegtlin et al. (1923) hypothesized that the MOA for Salvarsan involved a chemical binding between arsenoxide and the sulfhydryl group of glutathione or other cellular sulfhydryl compounds.
A general hypothesis in the 1920s and 1930s was that toxic chemicals selectively inhibited enzymes, which would lead to a pathologic effect. Rudolph Peters and colleagues (Peters et al., 1945), at the outset of World War II, were tasked to find an antidote for the arsenical warfare agent, β-chlorovinyldichloroarsine, also known as lewisite. Peters et al. (1945) had hypothesized that the pyruvate oxidase system was sensitive to arsenicals and that sulfhydryl groups within this system were attacked. Previously, Cohen et al. (1931) had shown that aromatic arsenicals bound to sulfhydryl groups were dissociable, which suggested the removal of arsenic from enzymes was achievable, thus decreasing its toxic effect. It was thought that excess monothiols could reverse the effect of arsenic on pyruvate oxidation. However, monothiols such as glutathione were ineffective antagonists. Stocken and Thompson (1946) incubated kerateine, a form of keratin with the disulfide linkages reduced (forming free sulfhydryl groups) with lewisite and arsenite. It was determined that the arsenic in lewisite was bound to two thiol groups, forming a stable 5-membered ring, whereas arsenite was bound to only one sulfhydryl group. From this work, a dithiol antidote to lewisite, 2,3-dimercaptopropanol (British anti-lewisite, BAL) was developed. Later it was determined that in the pyruvate oxidase system (later termed the pyruvate dehydrogenase complex [PDH]), that lipoic acid, which contains vicinal dithiols, was the sensitive moiety to which arsenicals would bind. Although arsenite can inhibit PDH by binding to vicinal sulfhydryl groups within this enzyme, as does phenylarsine oxide, studies by Samikkannu et al. (2003) suggest that arsenite inhibits the enzyme by generating ROS that inactivate the protein. This occurs at concentrations of arsenite much lower than concentrations required for inhibition by direct binding to the sulfhydryl group.
Interactions with Phosphate
Arsenate was observed to affect in vitro phosphate turnover in the early 1930s (Harden, 1932). Over the years, additional in vitro studies have shown that arsenate has inherent toxicological properties because of its interaction with phosphate. However, it is not known if there are in vivo effects that are a result of this interaction between arsenate and phosphate.
Arsenic and phosphorus are in Group 15 of the periodic table (nitrogen or nitrogen group) and have similar physicochemical properties. Arsenic acid (H3AsO4) and phosphoric acid (H3PO4), the fully protonated forms of arsenate and phosphate, respectively, have comparable structure and similar acid dissociation constants. Because of their similar properties, arsenate can substitute for phosphate in several biochemical reactions (Dixon, 1997). Like phosphate, arsenate forms ester linkages with its hydroxyl groups. However, the As–O bond is about 10% longer than the P–O bond (Dixon, 1997), rendering it less stable; the arsenate ester bond can easily dissociate. Arsenate uncouples formation of adenosine-5′-triphosphate (ATP) in vitro by a mechanism termed “arsenolysis.” Doudoroff et al. (1947) were the first to use this term when they observed that glucose-1-phosphate was hydrolyzed in vitro with addition of arsenate and sucrose phosphorylase. Arsenolysis can occur during glycolysis and oxidative phosphorylation in the presence of arsenate (Crane and Lipmann, 1953; Gresser, 1981). In the glycolytic pathway, arsenate can form the intermediate anhydride, 3-phosphoglyceroyl arsenate. In oxidative phosphorylation, arsenate can couple with adenosine-5’-diphosphate. Both reactions form unstable arsenate anhydrides, which hydrolyze easily. The overall result is that formation of ATP is diminished. At the cellular level, arsenate depletes ATP in rabbit (Delnomdedieu et al., 1994) and human (Winski and Carter, 1998) erythrocytes. The human erythrocytes died a few hours after arsenic exposure, presumably because of the loss of both ATP and plasma membrane integrity. Arsenite is ineffective in depleting ATP in human erythrocytes.
Reactive Oxygen Species
The formation of reactive oxygen and nitrogen species by arsenic is one of the most studied MOAs for arsenic toxicity today (Hughes and Kitchin, 2006; Kitchin and Ahmad, 2003; Kitchin and Conolly, 2010; Lantz and Hays, 2006; Shi et al., 2004). This has come about from studies in the 1980s that reported on the induction of “stress” proteins in vitro by arsenite (Johnston et al., 1980; Keyse and Tyrrell, 1989) and DNA strand breaks in mammals by DMAsV (Yamanaka et al., 1989). ROS formed by arsenic are involved in several of the proposed MOAs including genotoxicity, signal transduction, cell proliferation, and inhibition of DNA repair.
Reactive species are formed in vitro and in vivo in the presence of arsenic and include superoxide anion, hydroxyl radical, hydrogen peroxide, reactive nitrogen species, and arsenic-centered and arsenic peroxyl radicals (Kitchin 2001; Shi et al., 2004). Production of these species have been detected by measuring oxidative DNA damage (8-hydroxy-2′-deoxyguanosine), lipid peroxidation and stress response genes, the loss of antioxidant defenses (e.g., glutathione), and induction of heat shock proteins or the use of electron spin resonance and fluorescence spectroscopy (Del Razo et al., 2001; Liu et al., 2001; Nesnow et al., 2002; Shi et al., 2004; Yamanaka et al., 1990). Several studies have shown that the addition of antioxidants and radical scavengers decrease arsenic-induced ROS formation and related toxicity (Shi et al., 2004). However, this antagonism is not always observed in vivo (Wei et al., 2005). The mechanism of ROS formation by arsenic is not clear. It may occur during oxidation of arsenite to arsenate (Del Razo et al., 2001), by formation of arsine during arsenic metabolism (Yamanaka and Okada, 1994), stimulation of NADH or NADPH oxidase (Lynn et al., 2000; Smith et al., 2001; Straub et al., 2008), ferritin iron release by arsenic with active oxygen generated by the Fenton reaction (Ahmad et al., 2000), inhibition of redox enzymes such as glutathione reductase and thioredoxin reductase (Lin et al., 2001; Styblo et al., 1997), or may be secondary to arsenic-induced cytotoxicity (Wei et al., 2005).
Genotoxicity
Reports on the genotoxic effects of arsenic were first published in the mid-1970s. Nishioka (1975) screened the mutagenic activities of metal compounds using an assay (rec-assay) that detected inhibition of growth in wild-type (Rec+) and recombination-deficient (Rec−) strains of Bacillus subtilis. Arsenite, and less so arsenate, inhibited growth in Rec− relative to Rec+ cells, which suggested that arsenic induced DNA damage. However, in this same study, arsenite did not induce tryptophan reversions in Escherichia coli strains. Most studies published since the 1970s indicate that arsenic does not directly interact with DNA to cause point mutagens in bacterial or mammalian reversion assays (Basu et al., 2001). However, arsenic is comutagenic. Arsenite enhances the mutagenic effect of ultraviolet (UV) radiation in bacterial (Rossman, 1981) and mammalian cells (Li and Rossman, 1991) and that of direct-acting mutagens such as methyl methansulfonate (Lee et al., 1986) and N-methyl-N-nitrosourea (Li and Rossman, 1989a) in mammalian cells. Although not directly mutagenic, arsenic is genotoxic, inducing effects including deletion mutations, oxidative DNA damage, DNA strand breaks, sister chromatid exchanges, chromosomal aberrations, aneuploidy, and micronuclei (Basu et al., 2001; Hei et al., 1998; Rossman, 2003). Other effects of arsenic related to genotoxicity include gene amplification, transforming activity, and genomic instability (Rossman, 2003). These genotoxic effects of arsenic are observed in vitro in mammalian cells and in vivo in laboratory animals and humans (Basu et al., 2001; Rossman, 2003). For example, Beckman et al. (1977) observed chromosomal aberrations in lymphocytes in workers exposed to arsenic. Trivalent arsenicals, both inorganic and organic, are more potent genotoxins than the pentavalent arsenicals (Kligerman et al., 2003; Mass et al., 2001). The mechanism of genotoxic action of arsenic may result from generation of ROS, inhibition of DNA repair, and altered DNA methylation that may lead to genomic instability (Rossman, 2003).
Altered DNA Repair
Arsenic inhibits DNA repair in bacterial and mammalian cells. This inhibitory effect may account for the cogenotoxic effect of arsenic with N-methyl-N-nitrosourea and UV radiation (Rossman, 2003). One of the first studies examining the effect of arsenic on DNA repair used strains of E. coli with different DNA repair phenotypes (Rossman et al., 1977). Cells were exposed to UV radiation and then plated with or without arsenite. Cells competent for postreplicative DNA repair in the presence of arsenite were the most sensitive to the lethal effects of UV radiation. In nuclear extracts from Chinese hamster V79 cells, arsenite inhibited participation of DNA ligase II in N-methyl-N-nitrosourea induced DNA repair (Li and Rossman, 1989a). The inhibition, which required a high concentration of arsenite (50% decrease at 10mM) may have been due to binding of arsenite directly to ligase. However, later studies using human cell nuclear extracts suggested inhibition of DNA repair was an indirect effect of arsenic that occurred at a much lower concentration (50% decrease at 10μM), due to induction of ROS or altered cell signaling that changed gene expression (Hu et al., 1998).
Enzymes involved in nucleotide excision repair (NER) and base excision repair (BER) are also affected by arsenic (Hartwig et al., 2003; Rossman, 2003; Schoen et al., 2004; Sykora and Snow, 2008). In a small group of individuals exposed to arsenic in drinking water, toenail arsenic levels (a biomarker of arsenic exposure) were inversely correlated with the expression of three NER genes (Andrew et al., 2003). Trivalent arsenicals inhibit BER and NER activity by interacting with zinc finger motifs of proteins in these two DNA repair systems (Ding et al., 2009; Piatek et al., 2008). The catalytic function of zinc finger containing proteins depends on binding of zinc to cysteinyl residues. Arsenic may disrupt protein function by displacing zinc from its binding site or by inactivating the sulfhydryl groups of cysteine by oxidation.
In a recent review, Gentry et al. (2010) highlighted the role of inhibition of DNA repair by arsenic as a mode of action for its carcinogenic effect. They analyzed data on in vitro cellular and in vivo gene expression changes following exposure to inorganic arsenic. The analysis of the data suggests the key events in carcinogenicity of arsenic include inhibition of DNA repair under conditions of oxidative stress, inflammation, and proliferative signaling. This may lead to a condition in which mitosis proceeds without maintaining the integrity of the cellular DNA.
Signal Transduction
Signal transduction pathways transmit extracellular signals, via an intracellular series of signaling molecules (e.g., protein kinases), into alterations in gene expression. Cellular processes such as proliferation, differentiation, and apoptosis are directed and managed by these pathways or cascades. Arsenic can alter signal transduction, which leads to activation or inhibition of transcription factors, regulatory proteins which bind to DNA and regulate gene transcription (Bode and Dong, 2002; Druwe and Vaillancourt, 2010; Huang et al., 2004; Kumagi and Sumi, 2007; Leonard et al., 2004; Platanias, 2009). In the 1990s, some of the first studies examining the effects of arsenic on signal transduction were published. Rouse et al. (1994) observed that p38, a protein in the mitogen-activated protein kinase (MAPK) cascade, was activated by arsenite in vitro. The other arms of the MAPK pathway, extracellular-regulated protein kinases (ERKs), and the c-Jun N-terminal kinases (JNKs) are also activated by arsenic (Bode and Dong, 2002; Yang and Frenkel, 2002). Liu et al. (1996) reported that arsenite in vitro strongly activated JNK and p38 but to a lesser extent ERK. The activation process appeared to involve generation of oxidative stress, as the free radical scavenger N-acetylcysteine inhibited activation of the kinases (Liu et al., 1996).
Signal transduction via the MAPK pathway activates the transcription factor activator protein-1 (AP-1) (Bode and Dong, 2002; Kumagi and Sumi, 2007). Arsenite activates JNK and p38 in HeLa cells, which in turn stimulates AP-1 transcriptional activity, leading to increased expression of the proto-oncogenes c-jun and c-fos (Cavigelli et al., 1996). In mice exposed to arsenite in drinking water, activation of the MAPK pathway was correlated with hyperproliferation of bladder epithelium (Luster and Simeonova, 2004). This appeared to result from increased activation of AP-1 followed by expression of AP-1-related genes that have a role in cell proliferation. The pathway for this activation appears to involve c-Src and epidermal growth factor receptor (EGFR) signaling cascades (Simeonova et al., 2002). c-Scr is activated in primary porcine aortic endothelial cells at low arsenite concentrations (<5μM), which are also sufficient to induce cell proliferation and increase H2O2 and superoxide accumulation, H2O2-dependent tyrosine phosphorylation and NF-κB-dependent transcription (Barchowsky et al., 1999). MAP kinases, ERK, and p38 are not activated at these levels in the endothelial cells but at higher concentrations that can result in cell death. More recently, Andrew et al. (2009) reported that arsenic at levels relevant to human exposure activates the EGFR pathway signaling in the lung. In human bronchial epithelial cells, it appears that arsenic increases levels of EGFR ligand, heparin binding-EGF, and activates EGFR phosphorylation (downstream of EGFR effects included increased pERK and the cell cycle promoter cyclin D1 levels). In human lung tumor biopsies, levels of phosphorylated EGFR were higher in specimens from subjects with elevated toenail arsenic levels compared with those with lower exposures.
The transcription factors nuclear factor-κβ (NF-κβ) and nuclear factor erythroid-2-related factor 2 (Nrf2) are also affected by arsenic (Bode and Dong, 2002; Kumagi and Sumi, 2007). Inactive NF-κβ is bound in the cytosol to the protein inhibitory κβ (Iκβ). Upon a stimulus, Iκβ is phosphorylated by Iκβ kinase and NF-κβ is released. NF-κβ is translocated to the nucleus, binds to its promoter region, and expression of genes for cytokines and growth factors is stimulated. Arsenite appears to suppress tumor necrosis factor-α induced NF-κβ activation by modifying a reactive thiol in Iκβ kinase (Roussel and Barchowsky, 2000; Schumilla et al., 1998). However, arsenite has also been reported to activate NF-κβ via generation of ROS (Felix et al., 2005; Hu et al., 2002: Wijeweera et al., 2001). Barchowsky et al. (1996) were the first to demonstrate in cultured porcine aortic endothelial cells that noncytotoxic concentrations of arsenite (≤5μM) stimulated generation of ROS and the activation of NF-κB. For example, 5μM arsenite activates NF-κB in endothelial cells (Barchowsky et al., 1996), whereas 500μM arsenite in human bronchial epithelial or embryonic kidney cells inhibits NF-κB activation by directly blocking IκB kinase and subsequent phosphorylation and degradation of the inhibitor IκBα (Roussel and Barchowsky, 2000). These differing effects of arsenic on NF-κβ activation appear to be cell-specific and dose-dependent. Nrf2, by regulating gene expression, controls the cellular antioxidant response to exogenous insult. Activation of Nrf2 in vitro by tert-butylhydroquinone (tBHQ) and sulforaphone (SF) can protect a cell from trivalent arsenic-induced toxicity (Wang et al., 2007). Nrf2 null mice are more susceptible to arsenite-induced liver and bladder toxicity than Nrf2 homozygous mice (Jiang et al., 2009). Wang et al. (2008) reported that arsenite and MMAsIII activate Nrf2 by mechanism distinct from that of tBHQ or SF.
Arsenic is well known for its carcinogenic properties, but interestingly, this metalloid is also used to treat a specific form of cancer. Arsenic trioxide (which solubilizes to arsenite in water) is a treatment for cancer patients with all trans-retinoic acid-resistant acute promyelocytic leukemia (Bode and Dong, 2002; Platanias, 2009). Arsenic trioxide generates ROS, which activates JNK and upregulates pro-apoptotic proteins and downregulates anti-apoptotic proteins. Thus, the leukemic cells undergo arsenic-induced apoptosis and the patients enter a state of remission to the cancer. However, the cancer treatment with arsenic needs to be carefully monitored because of its acute toxicological effects.
Cellular Proliferation
A hallmark of arsenic toxicity in humans is hyperkeratosis. Cells may proliferate from mitogenic stimuli or compensatory regeneration due to cell toxicity and death (Cohen and Ellwein, 1990). Germolec and colleagues in the 1990s observed that inorganic arsenic stimulates overexpression of growth factors that could potentially mediate skin neoplasia. Primary human keratinocytes incubated with arsenite overexpress growth factors including granulocyte macrophage-colony stimulating factor (GM-CSF) and transforming growth factor-α (TGF-α) and the proinflammatory cytokine tumor necrosis factor-α (Germolec et al., 1996, 1997). These cells also proliferate in the presence of arsenite. The skin of Tg.AC mice (transgenic mouse, see below for details) displays hyperkeratosis following a 10-week drinking water exposure to arsenite (200 ppm). The expression of mRNAs for GM-CSF and TGF-α in skin of these arsenite-treated mice was significantly elevated (Germolec et al., 1998). More recently, Waalkes et al. (2008) have shown a role for Rac1 in epidermal hyperproliferation and skin carcinogenesis in arsenic-exposed animals. Rac1 is a signaling G protein that regulates the cell cycle, maintains epidermal stem cells, and other cellular processes. Rac1 gene transcripts and its protein are overexpressed in skin and tumors of Tg.AC mice treated with arsenite during gestation (days 8–18) followed by topical application of TPA through adulthood. The results suggest that the cancer response is associated with distorted signaling between skin tumor stem cells and altering population dynamics. Rac1 also appears to have a role in arsenite-induced NADPH oxidase generation of oxidants in porcine aortic endothelial cells (Smith et al., 2001) and arsenite-stimulated mouse hepatic sinusoidal endothelial cell differentiation and dysfunction (Straub et al., 2008).
The rat bladder urothelium shows increased cytotoxicity and cell proliferation following exposure to DMAsV in drinking water (Wanibuchi et al., 1996) or the diet (Arnold et al., 1999). Both studies showed increased bromodeoxyuridine labeling index, which is indicative of cell proliferation. The cytotoxicity and hyperplasia of the urothelium were reversible after removal of dietary DMAsV. Morphological examination of the rat urothelium following treatment of rats with dietary DMAsV (100 ppm) for 1–3 days showed focal cellular necrosis and after 7 days widespread necrosis (Cohen et al., 2001). Increased cell proliferation was observed in the urothelium after 7 days of exposure to DMAsV. Coadministering 2,3-dimercapto-1-propanesulfonic acid, a chelator of trivalent arsenic, in the diet with DMAsV to rats inhibited the necrosis and regenerative proliferation of the urothelium (Cohen et al., 2002). This and other evidence suggested to Cohen and colleagues that DMAsIII might be the toxic species. In vitro studies with rat urothelial cells suggest that the cytotoxicity of trivalent arsenicals may be a result of, at least in part, oxidative damage (Wei et al., 2005). Simeonova et al. (2000) observed hyperplasia of the bladder urothelium in female mice following exposure to 100 ppm sodium arsenite in drinking water for 4 weeks. After 16 weeks of exposure to arsenite, the hyperplasia was accompanied by an increase in DNA binding of the activating protein (AP)-1 transcription factor and inorganic arsenic in bladder tissue. In vitro studies with UROTSA cells, an immortalized human bladder epithelial cell line, showed increased proliferation and AP-1 DNA binding following a 72 h exposure to arsenite (2μM). A follow-up study by Simeonova et al. (2002) showed that arsenite induced a c-Src-dependent activation of the EGFR and mitogen-activated protein kinase pathway in UROTSA cells. In vivo exposure to arsenite (50 ppm, 8 weeks) to mice resulted in EGFR and extracellular signal-regulated kinase (ERK) activation with the involvement of c-Src interacting with EGFR. More recent work in As3mt knockout mice (Yokohira et al., 2010) and rat (Suzuki et al., 2010) also suggest that inorganic arsenic administered in the diet may be associated with a similar MOA (cytotoxicity and cell proliferation) for urothelial cytotoxicity.
Altered DNA Methylation
Epigenetic mechanisms such as altered DNA methylation have a role in arsenic toxicity and carcinogenicity. Gene transcription is regulated by DNA methylation. In the late 1990s, two separate laboratories reported that arsenic alters DNA methylation, with both hypo- and hypermethylation of DNA observed. Zhao et al. (1997) incubated a rat liver epithelial cell line (TRL 1215) with arsenite up to 18 weeks and reported global DNA hypomethylation, aberrant gene expression, and malignant transformation of the cells. Mass and Wang (1997) observed hypermethylation of a portion of the p53 promoter region of human lung adenocarcinoma A549 cells treated with arsenite. Arsenate was also effective but required higher doses; DMAsV was ineffective in A549 cells. The tumor suppressor protein p53 has a role in cell cycle regulation. Inhibition of its expression by hypermethylation of its promoter region could potentially lead to the development of cancer. A follow-up study showed that hypomethylation and hypermethylation of genomic DNA was associated with arsenite exposure in vitro in human cells using a method sensitive to DNA methylation alterations (Zhong and Mass, 2001). This suggests that the absolute level of genomic DNA methylation is less important than methylation within a specific DNA sequence. Benbrahim-Tallaa et al. (2005) observed genomic DNA hypomethylation in a human prostate epithelial cell line following a long-term exposure to arsenite that resulted in malignant transformation of the cells.
In some individuals chronically exposed to arsenic in drinking water, the p53 promoter region of DNA from whole blood shows a dose-dependent hypermethylation relative to control subjects (Chanda et al., 2006). However, some of the arsenic-exposed individuals showed hypomethylation of this promoter region. In this same study, the tumor suppressor gene p16 was also hypermethylated in individuals exposed to high levels of arsenic.
The mechanism of the effect of arsenic on DNA methylation is not clear. Hypomethylation may be due to nutritional factors or inhibition of DNA methyltransferase. Also, S-adenosylmethionine, which is the methyl donor for the methylation of both DNA and arsenic in its metabolism, may potentially be shunted to the latter with increased exposure to arsenic.
Pilsner et al. (2007, 2009, 2011) reported in several studies of Bangladeshi adults exposed to arsenic in drinking water on its effect on genomic methylation of PBL DNA. They observed that genomic PBL DNA methylation was positively associated with exposure to arsenic in a dose-dependent manner (Pilsner et al., 2007). Plasma folate modified this effect, needing to be ≥9 nmol/l for the hypermethylation to occur. Folate is an important cofactor in the transfer of methyl groups from SAM to DNA, arsenic, and other substrates. In a group of arsenic-exposed individuals that developed skin lesions, it was found that folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of PBL DNA were risk factors for the arsenic-induced skin lesions (Pilsner et al., 2009). Pilsner and colleagues suggested that the hypermethylation of PBL DNA that is associated with increased arsenic exposure might be an adaptive response because the hypomethylation of PBL DNA is associated with the risk for development of skin lesions. This same group reported that plasma selenium is inversely associated with genomic PBL DNA methylation. In addition, selenium may reduce the body of arsenic. Overall, the effect of arsenic on genomic DNA methylation is still not clear and requires further investigation.
Carcinogenicity of Arsenic in Animal Models
The use of animals for the study of cancer causing agents has been a scientific foundation for many years. This is so that chemicals with unknown carcinogenicity in humans can be tested or that a potential mechanism can be studied for chemicals known to be carcinogenic. Leitch and Kennaway (1922) conducted some of the first animal carcinogenicity studies with arsenic. They fed bread containing potassium arsenite (Fowler’s solution) to mice and rats 3 days per week. Most of the animals died from the treatment, and no tumors were detected. In a second experiment, the arsenical solution was directly applied to mouse skin three times per week. About two-thirds of the initial 100 animals died from the treatment, but a wart on one animal developed at the application site after 85 days of treatment. This wart eventually developed into a metastasizing squamous cell epithelioma. A follow-up study under similar conditions produced negative results (Leitch, 1923). These results by Leitch and Kennaway, of no carcinogenic effect with arsenic in one experiment, followed by a limited positive result, are similar to what has been published up to the 1990s. Many studies of experimental arsenic carcinogenesis using oral, dermal, or parenteral administration followed over the years. These studies included mice and rats (Baroni et al., 1963; Byron et al., 1967; Heuper and Payne, 1962; Kanisawa and Schroeder, 1969; Neubauer, 1947; Schroeder et al., 1968), dogs (Byron et al., 1967), rabbits and chickens (Neubauer, 1947), and monkeys (Thorgeirsson et al., 1994). Some studies involved classical initiation/promotion experiments with arsenic used as either the initiator (with the promoter croton oil) or promoter (with the initiator dimethylbenz(a)anthracene) (Boutwell, 1963). The results from these studies were largely negative or inconclusive. These carcinogenicity results as well as other findings up to this time led some to question the carcinogenic potency of arsenic in humans (Frost, 1967).
In the late 1970s and early 1980s, the potential for development of lung tumors in rats (Ishinishi et al., 1977) and Syrian Golden hamsters (Ishinishi et al., 1983; Pershagen et al., 1984; Pershagen and Bjorklund, 1985; Yamamoto et al., 1987) administered arsenic intratracheally was examined. Epidemiological studies had shown that workers in the nonferrous metal smelting and arsenical pesticide manufacturing industries were at risk of developing lung cancer (Lee and Franmeni, 1969; Ott et al., 1974). The experimental procedure was to instill arsenic (arsenic trioxide, arsenic trisulfide, or calcium arsenate) suspended in saline into the trachea of the animals. Animals received one dose per week for 15 weeks (total dose, 3.75 or 5.25 mg As) and then were monitored through their entire life span. Results for arsenic trioxide were problematic because up to 50% of the treated animals died. There was less mortality (10–14%) in animals treated with the other arsenicals. The results for arsenic trisulfide were inconclusive. Calcium arsenate and arsenic trioxide were weakly tumorigenic. The International Agency for Research on Cancer (IARC, 2004) considers that these studies provided limited evidence for carcinogenicity of inorganic arsenicals.
In the 1990s, carcinogenicity studies in animals shifted from inorganic arsenic to its main mammalian metabolite, dimethylarsinic acid (DMAsV). DMAsV is a clastogen and induces chromosomal aberrations in vitro (Dong and Lou, 1993; Endo et al., 1992). Yamamoto et al. (1995) assessed DMAsV in a multiorgan carcinogenesis bioassay. Rats were pretreated with five known carcinogens and then administered DMAsV in drinking water (50–400 ppm) for 24 weeks. DMAsV promoted tumors in urinary bladder, kidney, liver, and thyroid gland. Urinary bladder showed the strongest response. This result was important because urinary bladder is one of the target organs for arsenic in humans (NRC 1999, 2001). Studies continued investigating the tumor promoting effects of DMAsV in rats and mice in urinary bladder and lung, respectively (Yamanaka et al., 1996; Wanibuchi et al., 1996). DMAsV in drinking water (rats, 2–100 ppm; mice, 200–400 ppm) promoted the tumorigenic effects of two initiators (N-butyl-N-(4-hydroxybutyl) nitrosamine and 4-nitroquinolone 1-oxide). DMAsV alone in drinking water (400 ppm, 50 weeks) promoted lung tumor development in A/J mice, which spontaneously develop lung tumors (Hayashi et al., 1998). Dose-dependent cellular proliferation of urinary bladder epithelium was observed in rats administered DMAsV in drinking water (10–100 ppm) up to 23 weeks (Wanibuchi et al., 1996).
Studies then followed that assessed the complete carcinogenicity of DMAsV. Wei et al. (1999, 2002) administered DMAsV in drinking water (12.5–200 ppm) to male F344 rats for 2 years. They observed dose-related development of bladder tumors. Tumors were not observed in other organs or in the control or 12.5 ppm DMAsV groups. Arnold et al. (2006) administered DMAsV in the feed of B6C3F1 mice (8–500 ppm) and F344 rats (2–100 ppm) of both sexes for 2 years. Although no treatment-related tumors were observed in mice, rats developed urinary bladder tumors. Female rats were more sensitive than male rats to the effects of DMAsV in the diet. A 2-year dietary exposure of monomethylarsonic acid, a metabolic precursor of DMAsV, to F344 rats (50–1300 ppm) and B6C3F1 mice (10–400 ppm) yielded no tumors (Arnold et al., 2003). Cohen et al. (2006) proposed that the carcinogenic effect of DMAsV is a result of metabolism of DMAsV to dimethylarsinous acid (DMAsIII), which reacts with critical proteins in the urothelium or induces oxidative stress in the cells, resulting in cytotoxicity. This initiates cellular proliferation in the urothelium and eventual tumor development. A criticism of the DMAsV promoting and carcinogenicity studies are the high doses used and that there are species differences in arsenic pharmacokinetics. Rat hemoglobin uniquely binds DMAsIII (Lu et al., 2007), which extends the pharmacokinetic half-life of this arsenical relative to other mammals (Vahter et al., 1984). It is not known if the retention of arsenic by the rat affects development of urinary bladder tumors following exposure to DMAsV. Although these findings have been taken as evidence that DMAsV operates as a nonlinear carcinogen (Cohen et al., 2006), it has been noted that humans are unlikely to experience doses of DMAsV in the diet sufficient to induce cytotoxicity and subsequent carcinogenicity (U.S. EPA, 2006). As mentioned above, there is newer research investigating the role of inorganic arsenic-induced cytotoxicity and regenerative hyperplasia as an MOA for arsenic carcinogenesis. Suzuki et al. (2010) measured dose-dependent changes in the bladder epithelium of female F344 rats exposed to 0, 1, 10, 25, and 50 ppm sodium arsenite in feed (no effects were observed at 1 ppm or below). In this study, both trivalent methylated and other thiol-containing arsenic metabolites were hypothesized to account for the observed bladder cell cytotoxicity and regenerative hyperplasia. More recent studies find that even in mice that do not methylate arsenic (As3mt knockout mice), bladder cell cytotoxicity can be measured after exposure to inorganic arsenic. These studies indicate that inorganic arsenic itself, in the absence of metabolism to methylated trivalent species, is capable of causing cytotoxic damage and regenerative hyperplasia (Yokohira et al., 2010). However, there is a higher body burden of arsenic in the bladder of As3mt knockout mice than wild-type mice at steady state during repeated treatment with arsenate (Hughes et al., 2010). Inorganic arsenic was the predominant arsenic species in this tissue of As3mt knockout mice. In wild-type mice, the bladder contained a mixture of inorganic arsenic and methylated arsenicals.
The skin is a target organ in humans exposed to inorganic arsenic. Dermal effects of arsenic in humans include altered pigmentation, hyperkeratosis, and cancer. The transgenic mouse model, Tg.AC, contains the v-Ha ras oncogene. This strain has the characteristics of genetically initiated skin with a predisposition to papillomas. In an experiment conducted in the late 1990s, such transgenic mice were exposed to arsenite (200 ppm) in drinking water for 4 weeks. Then the mice were treated topically (two times per week for 2 weeks) with the promoter 12-O-tetradecanoyl phorbol-13-acetate (TPA). The number of papillomas increased in the arsenite plus TPA-treated Tg.AC mice (Germolec et al., 1997, 1998). Papillomas were not observed in Tg.AC mice treated with arsenite alone, control mice, or wild-type mice treated with arsenite and TPA. Results from this study and others led Germolec et al. (1997) to suggest that arsenic is a tumor enhancer.
In vitro studies have shown that arsenic is comutagenic with several chemicals and UV radiation (Li and Rossman, 1989a, 1989b, 1991). Rossman and colleagues examined the cocarcinogenic effect of arsenite in drinking water (1.25–10 ppm, 26 weeks) and exposure to solar UV radiation (1–1.7 KJ/m2, three times/week) in hairless Skh1 mice (Burns et al., 2004; Rossman et al., 2001). No tumors were observed in untreated mice or those exposed to arsenite alone. However, skin tumors including invasive squamous cell carcinomas developed in the arsenite plus UV radiation exposed group. Studies with a mouse keratinocyte cell line suggested that arsenite decreases the rate of DNA repair of some photoproducts and inhibits apoptosis induced by UV radiation (Wu et al., 2005). Skin carcinogenesis by UV radiation may be enhanced by arsenite decreasing DNA repair and inhibiting apoptosis of damaged cells.
The developing fetus can be highly sensitive to the adverse effects of chemicals. Metals such as nickel and lead are transplacental carcinogens in rodents (Diwan et al., 1992; Waalkes et al., 1995). The fetus can be exposed to arsenic because it crosses the placenta (Lindgren et al., 1984). With this information, Waalkes et al. (2003) conducted a gestational exposure study of arsenite in mice. Dams were exposed to arsenite at two dose levels (42.5 and 85 ppm) in drinking water from gestation day 8 to 18. The treatment did not affect the dams. Male and female offspring were observed through adulthood. There was an increase in hepatocellular carcinoma (dose-related) and liver tumor multiplicity in arsenic-exposed male offspring. In female offspring, there were dose-related increases in adrenal tumor incidence and multiplicity and incidence of ovarian tumors and lung carcinomas. The incidence of proliferative lesions in the uterus and oviduct were also increased by arsenite. This study was repeated (Waalkes et al., 2004) and included a group of offspring treated on the skin with TPA. In male offspring, similar results were observed with the 2003 study, and TPA treatment had no effect on the liver and adrenal tumors. TPA also had no effect on the hyperplasia of uterus and oviduct caused by arsenic. TPA promoted liver tumors in females and lung tumors in male and female mice. No skin tumors were detected. Gestational exposure of arsenic was also conducted with Tg.AC transgenic mouse (Waalkes et al., 2008). Following in utero exposure to arsenite in drinking water (42.5 and 85 ppm), offspring were treated with TPA through adulthood. Arsenic-alone treated animals were not affected. Papillomas and squamous cell carcinomas were observed in TPA-alone treated mice (no arsenite exposure). In arsenic and TPA-treated mice, there was a 30-fold increase in squamous cell carcinoma multiplicity. These carcinomas were also more invasive. Distorted skin tumor stem cell signaling and stem cell population dynamics were detected. Waalkes et al. (2008) suggested that arsenic targets stem cells as the fetal basis of skin cancer in adulthood.
The standard cancer bioassay is to treat animals with the chemical after they are weaned. However, humans are exposed to many chemicals, throughout their lifetime. Several recent epidemiology studies have reported that exposure to inorganic arsenic at an early age results in increased susceptibility to cancer in adulthood (Smith et al., 2006; Yuan et al., 2010). From this evidence and the results of their transplacental arsenic model, Waalkes and colleagues (Tokar et al., 2011a) conducted a lifetime arsenite drinking water (6–24 ppm) exposure study in mice. The exposure began before breeding (to the parents), during pregnancy and lactation and after weaning through adulthood. In offspring of both sexes, dose-related lung adenocarcinomas, hepatocellular carcinomas and adrenal tumors developed. Gall bladder tumors were detected in males and uterine tumors in females. In the whole life study, target sites were similar to the in utero alone exposures but occurred at lower doses. The tumors in the lifetime study were generally more aggressive and frequent. Also, an overabundance of cancer stem cells was found in the liver and lung tumors. This suggests that stem cells may be a target for arsenic. Based on the experimental results, it is also likely that interference with the normal functioning of the estrogen receptor by arsenic may be involved in carcinogenesis.
Overall, animal models, especially recently developed models, have advanced our understanding of arsenic carcinogenicity. A representative set of the oral arsenic carcinogenicity studies in animals is summarized in Table 3. As with many chemicals, differences in pharmacokinetics and pharmacodynamics among different animal species and the doses used in animal bioassays necessitates an examination of the human relevance of findings from animal studies.
TABLE 3.
Representative Oral Carcinogenicity Studies of Arsenic in Animals
| Study | Species | Sex | Arsenical species | Dose | Exposure period | Outcome | Notes |
| Kanisawa and Schroeder (1969) | Rat | Female and Male | Arsenite | 5 ppm, drinking water | Lifetime, started at weaning | No treatment-related effect | |
| Yamamoto et al. (1995) | Rat | Male | DMAsV | 50–100 ppm, drinking water | 6–30 weeks of age | Promoter of urinary bladder, kidney, liver, and thyroid gland tumors | Animals pretreated with five carcinogens |
| Wei et al. (1999) | Rat | Male | DMAsV | 12.5–200 ppm, drinking water | 10–104 weeks of age | Urinary bladder carcinogen | |
| Rossman et al. (2001) | Mouse | Female | Arsenite | 10 ppm, drinking water | 3–29 weeks of age | 2.4-fold increase in skin tumor yield in UV + arsenite-treated compared with UV alone; no tumors in arsenite alone | Mice irradiated with UV light, 3×/week |
| Waalkes et al. (2003) | Mouse | Female and male | Arsenite | 42.5 and 85 ppm, drinking water | Gestation day 8–18 | Female—ovarian and lung tumors | Transplacental exposure |
| Male—liver and adrenal tumors | |||||||
| Arnold et al. (2006) | Rat | Female and male | DMAsV | Rat: 2–100 ppm, diet | 104 weeks | Rat—urinary bladder carcinogen in both sexes | Female rats more sensitive than male rats |
| Mouse | Mouse: 8–500 ppm, diet | Mouse—no treatment-related tumors | |||||
| Tokar et al. (2011a) | Mouse | Female and male | Arsenite | 6–24 ppm, drinking water | Two weeks prior to breeding through adulthood | Female—lung, liver, adrenal, uterine, ovarian tumors | “Whole-life” exposure |
| Male—lung, liver, gall bladder, and adrenal tumors |
Epidemiologic Studies of Arsenic Exposure from Drinking Water
Elevated arsenic concentrations in water have been indicated as a public health hazard in many countries of the world where individuals rely on groundwater for drinking purposes (Smith et al., 2000). Since 1968, a series of epidemiologic reports from a small area in southwestern Taiwan have offered data on harmful effects of high levels (>300 μg/l) of arsenic in drinking water. Blackfoot disease, a unique peripheral disease in lower extremities that could result in spontaneous gangrene in the toes and feet, has been known in the area since the 1920s (Ch’i and Blackwell, 1968). In a landmark study, Tseng et al. (1968) documented in a cross-sectional study that high level of arsenic exposures at 300 μg/l and 600 μg/l were related to a high prevalence of skin cancer. Using the median well arsenic concentration within a village among 42 villages and ICD-8 codes on death certificates, Chen et al. (1988) subsequently revealed that there was a dose-response relationship of arsenic concentration and mortality due to internal cancers, including cancer of the lung, liver, bladder, and kidney, as well as all vascular diseases, peripheral vascular diseases, and cardiovascular diseases (Tseng et al., 1996). In areas of Argentina and Chile where drinking water was also naturally contaminated, studies have confirmed the associations between high levels of arsenic concentration with the risk of mortality from cancers of the bladder, lung, and kidney (Bates et al., 2004; Ferreccio et al., 2000; Hopenhayn-Rich et al., 1996, 1998; Marshall et al., 2007; Smith et al., 1998). Similar to the study conducted by Tseng et al., arsenic exposure was measured at the group level with an ecologic or partial ecologic study design in these studies. However, it is unlikely that biases or confounding could account for the observed associations, as the associations were strong and consistent across different populations, and the distribution of the exposure was not related to individuals’ lifestyles. In particular, the data from Taiwan have been used by the U.S. EPA for risk assessment; based in part on these data, in 2001, the U.S. EPA adopted a new standard for arsenic in drinking water at 10 μg/l, replacing the old standard of 50 μg/l (U.S. EPA, 2001).
In the two most recent decades, epidemiologic studies of arsenic exposure from drinking water have evolved using more modern epidemiology study designs and methodologies. Several case-control and cross-sectional studies have included measures of arsenic exposure at the level of the individual, including studies in the United States in states such as New Hampshire (Karagas et al., 2001, 2004) and Nevada (Steinmaus et al., 2003), as well as other countries such as West Bengal of India (Haque et al., 2003), Bangladesh (Ahsan et al., 2006; McCarty et al., 2006; Rahman et al., 1999; Sohel et al., 2009), Inner Mongolia of China (Mumford et al., 2007), and northeastern Taiwan (Chiou et al., 2001). These studies shed light on the chronic effects of arsenic, including total and cancer mortality (Sohel et al., 2009), bladder and skin cancer (Chen et al., 2010; Karagas et al., 2001, 2004; Steinmaus et al., 2003), skin lesions (Ahsan et al., 2006; Haque et al., 2003; McCarty et al., 2006), respiratory diseases (Mazumder et al., 2000; Parvez et al., 2010; von Ehrenstein et al., 2005), and cardiovascular effects (Mumford et al., 2007; Rahman et al., 1999), at levels of inorganic arsenic exposure <500 μg/l. The ascertainment of exposure at the individual level reduces exposure misclassification and enhances internal validity of the study; these are crucial considerations for studies in areas with varying levels of arsenic exposure, especially levels of arsenic concentrations <500 μg/l. Specifically, in some studies, the use of arsenic concentrations measured in urine (Ahsan et al., 2006; Navas-Acien et al., 2008), blood (Hall et al., 2006), and toenail samples (Karagas et al., 2001, 2004) as biomarkers for the internal dose of arsenic exposure have opened new research opportunities in molecular epidemiology as these studies often involved collection of biospecimens and detailed information on lifestyle factors. For instance, the case-control studies in New Hampshire gave suggestions of increased risk of bladder cancer and skin cancer associated with toenail arsenic, especially among smokers (Karagas et al., 2001, 2004). Another case-control study in this part of the United States, with information on specific cancer histologic types found that elevated toenail arsenic concentrations (≥0.114 μg/g) were associated with the risk of small-cell carcinomas of the lung (Heck et al., 2009). These studies, with collection of biospecimens, have also provided platforms for subsequent studies investigating the effects of arsenic on signal transduction, gene expression, and DNA repair functions using human samples (Andrew et al., 2006, 2008, 2009). The Health Effects of Arsenic Longitudinal Study (HEALS), a prospective cohort involving more than 20,000 participants in a rural area in Bangladesh, has recently reported that arsenic exposure, measured in both water (0.01–864 μg As/l) and urine (7–5000 μg As/g) samples, is related to all-cause, chronic-disease (Argos et al., 2010), and cardiovascular disease especially heart disease mortality in the population (Chen et al., 2011). The HEALS also creates an infrastructure for comprehensive evaluations of other health effects of arsenic exposure using an array of study designs (Chen et al., 2009), including case-controls, case-cohort, and intervention studies to evaluate genetic (Ahsan et al., 2007) and nutritional susceptibility (Chen et al., 2007; Gamble et al., 2006). These studies have provided valuable information on potential exposure-disease mechanisms using human samples.
Recent epidemiologic studies have extended the scope of research to outcomes beyond incidence or mortality of chronic diseases in adulthood (Smith and Steinmaus, 2009). In several comparisons between the arsenic-exposed and unexposed populations, studies in Chile indicated that individuals born in a high arsenic-exposure period with probable exposure in utero and early childhood experienced elevated risk of deaths due to acute myocardial infarction (Yuan et al., 2007), lung cancer, and bronchiectasis (Smith et al., 2006), suggesting the importance of early childhood or in utero exposure in disease causation. Several studies conducted in Bangladesh and West Bengal found that elevated arsenic concentration in groundwater was associated with increased risks of fetal loss and infant death (Rahman et al., 2007), spontaneous abortion and stillbirth (von Ehrenstein et al., 2006), decreased birth weight (Rahman et al., 2009), and decrements in intellectual testing in school-aged children (von Ehrenstein et al., 2007; Wasserman et al., 2004, 2007), although results were inconsistent and require further study. More recently, Yorifuji et al. (2010) reported elevated deaths and mortality ratios from several cancer types (pancreatic, leukemia) in an adult Japanese population following neonatal exposure to arsenic-contaminated milk powder. These studies give new insight on the potential early-life effects of arsenic exposure.
Taken together, over the past 50 years, epidemiologic studies have provided abundant data on health effects of arsenic exposure from drinking water. A representative set of these studies is summarized in Table 4. Over time, accuracy and reliability of measurements for the exposure in studies have been improved, with possibilities of using a life course approach to investigate the effects of in utero and prenatal exposure. Although early studies were mostly retrospective or cross-sectional in design, prospective studies with biorepository resources have been conducted to confirm previous associations and extended the assessment to the molecular level. Besides cancer, evidence has emerged for the adverse effects of arsenic exposure on cardiovascular disease, respiratory disease, neurological functions, reproductive outcomes, and children’s intelligence. In addition, studies began to identify mechanisms using biomarkers and preclinical abnormalities as endpoints and to assess susceptibility due to genetic, nutritional, and lifestyle factors. Although challenges of studying effects of lower level exposure relative to arsenicosis-endemic areas (e.g., Bangladesh) remain, we expect that epidemiologic studies in years to come will continue elucidating the etiology of arsenic-related health effects as well as providing information for prevention, risk assessment, and intervention.
TABLE 4.
Representative Human Epidemiology Studies in Arsenic Drinking Water-Exposed Populations
| Study | Population | Dose | Findings |
| Tseng et al. (1968) | Taiwan, >40,000 in survey | 0.01–1.82 ppm | Skin cancer (10.6/1000),a hyperpigmentation (183.5/1000), keratoses (71/1000), Blackfoot disease (8.9/1000); revalence rate of effects increased with age; Blackfoot disease associated with hyperpigmentation, keratosis, and skin cancer |
| Chen et al. (1988) | Taiwan, ∼900,000 person-years | 0-30 ppm, 30-59 ppm, ≥ 60 ppm | Significant dose-response relationship between As and age-adjusted mortality for bladder, kidney, skin, prostate, lung and liver cancer |
| Smith et al. (1998) | Chile, >400,000 person-years | <100 to 570 μg/l | SMR for bladder cancer: men, 6.0; women, 8.2; lung cancer: men 3.8; women 3.1; increased mortality for bladder, lung, kidney, and skin cancer |
| Karagas et al. (2001) | United States, ∼800 skin cancer cases + controls | Based on toenail As concentration (overall range, 0.01–2.57 μg/g) | Risk of skin cancer did not appear elevated at toenail As concentrations detected for most study subjects; odds ratios with toenail As concentration above 97th percentile were 2.07 for SCC and 1.44 for BCC. |
| Smith et al. (2006) | Antofagasta, Chile | ∼1 ppm for 12 years | In utero and early childhood exposure occurred between 1959 and 1971; lung cancer SMR: early childhood, 7.0; in utero + early childhood, 6.1; Bronchiectasis SMR: early childhood, 12.4, in utero + early childhood, 46.2 |
| Mumford et al. (2007) | Inner Mongolia, China; 313 subjects | ≤21–690 μg/l | Dose-dependent relationship between prevalence rates of QT prolongation and water As concentrations; measuring QT interval may detect early cardiac As cardiac toxicity |
| Argos et al. (2010) | Bangladesh, >10,000 in survey | 0.01–864 ppb | Chronic As exposure via drinking water associated with increased mortality rate. Mortality increased with As concentration in drinking water, As dose, and total As in urine |
Note. BCC, basal cell carcinoma; SCC, squamous cell carcinoma; SMR, standardized mortality rate.
Note. Prevalence rate.
Summary and Looking Forward
The knowledge base of the exposure and toxicological effects of arsenic has expanded greatly, particularly in the past 10–20 years. We know that exposure to arsenic for most people is an everyday occurrence because it is a natural component of the environment. The exposure pathways of arsenic to most people are dietary and drinking water and these exposures occur at relatively low levels. However, there are areas of the world, such as India, Bangladesh, and others, where the levels of arsenic in drinking water are naturally excessive, which has led to toxic manifestations in these populations. The effects of arsenic in drinking water on the U.S. population are less clear, which may be due to a lower arsenic exposure than in other areas of the world such as Bangladesh. Data from Karagas et al. (2001, 2004) has suggested, especially among smokers, an increased risk of bladder and skin cancer is associated with toenail arsenic.
Other types of exposure can come from soil contaminated with arsenic, from its occupational use as a pesticide or a by-product of metal ore smelting, from its use as a chemotherapeutic agent, and what interests many people, but occurs rarely, as a homicidal agent. With increases in analytical technology, what most likely will occur is the discovery of presently unknown forms of arsenic (e.g., arsenolipids) that we are exposed to, particularly in our diet.
The metabolic pathway of arsenic is now more clearly but not exactly defined. The discovery of arsenic (+3 oxidation state) methyltransferase has been a major breakthrough, particularly with the findings that there are polymorphisms in this enzyme. Several of these polymorphisms are associated with the toxic effects that develop from exposure to arsenic. Experimental use of the As3mt knockout mouse in the investigation of the metabolism and toxicity of arsenic may provide new knowledge. Finally, elucidation of the pathway of formation of the thiolated arsenic metabolites (e.g., dimethyldithioarsinic acid), some of which are toxicologically potent in vitro, will aid in the understanding of the toxicology of arsenic.
We know that arsenic causes acute and chronic dose-dependent effects, in many organ systems. A major unknown is the mode of action for the toxic effects of arsenic. Certainly, metabolism of arsenic has a role in this effect. However, if one or several metabolites are the putative toxic species is not known. Many MOA have been studied including oxidative stress, genotoxicity, altered DNA methylation, and others. Several of these MOA may be interrelated. With the advent of the “omics” age, toxic pathways of arsenic may soon be elucidated. Other recent advances are the development of an animal (mouse) model for arsenic carcinogenicity following transplacental and whole-life exposure to arsenic and findings in mice that arsenic may impact stem cell population dynamics, which ultimately lead to transformed cells (Tokar et al., 2011b). Also, there is important work in animal models that support a role for cytotoxicity and regenerative hyperplasia in the carcinogenic MOA for DMAsV.
More research is still needed to understand arsenic exposure, metabolism, effects, and MOA for cancer. Nevertheless, with recent findings and advances in technology, many of the unanswered questions regarding the toxicology of arsenic may soon be answered. This knowledge will lead to better protection of populations at risk from arsenic-related illnesses.
FUNDING
National Institute of Environmental Health Sciences (RO1ES017541, P30Es000260 to Y.C.); Intramural resources at U.S. EPA (to M.F.H. and D.J.T.).
Acknowledgments
We would like to thank Anna Engel of Gradient with assistance in researching the historical uses of arsenic. We also thank Drs Kirk Kitchin, Jane Ellen Simmons, and Erik Tokar for their helpful comments on an earlier version of this manuscript. Gradient, where B.D.B. and A.S.L. work, has conducted risk analyses for arsenic for a number of private and public sector clients. B.D.B. has been an expert in litigation matters involving arsenic. However, Gradient received no funding for preparation of this manuscript and the opinions are solely those of the author's.
References
- Agusa T, Iwata H, Fujihara J, Kunito T, Takeshita H, Minh TB, Trang PT, Viet PH, Tanabe S. Genetic polymorphisms in AS3MT and arsenic metabolism in residents of the Red River Delta, Vietnam. Toxicol. Appl. Pharmacol. 2009;236:131–141. doi: 10.1016/j.taap.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Kitchin KT, Cullen WR. Arsenic species that cause release of iron from ferritin and generation of activated oxygen. Arch. Biochem. Biophys. 2000;382:195–202. doi: 10.1006/abbi.2000.2023. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen Y, Kibriya MG, Slavkovich V, Parvez F, Jasmine F, Gamble MV, Graziano JH. Arsenic metabolism, genetic susceptibility, and risk of premalignant skin lesions in Bangladesh. Cancer Epidemiol. Biomarkers Prev. 2007;16:1270–1278. doi: 10.1158/1055-9965.EPI-06-0676. [DOI] [PubMed] [Google Scholar]
- Ahsan H, Chen H, Parvez F, Zablotska L, Argo M, Hussain I, Monotaj H, Levy D, Cheng Z, Stankovich V, et al. Arsenic exposure from drinking water and risk of premalignant skin lesions in Bangladesh: baseline results from the Health Effects of Arsenic Longitudinal Study. Am. J. Epidemiol. 2006;163:1138–1148. doi: 10.1093/aje/kwj154. [DOI] [PubMed] [Google Scholar]
- Andrew AS, Burgess JL, Meza MM, Demidenko E, Waugh M, Hamilton JW, Karagas MR. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environ. Health Perspect. 2006;114:1193–1198. doi: 10.1289/ehp.9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew AS, Jewell DA, Mason RA, Whitfield ML, Moore HJ, Karagas MR. Drinking-water arsenic exposure modulates gene expression in human lymphocytes from a U.S. population. Environ. Health Perspect. 2008;116:524–531. doi: 10.1289/ehp.10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew AS, Karagas MR, Hamilton JW. Decreased DNA repair gene expression among individuals exposed to arsenic in United States drinking water. Int. J. Cancer. 2003;104:263–268. doi: 10.1002/ijc.10968. [DOI] [PubMed] [Google Scholar]
- Andrew AS, Mason RA, Memoli V, Duell EJ. Arsenic activates EGFR pathway signaling in the lung. Toxicol. Sci. 2009;109:350–357. doi: 10.1093/toxsci/kfp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrewes P, DeMarini DM, Funasaka K, Wallace K, Lai VWM, Sun H, Cullen WR, Kitchin KT. Do arsenosugars pose a risk to human health? The comparative toxicities of a trivalent and pentavalent arsenosugar. Environ. Sci. Technol. 2004;38:4140–4148. doi: 10.1021/es035440f. [DOI] [PubMed] [Google Scholar]
- Antman KH. Introduction: the history of arsenic trioxide in cancer therapy. Oncologist. 2001;6(Suppl. 2):1–2. doi: 10.1634/theoncologist.6-suppl_2-1. [DOI] [PubMed] [Google Scholar]
- Aposhian HV. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu. Rev. Pharmacol. Toxicol. 1997;37:397–419. doi: 10.1146/annurev.pharmtox.37.1.397. [DOI] [PubMed] [Google Scholar]
- Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, Islam T, Ahmed A, Rakibuz-Zaman M, Hasan R, et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet. 2010;376:252–258. doi: 10.1016/S0140-6736(10)60481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold LL, Cano M, John MS, Eldan M, van Gemert M, Cohen SM. Effects of dietary dimethylarsinic acid on the urine and urothelium of rats. Carcinogenesis. 1999;20:2171–2179. doi: 10.1093/carcin/20.11.2171. [DOI] [PubMed] [Google Scholar]
- Arnold LL, Eldan M, Nyska A, van Gemert M, Cohen S. Dimethylarsinic acid: results of chronic toxicity/oncogenicity studies in F344 rats and B6C3F1 mice. Toxicology. 2006;223:82–100. doi: 10.1016/j.tox.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Arnold LL, Eldan M, van Gemert M, Capca C, Cohen SM. Chronic studies evaluating the carcinogenicity of monomethylarsonic acid in rats and mice. Toxicology. 2003;190:197–219. doi: 10.1016/s0300-483x(03)00165-3. [DOI] [PubMed] [Google Scholar]
- ATSDR. CERCLA Priority List of Hazardous Substances for 2007. Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services; 2007a. Available at: http://www.atsdr.cdc.gov/cercla/07list.html. Accessed June 9, 2011. [Google Scholar]
- ATSDR. Toxicological Profile for Arsenic. Atlanta, GA: 2007b. Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services, Atlanta, GA. Available at: http://www.atsdr.cdc.gov/ToxProfiles/tp2.pdf. Accessed July 27, 2011. [PubMed] [Google Scholar]
- Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Arsenic induces oxidant stress and NF-κβ activation in cultured aortic endothelial cells. Free Radic. Biol. Med. 1996;21:783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Toxicol. Appl. Pharmacol. 1999;159:65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]
- Baroni C, van Esch GJ, Saffiotti U. Carcinogenesis tests of two inorganic arsenicals. Arch. Environ. Health. 1963;7:54–60. doi: 10.1080/00039896.1963.10663598. [DOI] [PubMed] [Google Scholar]
- Bartrip P. A pennruth of arsenic for rat poison: the Arsenic Act, 1851 and the prevention of secret poisoning. Med. Hist. 1992;36:53–69. doi: 10.1017/s0025727300054624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Mahata J, Gupta S, Giri AK. Genetic toxicology of a paradoxical human carcinogen, arsenic: a review. Mutat. Res. 2001;488:171–194. doi: 10.1016/s1383-5742(01)00056-4. [DOI] [PubMed] [Google Scholar]
- Bates MN, Rey OA, Biggs ML, Hopenhayn C, Moore LE, Kalman D, Steinmaus C, Smith AH. Case-control study of bladder cancer and exposure to arsenic in Argentina. Am. J. Epidemiol. 2004;159:381–389. doi: 10.1093/aje/kwh054. [DOI] [PubMed] [Google Scholar]
- Beckman G, Beckman L, Nordenson I. Chromosome aberrations in workers exposed to arsenic. Environ. Health Perspect. 1977;19:145–146. doi: 10.1289/ehp.7719145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Waterland RA, Styblo M, Achanzar WE, Webber MM, Waalkes MP. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol. Appl. Pharmacol. 2005;206:288–289. doi: 10.1016/j.taap.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit. Rev. Oncol. Hematol. 2002;24:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- Borak J, Hosgood HD. Seafood arsenic: implications for human risk assessment. Regul. Toxicol. Pharmacol. 2007;47:204–212. doi: 10.1016/j.yrtph.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Boutwell RK. A carcinogenicity evaluation of potassium arsenite and arsanilic acid. J. Agric. Food Chem. 1963;11:381–385. [Google Scholar]
- Braman RS, Foreback CC. Methylated forms of arsenic in the environment. Science. 1973;182:1247–1249. doi: 10.1126/science.182.4118.1247. [DOI] [PubMed] [Google Scholar]
- Buchet JP, Lauwerys R. Study of inorganic arsenic methylation by rat liver in vitro: relevance for the interpretation of observations in man. Arch. Toxicol. 1985;57:125–129. doi: 10.1007/BF00343122. [DOI] [PubMed] [Google Scholar]
- Buchet JP, Lauwerys R, Roels H. Comparison of the urinary excretion of arsenic metabolites after a single oral dose of sodium arsenite, monomethylarsonate, or dimethylarsinate in man. Int. Arch. Occup. Environ. Health. 1981;48:71–79. doi: 10.1007/BF00405933. [DOI] [PubMed] [Google Scholar]
- Buck AH, Stedman TL. A Reference Handbook of the Medical Sciences Embracing the Entire Range of Scientific and Practical Medicine and Allied Science. Vol. III. New York, NY: William Wood and Company; 1904. [Google Scholar]
- Burns FJ, Uddin AN, Wu F, Nadas A, Rossman TG. Arsenic-induced enhancement of ultraviolet radiation carcinogenesis in mouse skin: a dose-response study. Environ. Health Perspect. 2004;112:559–602. doi: 10.1289/ehp.6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron WR, Bierbower GW, Brouwer JB, Hansen WH. Pathologic changes in rats and dogs from two-year feeding of sodium arsenite or sodium arsenate. Toxicol. Appl. Pharmacol. 1967;10:132–147. doi: 10.1016/0041-008x(67)90135-4. [DOI] [PubMed] [Google Scholar]
- Caudill DS. Arsenic and old chemistry: images of mad alchemists, experts attacking experts, and the crisis in forensic science. Boston Univ. J. Sci. Tech. Law. 2009;15 Villanova Law/Public Policy Research Paper no.2009-13. Available at: http:///ssrn.com/abstract=140275. Accessed on December 3, 2010. [Google Scholar]
- Cavigelli M, Li WW, Lin A, Su B, Yoshioka K, Karin M. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 1996;15:6269–6279. [PMC free article] [PubMed] [Google Scholar]
- Challenger F. Biological methylation. Sci. Prog. 1947;35:396–416. [PubMed] [Google Scholar]
- Challenger F. Biological methylation. Adv. Enzymol. Relat. Subj. Biochem. 1951;12:429–491. doi: 10.1002/9780470122570.ch8. [DOI] [PubMed] [Google Scholar]
- Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, Das S, Ghosh N, Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol. Sci. 2006;89:431–437. doi: 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Kuo TL, Wu MM. Arsenic and cancers. Lancet. 1988;1(8583):414–415. doi: 10.1016/s0140-6736(88)91207-x. [DOI] [PubMed] [Google Scholar]
- Chen CL, Chiou HY, Hsu LI, Hsueh YM, Wun MM, Wang YH, Chen CJ. Arsenic in drinking water and risk of urinary tract cancer: a follow-up study from northeastern Taiwan. Cancer Epidemiol. Biomarkers Prev. 2010;19:101–110. doi: 10.1158/1055-9965.EPI-09-0333. [DOI] [PubMed] [Google Scholar]
- Chen Y, Graziano JH, Parvez F, Liu M, Slavkovich V, Kalra T, Argos M, Islam T, Ahmed A, Rakibuz-Zaman M, et al. Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: prospective cohort study. BMJ. 2011 doi: 10.1136/bmj.d2431. doi: 10.1136/bmj.d2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Hall M, Graziano JH, Slavkovich V, van Geen A, Parvez F, Ahsan H. A prospective study of blood selenium levels and the risk of arsenic-related premalignant skin lesions. Cancer Epidemiol. Biomarkers Prev. 2007;17:207–213. doi: 10.1158/1055-9965.EPI-06-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Parvez F, Gamble M, Islam T, Ahmed A, Argos M, Graziano JH, Ahsan H. Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh. Toxicol. Appl. Pharmacol. 2009;239:184–192. doi: 10.1016/j.taap.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’i IC, Blackwell RQ. A controlled retrospective study of Blackfoot disease, an endemic peripheral gangrene disease in Taiwan. Am. J. Epidemiol. 1968;88:7–24. doi: 10.1093/oxfordjournals.aje.a120869. [DOI] [PubMed] [Google Scholar]
- Chiou HY, Chiou ST, Hsu YH, Chou YL, Tseng CH, Wei ML, Chen CJ. Incidence of transitional cell carcinoma and arsenic in drinking water: a follow-up study of 8,102 residents in an arseniasis-endemic area in northeastern Taiwan. Am. J. Epidemiol. 2001;153:411–418. doi: 10.1093/aje/153.5.411. [DOI] [PubMed] [Google Scholar]
- Chowdhury UK, Zakharyan RA, Hernandez A, Avram MD, Kopplin MJ, Aposhian HV. Glutathione-S-transferase-omega [MMA(V) reductase] knockout mice: enzyme and arsenic species concentrations in tissues after arsenate administration. Toxicol. Appl. Pharmacol. 2006;216:446–457. doi: 10.1016/j.taap.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Chung CJ, Hsueh YM, Bai CH, Huang YK, Huang YL, Yang MH, Chen CJ. Polymorphisms in arsenic metabolism genes, urinary arsenic methylation profile and cancer. Cancer Causes Control. 2009;20:1653–1661. doi: 10.1007/s10552-009-9413-0. [DOI] [PubMed] [Google Scholar]
- Clements R, Tree HG, Ewing PL, Emerson GA. Conversion by tissues of inorganic arsenicals to forms not recoverable by the usual assay methods. Tex. Rep. Biol. Med. 1951;9:27–33. [PubMed] [Google Scholar]
- Cohen SM, Arnold LL, Eldan M, Lewis AS, Beck BD. Methylated arsenicals: the implications of metabolism and carcinogenicity studies in rodents to human risk assessment. Crit. Rev. Toxicol. 2006;36:99–133. doi: 10.1080/10408440500534230. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Arnold LL, Uzvolgyi E, St. John M, Yamamoto S, Lu X, Le XC. Possible role of dimethylarsinous acid in dimethylarsinic acid-induced urothelial toxicity and regeneration in the rat. Chem. Res. Toxicol. 2002;15:1150–1157. doi: 10.1021/tx020026z. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Ellwein LB. Cell proliferation in carcinogenesis. Science. 1990;249:1007–1011. doi: 10.1126/science.2204108. [DOI] [PubMed] [Google Scholar]
- Cohen A, King H, Strangeways WI. CCCCXXI. Trypanocidal action and chemical constitution. Part X. Arylthioarsinates. J. Chem. Soc. 1931:3043–3057. [Google Scholar]
- Cohen SM, Yamamoto S, Cano M, Arnold LL. Urothelial cytotoxicity and regeneration induced by dimethylarsinic acid in rats. Toxicol. Sci. 2001;59:68–74. doi: 10.1093/toxsci/59.1.68. [DOI] [PubMed] [Google Scholar]
- Crane RK, Lipmann R. The effect of arsenate on aerobic phosphorylation. J. Biol. Chem. 1953;201:235–243. [PubMed] [Google Scholar]
- Crecelius EA. Changes in the chemical speciation of arsenic following ingestion by man. Environ. Health Perspect. 1977;19:147–150. doi: 10.1289/ehp.7719147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen WR. Is Arsenic an Aphrodisiac? The Sociochemistry of an Element. Cambridge, U.K: Royal Society of Chemistry; 2008. [Google Scholar]
- Cullen WR, Bentley R. The toxicity of trimethylarsine: an urban myth. J. Environ. Monit. 2005;7:11–15. doi: 10.1039/b413752n. [DOI] [PubMed] [Google Scholar]
- De Chaudhuri S, Ghosh P, Sarma N, Majumdar P, Sau TJ, Basu S, Roychoudhury S, Ray K, Giri AK. Genetic variants associated with arsenic susceptibility: study of purine nucleoside phosphorylase, arsenic (+3) methyltransferase, and glutathione S-transferase omega genes. Environ. Health Perspect. 2008;116:501–505. doi: 10.1289/ehp.10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delnomdedieu M, Basti MM, Styblo M, Otvos JD, Thomas DJ. Complexation of arsenic species in rabbit erythrocytes. Chem. Res. Toxicol. 1994;7:621–627. doi: 10.1021/tx00041a006. [DOI] [PubMed] [Google Scholar]
- Del Razo LM, Quintanilla-Vega B, Brambila-Colombres E, Calderon-Avande ES, Manno M, Albores A. Stress proteins induced by arsenic. Toxicol. Appl. Pharmacol. 2001;177:132–148. doi: 10.1006/taap.2001.9291. [DOI] [PubMed] [Google Scholar]
- Ding W, Liu W, Cooper KL, Qin XJ, de Souza Bergo PL, Hudson LG, Liu KJ. Inhibition of poly(ADP-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J. Biol. Chem. 2009;384:6809–6817. doi: 10.1074/jbc.M805566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan BA, Kasprzak KS, Rice JM. Transplacental carcinogenic effects of nickel (II) acetate in the renal cortex, renal pelvis and adenohypophysis in F344/NCr rats. Carcinogenesis. 1992;13:1351–1357. doi: 10.1093/carcin/13.8.1351. [DOI] [PubMed] [Google Scholar]
- Dixon HBF. The biochemical action of arsenic acids especially as phosphate analogues. Adv. Inorg. Chem. 1997;44:191–227. [Google Scholar]
- Dong J-T, Lou X-M. Arsenic-induced DNA-strand breaks associated with DNA-protein crosslinks in human fetal lung fibroblasts. Mutat. Res. 1993;302:97–102. doi: 10.1016/0165-7992(93)90010-s. [DOI] [PubMed] [Google Scholar]
- Doudoroff M, Barker HA, Hassid WZ. Studies with bacterial sucrose phosphorylase. III. Arsenolytic decomposition of sucrose and of glucose-1-phosphate. J. Biol. Chem. 1947;170:147–150. [Google Scholar]
- Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Herbin-Davis K, Styblo M, Le XC, Creed JT, Maeda N, Hughes MF, et al. Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem. Res. Toxicol. 2009;22:1713–1720. doi: 10.1021/tx900179r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobna Z, Waters SB, Devesa V, Harmon AW, Thomas DJ, Styblo M. Metabolism and toxicity of arsenic in human urothelial cells expressing rat arsenic (+3 oxidation state)-methyltransferase. Toxicol. Appl. Pharmacol. 2005;207:147–159. doi: 10.1016/j.taap.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobna Z, Waters SB, Walton FS, LeCluyse EL, Thomas DJ, Styblo M. Interindividual variation in the metabolism of arsenic in cultured primary human hepatocytes. Toxicol. Appl. Pharmacol. 2004;201:166–177. doi: 10.1016/j.taap.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Drobna Z, Xing W, Thomas DJ, Styblo M. shRNA silencing of AS3MT expression minimizes arsenic methylation capacity of HepG2 cells. Chem. Res. Toxicol. 2006;19:894–898. doi: 10.1021/tx060076u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druwe IL, Vaillancourt RR. Influence of arsenate and arsenite on signal transduction pathways: an update. Arch. Toxicol. 2010;84:585–596. doi: 10.1007/s00204-010-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap LG. Perforations of the nasal septum due to inhalation of arsenous oxide. JAMA. 1921;76:568–569. [Google Scholar]
- El-Masri HA, Kenyon EM. Development of a human physiologically based pharmacokinetic (PBPK) model for inorganic arsenic and its mono- and di-methylated metabolites. J. Pharmacokinet. Pharmacodyn. 2008;35:31–68. doi: 10.1007/s10928-007-9075-z. [DOI] [PubMed] [Google Scholar]
- Endo G, Kuroda K, Okamoto A, Hiriguchi S. Dimethylarsenic acid induces tetraploids in Chinese hamster cells. Bull. Environ. Contam. Toxicol. 1992;48:131–137. doi: 10.1007/BF00197495. [DOI] [PubMed] [Google Scholar]
- Enterline PE, Day R, Marsh GM. Cancers related to exposure to arsenic at a copper smelter. Occup. Environ. Med. 1995;52:28–32. doi: 10.1136/oem.52.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Commission. Ambient Air Pollution by AS, CD and NI compounds (Position Paper—Final) 2000. Available at: http://ec.europa.eu/environment/air/pdf/pp_as_cd_ni.pdf, pp. 318. Accessed on March 22, 2011. [Google Scholar]
- European Food Safety Authority (EFSA) Scientific opinion on arsenic in food. EFSA J. 2009;7:1351. [Google Scholar]
- Evans MV, Dowd SM, Kenyon EM, Hughes MF, El-Masri HA. A physiologically based pharmacokinetic model for intravenous and ingested dimethylarsinic acid in mice. Toxicol. Sci. 2008;104:250–260. doi: 10.1093/toxsci/kfn080. [DOI] [PubMed] [Google Scholar]
- Feldman RG, Niles CA, Kelly-Hayes M, Sax DS, Dixon WJ, Thompson DJ, Laundau E. Peripheral neuropathy in arsenic smelter workers. Neurology. 1979;29:939–944. doi: 10.1212/wnl.29.7.939. [DOI] [PubMed] [Google Scholar]
- Felix K, Manna SK, Wise K, Barr J, Ramesh GT. Low levels of arsenic activates nuclear factor kappaβ and activator protein-1 in immortalized mesencephalic cells. J. Biochem. Mol. Toxicol. 2005;19:67–77. doi: 10.1002/jbt.20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreccio C, Gonzalez C, Milosavjlevic V, Marshall G, Sancha AM, Smith AH. Lung cancer and arsenic concentrations in drinking water in Chile. Epidemiology. 2000;11:673–679. doi: 10.1097/00001648-200011000-00010. [DOI] [PubMed] [Google Scholar]
- Focazio MJ, Welch AH, Watkins SA, Helsel DR, Horn MA. A Retrospective Analysis on the Occurrence of Arsenic in Ground-Water Resources of the United States and Limitations in Drinking Water Supply Characterizations. USGS Water-Resources Investigations Report 99-4279, pp.27. 1999. p. 27. Available at: http://pubs.usgs.gov/wri/wri994279/pdf/wri994279.pdf. Accessed on March 23, 2011. [Google Scholar]
- Fomenko DE, Xing W, Adair BM, Thomas DJ, Gladyshev VN. High-throughput identification of catalytic redox-active cysteine residues. Science. 2007;315:387–389. doi: 10.1126/science.1133114. [DOI] [PubMed] [Google Scholar]
- Frisbie WS. Federal control of spray residues on fruits and vegetables. Am. J. Public Health. 1936;26:369–373. doi: 10.2105/ajph.26.4.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost DV. Arsenicals in biology—retrospect and prospect. Fed. Proc. 1967;26:194–207. [PubMed] [Google Scholar]
- Fujihara J, Kunito T, Agusa T, Yasuda T, Iida R, Fujii Y, Takeshita H. Population differences in the human arsenic (+3 oxidation state) methyltransferase (AS3MT) gene polymorphism detected by using genotyping method. Toxicol. Appl. Pharmacol. 2007;225:251–254. doi: 10.1016/j.taap.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Gamble MV, Liu X, Ahsan H, Pilsner JR, Ilievski V, Slavkovich V, Parvez F, Chen Y, Levy P, Factor-Litvak P, et al. Folate and arsenic metabolism: a double-blind, placebo-controlled folic acid-supplementation trial in Bangladesh. Am. J. Clin. Nutr. 2006;84:1093–1101. doi: 10.1093/ajcn/84.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry PR, Covington TR, Mann S, Shipp AM, Yager JW, Clewell HJ., 3rd Physiologically based pharmacokinetic modeling of arsenic in the mouse. J. Toxicol. Environ. Health A. 2004;67:43–71. doi: 10.1080/15287390490253660. [DOI] [PubMed] [Google Scholar]
- Gentry PR, McDonald TB, Sullivan DE, Shipp AM, Yager JW, Clewell HJ., III Analysis of genomic dose-response information on arsenic to inform key events in a mode of action for carcinogenicity. Environ. Molec. Mutagen. 2010;51:1–14. doi: 10.1002/em.20505. [DOI] [PubMed] [Google Scholar]
- Germolec DR, Spalding J, Boorman GA, Wilmer JL, Yoshida T, Simeonova PP, Bruccoleri A, Kayama F, Gaido K, Tennant R, et al. Arsenic can mediate skin neoplasia by chronic stimulation of keratinocyte-derived growth factors. Mutat. Res. 1997;386:209–218. doi: 10.1016/s1383-5742(97)00006-9. [DOI] [PubMed] [Google Scholar]
- Germolec DR, Spalding J, Yu H-S, Chen GS, Simeonova PP, Humble MC, Bruccoleri A, Boorman GA, Foley JF, Yoshida T, et al. Arsenic enhancement of skin neoplasia by chronic stimulation of growth factors. Am. J. Pathol. 1998;153:1775–1785. doi: 10.1016/S0002-9440(10)65692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germolec DR, Yoshida T, Gaido K, Wilmer JL, Simeonova PP, Kayama F, Burleson F, Dong W, Lange RW, Luster MI. Arsenic induces overexpression of growth factors in human keratinocytes. Toxicol. Appl. Pharmacol. 1996;141:308–318. doi: 10.1006/taap.1996.0288. [DOI] [PubMed] [Google Scholar]
- Gomez-Rubio P, Meza-Montenegro MM, Cantu-Soto E, Klimecki WT. Genetic association between intronic variants in AS3MT and arsenic methylation efficiency is focused on a large linkage disequilibrium cluster in chromosome 10. J. Appl. Toxicol. 2010;30:260–270. doi: 10.1002/jat.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Lu X, Cullen WR, Le XC. Unstable trivalent arsenic metabolites, monomethylarsonous acid and dimethylarsinous acid. J. Anal. At. Spectrom. 2001;16:1409–1413. [Google Scholar]
- Gregus Z, Roos G, Geerlings P, Németi B. Mechanism of thiol-supported arsenate reduction mediated by phosphorolytic-arsenolytic enzymes: II. Enzymatic formation of arsenylated products susceptible for reduction to arsenite by thiols. Toxicol. Sci. 2009;110:282–292. doi: 10.1093/toxsci/kfp113. [DOI] [PubMed] [Google Scholar]
- Gresser MJ. ADP-arsenate. J. Biol. Chem. 1981;256:5981–5983. [PubMed] [Google Scholar]
- Hall M, Chen Y, Ahsan H, Slavkovich J, van Geen A, Parvez F, Graziano J. Blood arsenic as a biomarker of arsenic exposure: results from a prospective study. Toxicology. 2006;225:225–233. doi: 10.1016/j.tox.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Haque R, Mazumder DN, Samanta S, Ghosh N, Kalman D, Smith MM, Mitra S, Santra A, Lahiva S, Das S, et al. Arsenic in drinking water and skin lesions: dose-response data from West Bengal, India. Epidemiology. 2003;14:174–182. doi: 10.1097/01.EDE.0000040361.55051.54. [DOI] [PubMed] [Google Scholar]
- Harden A. Alcoholic Fermentation. Monographs on Biochemistry. London, UK: Longmans, Green & Co; 1932. pp. 243. Longmans, Green & Co, London, U.K. [Google Scholar]
- Hartwig A, Blessing H, Schwerdtle T, Walter I. Modulation of DNA repair process by arsenic and selenium compounds. Toxicology. 2003;193:161–169. doi: 10.1016/j.tox.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Kanisawa M, Yamanaka K, Ito T, Udaka N, Ohji H, Okudela K, Okada S, Kitamura H. Dimethylarsinic acid, a main metabolite of inorganic arsenics, has tumorigenicity and progression effects in the pulmonary tumors of A/J mice. Cancer Lett. 1998;125:83–88. doi: 10.1016/s0304-3835(97)00484-9. [DOI] [PubMed] [Google Scholar]
- Heck JE, Andrew AS, Onega T, Rigas JR, Jackson BP, Karagas MR, Duell EJ. Lung cancer in a U.S. population with low to moderate arsenic exposure. Environ. Health Perspect. 2009;117:1718–1723. doi: 10.1289/ehp.0900566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hei TK, Liu SX, Waldren C. Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A. 1998;95:8103–8107. doi: 10.1073/pnas.95.14.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez A, Xamena N, Sekaran C, Tokunaga H, Sampayo-Reyes A, Quinteros D, Creus A, Marcos R. High arsenic metabolic efficiency in AS3MT287Thr allele carriers. Pharmacogenet. Genomics. 2008;18:349–355. doi: 10.1097/FPC.0b013e3282f7f46b. [DOI] [PubMed] [Google Scholar]
- Hernandez A, Xamena N, Surrallés J, Sekaran C, Tokunaga H, Quinteros D, Creus A, Marcos R. Role of the Met(287)Thr polymorphism in the AS3MT gene on the metabolic arsenic profile. Mutat. Res. 2007;637:80–92. doi: 10.1016/j.mrfmmm.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Heuper WC, Payne WW. Experimental studies in metal carcinogenesis. Arch. Environ. Health. 1962;5:445–462. doi: 10.1080/00039896.1962.10663311. [DOI] [PubMed] [Google Scholar]
- Hood E. The apple bites back: claiming old orchards for residential development. Environ. Health Perspect. 2006;114:A470–A476. doi: 10.1289/ehp.114-a470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopenhayn-Rich C, Biggs ML, Fuchs A, Bergoglio R, Tello EE, Nicolli H, Smith AH. Bladder cancer mortality associated with arsenic in drinking water in Argentina. Epidemiology. 1996;7:117–124. doi: 10.1097/00001648-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Hopenhayn-Rich C, Biggs ML, Smith AH. Lung and kidney cancer mortality associated with arsenic in drinking water in Cordoba, Argentina. Int. J. Epidemiol. 1998;27:561–569. doi: 10.1093/ije/27.4.561. [DOI] [PubMed] [Google Scholar]
- Hu Y, Jin X, Snow ET. Effect of arsenic on transcription factor AP-1 and NF-kappaβ DNA binding activity and related gene expression. Toxicol. Lett. 2002;133:33–45. doi: 10.1016/s0378-4274(02)00083-8. [DOI] [PubMed] [Google Scholar]
- Hu Y, Su L, Snow ET. Arsenic toxicity is enzyme specific and its affects on ligation are not caused by the direct inhibition of DNA repair enzymes. Mutat. Res. 1998;408:203–218. doi: 10.1016/s0921-8777(98)00035-4. [DOI] [PubMed] [Google Scholar]
- Huang C, Ke Q, Costa M, Shi X. Molecular mechanisms of arsenic carcinogenesis. Mol. Cell. Biochem. 2004;25:57–66. doi: 10.1023/b:mcbi.0000007261.04684.78. [DOI] [PubMed] [Google Scholar]
- Hughes MF, Edwards BC, Herbin-Davis KM, Saunders J, Styblo M, Thomas DJ. Arsenic (+3 oxidation state) methyltransferase genotype affects steady-state distribution and clearance of arsenic in arsenate-treated mice. Toxicol. Appl. Pharmacol. 2010;249:217–223. doi: 10.1016/j.taap.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Hughes MF, Kitchin KT. Arsenic, oxidative stress and carcinogenesis. In: Singh KK, editor. Oxidative Stress, Disease and Cancer. London, U.K: Imperial College Press; 2006. pp. 825–850. [Google Scholar]
- Hyson JM., Jr. A history of arsenic in dentistry. J. Calif. Dent. Assoc. 2007;35:135–139. [PubMed] [Google Scholar]
- IARC. Some Drinking-Water Disinfectants and Contaminants, Including Arsenic. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Lyon, France: International Agency for Research on Cancer, WHO; 2004. [PMC free article] [PubMed] [Google Scholar]
- Ishinishi N, Kodama Y, Nobotomo K, Hisanaga A. Preliminary experimental study on carcinogenicity of arsenic trioxide in rat lung. Environ. Health Perspect. 1977;19:191–196. doi: 10.1289/ehp.7719191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishinishi N, Yamamoto A, Hisanaga A, Inamasu T. Tumorigenicity of arsenic trioxide to the lung in Syrian Golden hamsters by intermittent instillations. Cancer Lett. 1983;21:141–147. doi: 10.1016/0304-3835(83)90200-8. [DOI] [PubMed] [Google Scholar]
- Jarup L, Pershagen G, Wall S. Cumulative arsenic exposure and lung cancer in smelter workers: a dose-response study. Am. J. Ind. Med. 1989;15:31–41. doi: 10.1002/ajim.4700150105. [DOI] [PubMed] [Google Scholar]
- Jiang T, Huang Z, Chan JY, Zhang DD. Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol. Appl. Pharmacol. 2009;250:8–14. doi: 10.1016/j.taap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Oppermann H, Jackson J, Levinson W. Induction of four proteins in chick embryo cells by sodium arsenite. J. Biol. Chem. 1980;25:6975–6980. [PubMed] [Google Scholar]
- Jolliffe DM. A history of the use of arsenicals in man. J. R. Soc. Med. 1993;86:287–289. doi: 10.1177/014107689308600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaise T, Oya-Ohta Y, Ochi T, Okubo T, Hanaoka K, Irgolic KJ, Sakurai T, Matsubara C. Toxicological study of organic arsenic compound in marine algae using mammalian cell culture technique. J. Food Hyg. Soc. Jpn. 1996;37:135–141. [Google Scholar]
- Kanisawa M, Schroeder HA. Life term studies on the effect of trace elements on spontaneous tumors in mice and rats. Cancer Res. 1969;29:892–895. [PubMed] [Google Scholar]
- Karagas MR, Stokel TA, Morris JS, Tosteson TD, Weiss JE, Spencer SK, Greenberg ER. Skin cancer risk in relation to toenail arsenic concentrations in a US population based case-control study. Am. J. Epidemiol. 2001;153:559–565. doi: 10.1093/aje/153.6.559. [DOI] [PubMed] [Google Scholar]
- Karagas MR, Tosteson TD, Morris JS, Demidenko E, Mott LA, Heaney J, Schned A. Incidence of transitional cell carcinoma of the bladder and arsenic exposure in New Hampshire. Cancer Causes Control. 2004;15:465–472. doi: 10.1023/B:CACO.0000036452.55199.a3. [DOI] [PubMed] [Google Scholar]
- Kenyon EM, Hughes MF, Levander OA. Influence of dietary selenium on the disposition of arsenate in the female B6C3F1 mouse. J. Toxicol. Environ. Health. 1997;51:279–299. doi: 10.1080/00984109708984027. [DOI] [PubMed] [Google Scholar]
- Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. U. S. A. 1989;86:99–103. doi: 10.1073/pnas.86.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated metabolites. Toxicol. Appl. Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol. Lett. 2003;137:3–13. doi: 10.1016/s0378-4274(02)00376-4. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Conolly R. Arsenic-induced carcinogenesis—oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Chem. Res. Toxicol. 2010;23:327–335. doi: 10.1021/tx900343d. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Wallace K. Evidence against the nuclear in situ binding of arsenicals—oxidative stress theory of arsenic carcinogenesis. Toxicol. Appl. Pharmacol. 2008;232:252–257. doi: 10.1016/j.taap.2008.06.021. [DOI] [PubMed] [Google Scholar]
- Kligerman AD, Doerr CL, Tennant AH, Harrington-Brock K, Allen JW, Winkfield E, Poorman-Allen P, Kundu B, Funasaka K, Roop BC, et al. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: induction of chromosomal mutations but not gene mutations. Environ. Mol. Mutagen. 2003;42:192–205. doi: 10.1002/em.10192. [DOI] [PubMed] [Google Scholar]
- Kubachka KM, Kohan MJ, Herbin-Davis K, Creed JT, Thomas DJ. Exploring the in vitro formation of trimethylarsine sulfide from dimethylthioarsinic acid in anaerobic microflora of mouse cecum using HPLC-ICP-MS and HPLC-ESI-MS. Toxicol. Appl. Pharmacol. 2009;239:137–143. doi: 10.1016/j.taap.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Kumagi Y, Sumi D. Arsenic: signal transduction, transcription factor and biotransformation involved in cellular response and toxicity. Annu. Rev. Pharmacol. Toxicol. 2007;47:243–262. doi: 10.1146/annurev.pharmtox.47.120505.105144. [DOI] [PubMed] [Google Scholar]
- Lagerkvist B, Linderholm H, Nordberg GF. Vasospastic tendency and Raynaud’s phenomenon in smelter workers exposed to arsenic. Environ. Res. 1986;39:465–474. doi: 10.1016/s0013-9351(86)80070-6. [DOI] [PubMed] [Google Scholar]
- Landner L. The biological alkylation of mercury. Biochem. J. 1972;130:67P–69P. doi: 10.1042/bj1300067p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz RC, Hays AM. Role of oxidative stress in arsenic-induced toxicity. Drug Metab. Rev. 2006;38:791–804. doi: 10.1080/03602530600980108. [DOI] [PubMed] [Google Scholar]
- Lee AM, Franmeni JF. Arsenic and respiratory cancer in man. An occupational study. J. Natl. Cancer Inst. 1969;42:1045–1052. [PubMed] [Google Scholar]
- Lee T-C, Wang-Wuu S, Huang RY, Lee KCC, Jan KY. Differential effects of pre- and posttreatment of sodium arsenite on the genotoxicity of methyl methanesulfonate in Chinese hamster ovary cells. Cancer Res. 1986;46:1854–1857. [PubMed] [Google Scholar]
- Leitch AL. The experimental inquiry into the causes of cancer. Br. Med. J. 1923;2:1–7. doi: 10.1136/bmj.2.3262.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch AL, Kennaway EL. Experimental production of cancer by arsenic. Br. Med. J. 1922;2:1107–1108. [Google Scholar]
- Leonard SS, Harris GK, Shi X. Metal-induced oxidative stress and signal transduction. Free Radic. Biol. Med. 2004;37:1921–1942. doi: 10.1016/j.freeradbiomed.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Lewis DR, Southwick JW, Ouellet-Hellstrom R, Rench J, Calderon RL. Drinking water arsenic in Utah: a cohort mortality study. Environ. Health Perspect. 1999;107:359–365. doi: 10.1289/ehp.99107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-H, Rossman TG. Mechanism of comutagenesis of sodium arsenite with N-methyl-N-nitrosourea. Biol. Trace Element Res. 1989a;28:373–381. doi: 10.1007/BF02917278. [DOI] [PubMed] [Google Scholar]
- Li J-H, Rossman TG. Inhibition of DNA ligase activity by arsenite: a possible mechanism of its comutagenesis. Mol. Toxicol. 1989b;2:1–9. [PubMed] [Google Scholar]
- Li J-H, Rossman TG. Comutagenesis of sodium arsenite with ultraviolet radiation in Chinese hamster V79 cells. Biol. Metals. 1991;4:197–200. doi: 10.1007/BF01141180. [DOI] [PubMed] [Google Scholar]
- Liao CM, Lin TL, Chen SC. A Weibull-PBPK model for assessing risk of arsenic-induced skin lesions in children. Sci. Total Environ. 2008;392:203–217. doi: 10.1016/j.scitotenv.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Lin GF, Du H, Chen JG, Lu HC, Kai JX, Zhou YS, Guo WC, Zhang XJ, Lu DR, Golka K, et al. Glutathione S-transferases M1 and T1 polymorphisms and arsenic content in hair and urine in two ethnic clans exposed to indoor combustion of high arsenic coal in Southwest Guizhou, China. Arch. Toxicol. 2007;81:545–551. doi: 10.1007/s00204-007-0187-4. [DOI] [PubMed] [Google Scholar]
- Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem. Res. Toxicol. 2001;14:305–311. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- Lin S, Shi Q, Nix FB, Styblo M, Beck MA, Herbin-Davis KM, Hall LL, Simeonsson JB, Thomas DJ. A novel S-adenosyl-L-methionine: arsenic(III) methyltransferase from rat liver cytosol. J. Biol. Chem. 2002;277:10795–10803. doi: 10.1074/jbc.M110246200. [DOI] [PubMed] [Google Scholar]
- Lindberg AL, Kumar R, Goessler W, Thirumaran R, Gurzau E, Koppova K, Rudnai P, Leonardi G, Fletcher T, Vahter M. Metabolism of low-dose inorganic arsenic in a central European population: influence of sex and genetic polymorphisms. Environ. Health Perspect. 2007;115:1081–1086. doi: 10.1289/ehp.10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren A, Danielsson RG, Dencker W, Vahter M. Embryotoxicity of arsenite and arsenate: distribution in pregnant mice and monkeys and effects on embryonic cells in vitro. Acta Pharmacol. Toxicol. 1984;54:311–320. doi: 10.1111/j.1600-0773.1984.tb01936.x. [DOI] [PubMed] [Google Scholar]
- Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Guyton KZ, Gorospe M, Xu Q, Lee JC, Holbrook NJ. Differential activation of ERK, JNK/SAPK and p38/CSBP/RK MAP kinase family members during the cellular response to arsenite. Free Radic. Biol. Med. 1996;21:771–781. doi: 10.1016/0891-5849(96)00176-1. [DOI] [PubMed] [Google Scholar]
- Lloyd NC, Morgan HW, Nicholson BK, Ronimus RS. The composition of Ehrlich’s Salvarsan: resolution of a century-old debate. Angew. Chem. Int. Ed. 2005;44:941–944. doi: 10.1002/anie.200461471. [DOI] [PubMed] [Google Scholar]
- Lu M, Wang H, Li X-F, Arnold LL, Cohen SM, Le XC. Binding of dimethylarsinous acid to Cys-13α of rat hemoglobin is responsible for the retention of arsenic in rat blood. Chem. Res. Toxicol. 2007;20:27–37. doi: 10.1021/tx060195+. [DOI] [PubMed] [Google Scholar]
- Luster MI, Simeonova PP. Arsenic and urinary bladder cell proliferation. Toxicol. Appl. Pharmacol. 2004;198:419–423. doi: 10.1016/j.taap.2003.07.017. [DOI] [PubMed] [Google Scholar]
- Lynn S, Gurr JR, Lai HT, Jan KY. NADH oxidase activation is involved in arsenite-induced oxidative DNA damage in human vascular smooth muscle cells. Circ. Res. 2000;85:514–519. doi: 10.1161/01.res.86.5.514. [DOI] [PubMed] [Google Scholar]
- Mabuchi K, Lilienfeld AM, Snell LM. Cancer and occupational exposure to arsenic: a study of pesticide workers. Prev. Med. 1980;9:51–77. doi: 10.1016/0091-7435(80)90059-6. [DOI] [PubMed] [Google Scholar]
- Maine Rural Health. Maine Rural Health Association 2003 Outstanding Service Award. 2008. Available at: http://www.maineruralhealth.org/award.htm. Accessed December 3, 2010. [Google Scholar]
- Mann S, Droz PO, Vahter M. A physiologically based pharmacokinetic model for arsenic exposure. I. Development in hamsters and rabbits. Toxicol. Appl. Pharmacol. 1996a;137:8–22. doi: 10.1006/taap.1996.0052. [DOI] [PubMed] [Google Scholar]
- Mann S, Droz PO, Vahter M. A physiologically based pharmacokinetic model for arsenic exposure. II. Validation and application in humans. Toxicol. Appl. Pharmacol. 1996b;140:471–486. doi: 10.1006/taap.1996.0244. [DOI] [PubMed] [Google Scholar]
- Marafante E, Vahter M, Norin H, Envall J, Sandström M, Christakopoulos A, Ryhage R. Biotransformation of dimethylarsinic acid in mouse, hamster and man. J. Appl. Toxicol. 1987;7:111–117. doi: 10.1002/jat.2550070207. [DOI] [PubMed] [Google Scholar]
- Marsh GM, Stone RA, Esmen NA, Gula MJ, Gause CK, Petersen NJ, Meaney FJ, Rodney S, Prybylski D. A case-control study of lung cancer mortality in six Gila basin, Arizona smelter towns. Environ. Res. 1997;75:56–72. doi: 10.1006/enrs.1997.3768. [DOI] [PubMed] [Google Scholar]
- Marsh J. Separation of arsenic. In: Carson J, editor. The American Journal of Pharmacy. Vol. II. Philadelphia, PA: Merriew and Gunn; 1837. pp. 307–314. [Google Scholar]
- Marshall G, Ferreccio C, Yuan Y, Bates MN, Steinmaus C, Selvin S, Liaw J, Smith AH. Fifty-year study of lung and bladder cancer mortality in Chile related to arsenic in drinking water. J. Natl. Cancer Inst. 2007;99:920–928. doi: 10.1093/jnci/djm004. [DOI] [PubMed] [Google Scholar]
- Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, Kligerman AD. Methylated trivalent arsenic species are genotoxic. Chem. Res. Toxicol. 2001;14:355–361. doi: 10.1021/tx000251l. [DOI] [PubMed] [Google Scholar]
- Mass MJ, Wang L. Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat. Res. 1997;386:263–277. doi: 10.1016/s1383-5742(97)00008-2. [DOI] [PubMed] [Google Scholar]
- Mazumder DN, Haque R, Ghosh N, De BK, Chakraborti D, Smith AH. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int. J. Epidemiol. 2000;29:1047–1052. doi: 10.1093/ije/29.6.1047. [DOI] [PubMed] [Google Scholar]
- McCarty KM, Chen YC, Quamruzzaman Q, Rahman M, Mahiuddin G, Hsueh YM, Su L, Smith T, Ryan L, Christiani DC. Arsenic methylation, GSTT1, GSTM1, GSTP1 polymorphisms, and skin lesions. Environ. Health Perspect. 2007a;115:341–345. doi: 10.1289/ehp.9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty KM, Houseman EA, Quamruzzaman Q, Rahman M, Mahsuddin G, Smith T, Ryan L, Christiani DC. The impact of diet and betel nut use on skin lesions associated with drinking-water arsenic in Pabna, Bangladesh. Environ. Health Perspect. 2006;114:334–340. doi: 10.1289/ehp.7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty KM, Ryan L, Houseman EA, Williams PL, Miller DP, Quamruzzaman Q, Rahman M, Mahiuddin G, Smith T, Gonzalez E, et al. A case-control study of GST polymorphisms and arsenic related skin lesions. Environ. Health. 2007b;6:5. doi: 10.1186/1476-069X-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza MM, Yu L, Rodriguez YY, Guild M, Thompson D, Gandolfi AJ, Klimecki WT. Developmentally restricted genetic determinants of human arsenic metabolism: association between urinary methylated arsenic and CYT19 polymorphisms in children. Environ. Health Perspect. 2005;113:775–781. doi: 10.1289/ehp.7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozier NM, McConnell KP, Hoffman JL. S-adenosyl-L-methionine: thioether S-methyltransferase, a new enzyme in sulfur and selenium metabolism. J. Biol. Chem. 1988;263:4527–4531. [PubMed] [Google Scholar]
- Mumford JL, Wu K, Xia Y, Kwok R, Yang Z, Foster J, Sanders WE. Chronic arsenic exposure and cardiac repolarization abnormalities with QT interval prolongation in a population-based study. Environ. Health Perspect. 2007;115:690–694. doi: 10.1289/ehp.9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgo AJ. Clinical trials of arsenic trioxide in hematologic and solid tumors: overview of the National Cancer Institute Cooperative Research and Development Studies. Oncologist. 2001;6:22–28. doi: 10.1634/theoncologist.6-suppl_2-22. [DOI] [PubMed] [Google Scholar]
- Navas-Acien A, Silbergeld EK, Pastor-Barriuso R, Guallar E. Arsenic exposure and prevalence of type 2 diabetes in US adults. JAMA. 2008;300:814–822. doi: 10.1001/jama.300.7.814. [DOI] [PubMed] [Google Scholar]
- Nelson WC, Lykins MH, Mackey J, Newill VA, Finklea JF, Hammer DI. Mortality among orchard workers exposed to lead arsenate spray: a cohort study. J. Chronic Dis. 1973;26:105–118. doi: 10.1016/0021-9681(73)90009-x. [DOI] [PubMed] [Google Scholar]
- Nemeti B, Regonesi ME, Tortora P, Gregus Z. Polynucleotide phosphorylase and mitochondrial ATP synthase mediate reduction of arsenate to the more toxic arsenite by forming arsenylated analogues of ADP and ATP. Toxicol. Sci. 2010;117:270–281. doi: 10.1093/toxsci/kfq141. [DOI] [PubMed] [Google Scholar]
- Nesnow S, Roop BC, Lambert G, Kadiiska M, Mason RP, Cullen WR, Mass MJ. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. Chem. Res. Toxicol. 2002;14:1627–1634. doi: 10.1021/tx025598y. [DOI] [PubMed] [Google Scholar]
- Neubauer O. Arsenical cancer: a review. Br. J. Cancer. 1947;1:192–251. doi: 10.1038/bjc.1947.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton DE. Forensic Chemistry. New York, NY: Infobase Publishing; 2007. [Google Scholar]
- Nishioka H. Mutagenic activities of metal compounds in bacteria. Mutat. Res. 1975;31:185–189. doi: 10.1016/0165-1161(75)90088-6. [DOI] [PubMed] [Google Scholar]
- Nordstrom DK. Worldwide occurrences of arsenic in ground water. Science. 2002;296:2143–2145. doi: 10.1126/science.1072375. [DOI] [PubMed] [Google Scholar]
- NRC. Arsenic in Drinking Water. Washington, DC: National Research Council, National Academy Press; 1999. [Google Scholar]
- NRC. Arsenic in Drinking Water. Update to the 1999 Arsenic in Drinking Water Report. Washington, DC: National Research Council, National Academy Press; 2001. [Google Scholar]
- Ott MG, Holder BB, Gordon HL. Respiratory cancer and occupational exposure to arsenicals. Arch. Environ. Health. 1974;29:250–255. doi: 10.1080/00039896.1974.10666582. [DOI] [PubMed] [Google Scholar]
- Paiva L, Hernández A, Martínez V, Creus A, Quinteros D, Marcos R. Association between GSTO2 polymorphism and the urinary arsenic profile in copper industry workers. Environ. Res. 2010;110:463–468. doi: 10.1016/j.envres.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Parascandola J. Carl Voegtlin and the ‘arsenic receptor’ in chemotherapy. J. Hist. Med. Allied Sci. 1977;23:151–171. doi: 10.1093/jhmas/xxxii.2.151. [DOI] [PubMed] [Google Scholar]
- Parvez F, Chen Y, Brandt-Rauf PW, Slavkovich V, Islam T, Ahmed A, Argos M, Hassan R, Yunus M, Haque SE, et al. A prospective study of respiratory symptoms associated with chronic arsenic exposure in Bangladesh: findings from the Health Effects of Arsenic Longitudinal Study (HEALS) Thorax. 2010;65:528–533. doi: 10.1136/thx.2009.119347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershagen G, Bjorklund N-E. On the pulmonary tumorigenicity of arsenic trisulfide and calcium arsenate in hamsters. Cancer Lett. 1985;27:99–104. doi: 10.1016/0304-3835(85)90013-8. [DOI] [PubMed] [Google Scholar]
- Pershagen G, Nordberg G, Bjorklund N-E. Carcinomas of the respiratory tract in hamsters given arsenic trioxide and/or benzo(a)pyrene by the pulmonary route. Environ. Res. 1984;34:227–241. doi: 10.1016/0013-9351(84)90091-4. [DOI] [PubMed] [Google Scholar]
- Peryea FJ. Historical use of lead arsenate insecticides, resulting soil contamination and implications for soil remediation. 1998 Proceedings, 16th World Congress of Soil Science, 20–26 August 1998, Montpelllier, France, pp. 7. Available at: http://soils.tfrec.wsu.edu/leadhistory.htm. [Google Scholar]
- Peters RA. The study of enzymes in relation to selective toxicity in animal tissues. Symp. Soc. Exper. Biol. 1949;3:36–59. [Google Scholar]
- Peters RA, Stocken LA, Thompson RHS. British anti-lewisite. Nature. 1945;156:616–619. doi: 10.1038/156616a0. [DOI] [PubMed] [Google Scholar]
- Petito Boyce C, Lewis AS, Sax SN, Eldan M, Cohen SM, Beck BD. Probabilistic analysis of human health risks associated with background concentrations of inorganic arsenic: use of a margin exposure approach. Hum. Ecol. Risk Assess. 2008;14:1159–1201. [Google Scholar]
- Piatek K, Schwerdtle T, Hartwig A, Bal W. Monomethylarsonous acid destroys a tetrathiolate zinc finger much more efficiently than inorganic arsenite: mechanistic considerations and consequences for DNA repair inhibition. Chem. Res. Toxicol. 2008;21:600–606. doi: 10.1021/tx7003135. [DOI] [PubMed] [Google Scholar]
- Pilsner JR, Hall MN, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect. 2007;117:254–260. doi: 10.1289/ehp.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsner JR, Hall MN, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Associations of plasma selenium with arsenic and genomic methylation of leukocyte DNA in Bangladesh. Environ. Health Perspect. 2011;119:113–118. doi: 10.1289/ehp.1001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am. J. Clin. Nutr. 2007;86:1179–1186. doi: 10.1093/ajcn/86.4.1179. [DOI] [PubMed] [Google Scholar]
- Pilsner J R, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano J H, Gamble M V. Folate deficiency, hyperhomocysteinemia, low urinary creatinine and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect. 2009;117:254–260. doi: 10.1289/ehp.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto SS, Enterline PE, Henderson V, Varner MO. Mortality experience in relation to a measured arsenic trioxide exposure. Environ. Health Perspect. 1977;19:127–130. doi: 10.1289/ehp.7719127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinyayev TS, Kohan MJ, Herbin-Davis K, Creed JT, Thomas DJ. Preabsorptive metabolism of sodium arsenate by anaerobic microbiota of mouse cecum forms a variety of methylated and thiolated arsenicals. Chem. Res. Toxicol. 2011;24:475–477. doi: 10.1021/tx200040w. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Biological responses to arsenic compounds. J. Biol. Chem. 2009;284:18583–18587. doi: 10.1074/jbc.R900003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KE, Basu A, Hubbard AE, Bates MN, Kalman D, Rey O, Smith A, Smith MT, Steinmaus C, Skibola CF. Association of genetic variation in cystathionine-beta-synthase and arsenic metabolism. Environ. Res. 2010;110:580–587. doi: 10.1016/j.envres.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Vahter M, Ekstrom EC, Rahman M, Golan Mustafa AH, Waheed MA, Yunus M, Persson LA. Association of arsenic exposure during pregnancy with fetal loss and infant death: a cohort study in Bangladesh. Am. J. Epidemiol. 2007;165:1389–1396. doi: 10.1093/aje/kwm025. [DOI] [PubMed] [Google Scholar]
- Rahman A, Vahter M, Smith AH, Nermell B, Yunus M, El Arifeen S, Persson LA, Ekstrom EC. Arsenic exposure during pregnancy and size at birth: a prospective cohort study in Bangladesh. Am. J. Epidemiol. 2009;169:304–312. doi: 10.1093/aje/kwn332. [DOI] [PubMed] [Google Scholar]
- Rahman M, Tondel M, Ahmad SA, Chowdhury IA, Faruquee MH, Axelson O. Hypertension and arsenic exposure in Bangladesh. Hypertension. 1999;33:74–78. doi: 10.1161/01.hyp.33.1.74. [DOI] [PubMed] [Google Scholar]
- Riethmiller S. From Atoxyl to Salvarsan: searching for the magic bullet. Chemotherapy. 2005;51:235–242. doi: 10.1159/000087453. [DOI] [PubMed] [Google Scholar]
- Roberts SM, Weimar WR, Vinson JRT, Munson JW, Bergeron RJ. Measurement of arsenic bioavailability in soil using a primate model. Toxicol. Sci. 2002;67:303–310. doi: 10.1093/toxsci/67.2.303. [DOI] [PubMed] [Google Scholar]
- Rohe G H. Arsenic. In Reference-Book of Practical Therapeutics, 1896 (F. P. Foster, Ed.), Vol.1, pp. 142--146. D. Appleton and Company, New York. Available at: http://www.archive.org/details/cu31924104225978. Accessed July 27, 2011. [Google Scholar]
- Rosenthal SM. Action of arsenic upon the fixed sulphydryl groups of proteins. Public Health Rep. 1932;47:241–256. [Google Scholar]
- Rossman TG. Enhancement of UV-mutagenesis by low concentrations of arsenite in E. coli. Mutat. Res. 1981;91:207–211. doi: 10.1016/0165-7992(81)90032-4. [DOI] [PubMed] [Google Scholar]
- Rossman TG. Mechanisms of arsenic carcinogenesis. An integrated approach. Mutat. Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Rossman TG, Meyn MS, Troll W. Effects of arsenite on DNA repair in Escherichia coli. Environ. Health Perspect. 1977;19:229–233. doi: 10.1289/ehp.7719229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FS, Bosland MC. Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin. An animal model for arsenic carcinogenesis. Toxicol. Appl. Pharmacol. 2001;176:64–71. doi: 10.1006/taap.2001.9277. [DOI] [PubMed] [Google Scholar]
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llaamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- Roussel RR, Barchowsky A. Arsenic inhibits NF-kappaβ-mediated gene transcription by blocking Ikappaβ phosphorylation and degradation. Arch. Biochem. Biophys. 2000;377:204–212. doi: 10.1006/abbi.2000.1770. [DOI] [PubMed] [Google Scholar]
- Rust DM, Soignet SL. Risk/benefit profile of arsenic trioxide. Oncologist. 2001;6(Suppl. 2):29–32. doi: 10.1634/theoncologist.6-suppl_2-29. [DOI] [PubMed] [Google Scholar]
- Samikkannu T, Chen CH, Yih LH, Wang AS, Lin SY, Chen TC, Jan KY. Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem. Res. Toxicol. 2003;16:409–414. doi: 10.1021/tx025615j. [DOI] [PubMed] [Google Scholar]
- Sampayo-Reyes A, Hernandez A, El-Yamani N, Lopez-Campos C, Mayet-Machado E, Rincon-Castaneda CB, Limones-Aguilar Mde L, Lopez-Campos JE, de Leon MB, González-Hernández S, et al. Arsenic induces DNA damage in environmentally exposed Mexican children and adults. Influence of GSTO1 and AS3MT polymorphisms. Toxicol. Sci. 2010;117:63–71. doi: 10.1093/toxsci/kfq173. [DOI] [PubMed] [Google Scholar]
- Saxe JK, Bowers TS, Reid KR. Arsenic. In: Morrison RD, Murphy BL, editors. Environmental Forensics: Contaminant Specific Guide. Burlington, MA: Academic Press; 2006. pp. 279–292. [Google Scholar]
- Scheindlin S. The duplicitous nature of inorganic arsenic. Mol. Interv. 2005;5:60–64. doi: 10.1124/mi.5.2.1. [DOI] [PubMed] [Google Scholar]
- Schläwicke Engström K, Broberg K, Concha G, Nermell B, Warholm M, Vahter M. Genetic polymorphisms influencing arsenic metabolism: evidence from Argentina. Environ. Health Perspect. 2007;115:599–605. doi: 10.1289/ehp.9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser E, Goessler W, Francesconi KA. Human metabolism of arsenolipids present in cod liver. Anal. Bioanal. Chem. 2006;385:367–376. doi: 10.1007/s00216-006-0401-x. [DOI] [PubMed] [Google Scholar]
- Schoen A, Beck B, Jharma R, Dube E. Arsenic toxicity of low doses: epidemiology and mode of action consideration. Toxicol. Appl. Pharmacol. 2004;198:253–267. doi: 10.1016/j.taap.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Schoof RA, Eickhoff J, Yost LJ, Crecelius EA, Cragin DW, Meacher DM, Menzel DB. Dietary exposure to inorganic arsenic. In: Chappell WR, Abernathy CO, Calderon RL, editors. Arsenic Exposure and Health Effects. Amsterdam, The Netherlands: Elsevier Science B.V.; 1999. pp. 81–88. [Google Scholar]
- Schroeder HA, Kanisawa M, Frost DV, Mitchener M. Germanium, tin and arsenic in rats: effects on growth, survival, pathological lesions and life span. J. Nutrition. 1968;96:37–45. [Google Scholar]
- Schumacher-Wolz U, Dieter HH, Klein D, Schneider K. Oral exposure to inorganic arsenic: evaluation of its carcinogenic and non-carcinogenic effects. Crit. Rev. Toxicol. 2009;39:271–298. doi: 10.1080/10408440802291505. [DOI] [PubMed] [Google Scholar]
- Schumilla JA, Wetterhahn KE, Barchowsky A. Inhibition of NF-kappa β binding to DNA by chromium, cadmium, mercury, zinc, and arsenite in vitro: evidence of a thiol mechanism. Arch. Biochem. Biophys. 1998;349:356–362. doi: 10.1006/abbi.1997.0470. [DOI] [PubMed] [Google Scholar]
- Sekeres MS. New data with arsenic trioxide in leukemias and myelodysplastic syndromes. Clin. Lymphoma Myeloma. 2007;8(Suppl. 1):S7–S12. doi: 10.3816/clm.2007.s.027. [DOI] [PubMed] [Google Scholar]
- Shacklette HT, Boerngen JG. Element Concentrations in Soils and Other Surficial Materials of the Conterminous United States. 1984. U.S. Government Printing Office US Geological Survey Professional Paper 1270, pp. 15. U.S. Government Printing Office, Washington, DC. Available at: http://pubs.usgs.gov/pp/pdf/1270/PP1270_508.pdf. Accessed July 27, 2011. [Google Scholar]
- Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004;255:67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Hulderman T, Luster MI. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic. J. Biol. Chem. 2002;277:2945–2950. doi: 10.1074/jbc.M109136200. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, et al. Arsenic mediates cell proliferation and gene expression in the bladder epithelium: association with activating protein-1 transactivation. Cancer Res. 2000;60:2445–3453. [PubMed] [Google Scholar]
- Smith AH, Goycolea M, Haque R, Biggs ML. Marked increase in bladder and lung cancer mortality in a region of Northern Chile due to arsenic in drinking water. Am. J. Epidemiol. 1998;147:660–669. doi: 10.1093/oxfordjournals.aje.a009507. [DOI] [PubMed] [Google Scholar]
- Smith KR, Klei LR, Barchowsky A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;280:L442–L449. doi: 10.1152/ajplung.2001.280.3.L442. [DOI] [PubMed] [Google Scholar]
- Smith AH, Lingas EO, Rahman M. Contamination of drinking-water by arsenic in Bangladesh: a public health emergency. Bull. World Health Organ. 2000;78:1093–1103. [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein O, Steinmaus C, Bates MN, Selvin S. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ. Health Perspect. 2006;114:1293–1296. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Steinmaus CM. Health effects of arsenic and chromium in drinking water: recent human findings. Annu. Rev. Public Health. 2009;30:107–122. doi: 10.1146/annurev.publhealth.031308.100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohel N, Persson LA, Rahman M, Streatfield PK, Yunus M, Ekström EC, Vahter M. Arsenic in drinking water and adult mortality: a population-based cohort study in rural Bangladesh. Epidemiology. 2009;20:824–830. doi: 10.1097/EDE.0b013e3181bb56ec. [DOI] [PubMed] [Google Scholar]
- Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011;9:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- Steinmaus C, Moore LE, Shipp M, Kalman D, Rey OA, Biggs ML, Hopenhayn C, Bates MN, Zheng S, Wiencke JK, et al. Genetic polymorphisms in MTHFR 677 and 1298, GSTM1 and T1, and metabolism of arsenic. J. Toxicol. Environ. Health A. 2007;70:159–170. doi: 10.1080/15287390600755240. [DOI] [PubMed] [Google Scholar]
- Steinmaus C, Yuan Y, Bates MN, Smith AH. Case-control study of bladder cancer and drinking water arsenic in the western United States. Am. J. Epidemiol. 2003;158:1193–2001. doi: 10.1093/aje/kwg281. [DOI] [PubMed] [Google Scholar]
- Stocken LA, Thompson RHS. British anti-lewisite. 1. Arsenic derivatives of thiol proteins. Biochem. J. 1946;40:529–535. doi: 10.1042/bj0400529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub AC, Clark KA, Ross MA, Chandra AG, Li S, Gao X, Pagano PJ, Stolz DB, Barchowsky A. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J. Clin. Invest. 2008;118:3980–3989. doi: 10.1172/JCI35092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styblo M, Serves SV, Cullen WR, Thomas DJ. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem. Res. Toxicol. 1997;10:27–33. doi: 10.1021/tx960139g. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Arnold LL, Pennington KL, Chen B, Naranmandura H, Le XC, Cohen SM. Dietary administration of sodium arsenite to rats: relations between dose and urinary concentrations of methylated and thio-metabolites and effects on the rat urinary bladder epithelium. Toxicol. Appl. Pharmacol. 2010;244:99–105. doi: 10.1016/j.taap.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Sykora P, Snow ET. Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite. Toxicol. Appl. Pharmacol. 2008;228:385–394. doi: 10.1016/j.taap.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Tapio S, Grosche B. Arsenic in the aetiology of cancer. Mutat. Res. 2006;612:215–246. doi: 10.1016/j.mrrev.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Taylor AS. The Principles and Practice of Medical Jurisprudence (A. S. Taylor Ed.) 2nd ed. Vol. 1. London, U.K: J & A Churchill; 1873. Metallic Irritants; pp. 250–279. [Google Scholar]
- Thomas DJ, Li J, Waters SB, Xing W, Adair BM, Drobna Z, Devesa V, Styblo M. Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp. Biol. Med. (Maywood) 2007;232:3–13. [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson UP, Dalgard DW, Reeves J, Adamson RH. Tumor incidence in a chemical carcinogenesis study of nonhuman primates. Regul. Toxicol. Pharmacol. 1994;19:130–151. doi: 10.1006/rtph.1994.1013. [DOI] [PubMed] [Google Scholar]
- Tokar EJ, Diwan BA, Ward JM, Delker DA, Waalkes MP. Carcinogenic effects of “whole life” exposure to inorganic arsenic in CD1 mice. Toxicol. Sci. 2011a;119:73–83. doi: 10.1093/toxsci/kfq315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokar EJ, Qu W, Waalkes MP. Arsenic, stem cells and the developmental basis of adult cancer. Toxicol. Sci. 2011b;120(Suppl. 1):S192–S203. doi: 10.1093/toxsci/kfq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollestrup K, Daling JR, Allard J. Mortality in a cohort of orchard workers exposed to lead arsenate pesticide spray. Arch. Environ. Health. 1995;50:221–229. doi: 10.1080/00039896.1995.9940391. [DOI] [PubMed] [Google Scholar]
- Tollestrup K, Frost FJ, Harter LC, McMillan GP. Mortality among children residing near the American Smelting and Refining Company (ASARCO) copper smelter in Ruston, Washington. Arch. Environ. Health. 2003;58:683–691. doi: 10.3200/AEOH.58.11.683-691. [DOI] [PubMed] [Google Scholar]
- Tseng CH, Chong CK, Chen CJ, Tai TY. Dose-response relationship between peripheral vascular disease and ingested inorganic arsenic among residents in blackfoot disease endemic villages in Taiwan. Atherosclerosis. 1996;120:125–133. doi: 10.1016/0021-9150(95)05693-9. [DOI] [PubMed] [Google Scholar]
- Tseng WP, Chu HM, How SW, Fong JM, Lin CS, Yeh S. Prevalence of skin cancer in an endemic area of chronic arsenicism in Taiwan. J. Natl. Cancer Inst. 1968;40:453–463. [PubMed] [Google Scholar]
- U.S. EPA. Special Report on Ingested Inorganic Arsenic. Skin Cancer; Nutritional Essentiality, 1988. EPA/625/3-87/013. Risk Assessment Forum, Washington DC. [Google Scholar]
- U.S. EPA. Risk Assessment Guidance for Superfund (RAGS) Volume I: Human Health Evaluation Manual (Part E, Supplemental Guidance for Dermal Risk Assessment, Interim) (Draft). EPA/540/R.99/005; OSWER 9284.7–102.EP. 2001. Available at: http://www.epa.gov/oswer/riskassessment/ragse/index.htm. Accessed November 29, 2010. [Google Scholar]
- U.S. EPA. (2006). Revised Reregistration Eligibility Decision for MSMA, DSMA, CAMA and Cacodylic Acid. EPA-738-R-06-021. Available at: http://www.epa.gov/oppsrrd1/REDs/organic_arsenicals_red.pdf. Accessed July 27, 2011. [Google Scholar]
- U.S. EPA. Letter to S. Johnson (U.S. EPA) re: Advisory on EPA’s Assessments of Carcinogenic Effects of Organic and Inorganic Arsenic: A report of the U.S. EPA Science Advisory Board. EPA-SAB-07–008. 2007. Available at: http://epa.gov/sab/pdf/sab-07-008.pdf. [Google Scholar]
- U.S. EPA. Reregistration Eligibility Decision for Chromated Arsenicals. EPA 739-R-08–1006. 2008. p. 90. Available at: http://www.epa.gov/oppsrrd1/REDs/cca_red.pdf. Accessed July 27, 2011. [Google Scholar]
- U.S. EPA. Amendment to Organic Arsenicals RED. 2009. Available at: http://www.epa.gov/oppsrrd1/REDs/organic-arsenicals-amended.pdf. Accessed on March 31, 2011. [Google Scholar]
- Uthus EO. Evidence for arsenic essentiality. Environ. Geochem. Health. 1992;14:55–58. doi: 10.1007/BF01783629. [DOI] [PubMed] [Google Scholar]
- Uthus EO. Arsenic essentiality. A role affecting methionine essentiality. J. Trace Elem. Exp. Med. 2003;16:345–355. [Google Scholar]
- Vahidnia A, van der Voet GB, de Wolff FA. Arsenic neurotoxicity—a review. Hum. Exp. Toxicol. 2007;26:823–832. doi: 10.1177/0960327107084539. [DOI] [PubMed] [Google Scholar]
- Vahter M, Marafante E. Effects of low dietary intake of methionine, choline or proteins on the biotransformation of arsenite in the rabbit. Toxicol. Lett. 1987;37:41–46. doi: 10.1016/0378-4274(87)90165-2. [DOI] [PubMed] [Google Scholar]
- Vahter M, Marafante E, Dencker L. Tissue distribution and retention of 74As-dimethylarsinic acid in mice and rats. Arch. Environ. Contam. Toxicol. 1984;13:259–264. doi: 10.1007/BF01055275. [DOI] [PubMed] [Google Scholar]
- Valenzuela OL, Drobna Z, Hernandez-Castellanos E, Sanchez-Pena LC, García-Vargas GG, Borja-Aburto VH, Styblo M, Del Razo LM. Association of AS3MT polymorphisms and the risk of premalignant arsenic skin lesions. Toxicol. Appl. Pharmacol. 2009;239:200–207. doi: 10.1016/j.taap.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaanderen J, Moore LE, Smith MT, Lan Q, Zhang L, Skibola CF, Rothman N, Vermeulen R. Application of OMICS technologies in occupational and environmental health research; current status and projections. Occup. Environ. Med. 2010;67:136–143. doi: 10.1136/oem.2008.042788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegtlin C. The pharmacology of arsphenamine (Salvarsan) and related compounds. Physiol. Rev. 1925;5:563–594. [Google Scholar]
- Voegtlin C, Dyer HA, Leonard CS. On the mechanism of the action of arsenic upon protoplasm. Public Health Rep. 1923;38:1882–1912. [Google Scholar]
- von Ehrenstein OS, Guha Mazumder DN, Hira-Smith M, Ghosh N, Yuan Y, Windam G, Gosh A, Haque R, Lahir S, Kalman D, et al. Pregnancy outcomes, infant mortality, and arsenic in drinking water in West Bengal, India. Am. J. Epidemiol. 2006;163:662–669. doi: 10.1093/aje/kwj089. [DOI] [PubMed] [Google Scholar]
- von Ehrenstein OS, Mazumder DN, Yuan Y, Samanta S, Balmes J, Sil A, Ghosh N, Hira-Smith M, Haque R, Purushothaman R, et al. Decrements in lung function related to arsenic in drinking water in West Bengal, India. Am. J. Epidemiol. 2005;162:533–541. doi: 10.1093/aje/kwi236. [DOI] [PubMed] [Google Scholar]
- von Ehrenstein OS, Poddar S, Yuan Y, Mazumder DG, Eskenazi B, Basu A, Hira-Smith M, Gosh N, Labiri S, Haque R, et al. Children’s intellectual function in relation to arsenic exposure. Epidemiology. 2007;18:44–51. doi: 10.1097/01.ede.0000248900.65613.a9. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Diwan BA, Ward JM, Devor DE, Goyer RA. Renal tubular tumors and atypical hyperplasia in B6C3F1 mice exposed to lead acetate during gestation and lactation occur with minimal chronic nephropathy. Cancer Res. 1995;55:5265–5271. [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Germolec DR, Trempus CS, Cannon RE, Tokar EJ, Tennant RW, Ward JM, Diwan BA. Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 2008;68:8278–8285. doi: 10.1158/0008-5472.CAN-08-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Diwan BA. Induction of tumors of the liver, lung, ovary and adrenal in adult mice after brief maternal gestational exposure to inorganic arsenic: promotional effects of postnatal phorbol ester exposure on hepatic and pulmonary, but not dermal cancers. Carcinogenesis. 2004;25:133–141. doi: 10.1093/carcin/bgg181. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward J, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic ovarian, pulmonary and adrenal tumors in mice. Toxicol. Appl. Pharmacol. 2003;186:7–17. doi: 10.1016/s0041-008x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- Walton FS, Waters SB, Jolley SL, LeCluyse EL, Thomas DJ, Styblo M. Selenium compounds modulate the activity of recombinant rat AsIII-methyltransferase and the methylation of arsenite by rat and human hepatocytes. Chem. Res. Toxicol. 2003;16:261–265. doi: 10.1021/tx025649r. [DOI] [PubMed] [Google Scholar]
- Wang X-J, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol. Appl. Pharmacol. 2007;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-J, Sun Z, Chen W, Li Y, Villeneuve NF, Zhang DD. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: enhanced Keap1-Cul3 interaction. Toxicol. Appl. Pharmacol. 2008;230:383–389. doi: 10.1016/j.taap.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, Yeh SD, Shen KH, Shen CH, Juang GD, Hsu LI, Chiou HY, Chen CJ. A significantly joint effect between arsenic and occupational exposures and risk genotypes/diplotypes of CYP2E1, GSTO1 and GSTO2 on risk of urothelial carcinoma. Toxicol. Appl. Pharmacol. 2009;241:111–118. doi: 10.1016/j.taap.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang J. Abundant indispensable redundancies in cellular metabolic networks. Genome Biol. Evol. 2009;1:23–33. doi: 10.1093/gbe/evp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanibuchi H, Yamamoto S, Chen H, Yoshida K, Endo G, Hori T, Fukushima S. Promoting effects of dimethylarsinic acid on N-butyl-N-(4-hydroxybutyl)nitrosamine-induced urinary bladder carcinogenesis in rats. Carcinogenesis. 1996;17:2435–2439. doi: 10.1093/carcin/17.11.2435. [DOI] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Parvez F, Ahsan H, Factor-Litvak P, Kline J, van Geen A, Slavkovich V, Lolacono NJ, Levy D, et al. Water arsenic exposure and intellectual function in 6-year-old children in Araihazar, Bangladesh. Environ. Health Perspect. 2007;115:285–289. doi: 10.1289/ehp.9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Parvez F, Ahsan H, Factor-Litvak P, van Geen A, Slavkovich V, Lolacono NJ, Cheng Z, Hussain I, et al. Water arsenic exposure and children’s intellectual function in Araihazar, Bangladesh. Environ. Health Perspect. 2004;112:1329–1333. doi: 10.1289/ehp.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters SB, Devesa V, Del Razo LM, Styblo M, Thomas DJ. Endogenous reductants support the catalytic function of recombinant rat cyt19, an arsenic methyltransferase. Chem. Res. Toxicol. 2004a;17:404–409. doi: 10.1021/tx0342161. [DOI] [PubMed] [Google Scholar]
- Waters SB, Devesa V, Fricke MW, Creed JT, Stýblo M, Thomas DJ. Glutathione modulates recombinant rat arsenic (+3 oxidation state) methyltransferase-catalyzed formation of trimethylarsine oxide and trimethylarsine. Chem. Res. Toxicol. 2004b;17:1621–1629. doi: 10.1021/tx0497853. [DOI] [PubMed] [Google Scholar]
- Waxman S, Anderson KC. History of the development of arsenic derivatives in cancer therapy. Oncologist. 2001;6:3–10. doi: 10.1634/theoncologist.6-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- Wei M, Cohen SM, Cao M, Arnold LL. Effects of co-administration of antioxidants and arsenicals on the rat urinary bladder epithelium. Toxicol. Sci. 2005;83:237–245. doi: 10.1093/toxsci/kfi033. [DOI] [PubMed] [Google Scholar]
- Wei M, Wanibuchi H, Morimura K, Iwai S, Yoshida K, Endo G, Nakae D, Fukushima S. Carcinogenicity of dimethylarsinic acid in male F344 rats and genetic alterations in induced urinary bladder tumors. Carcinogenesis. 2002;23:1387–1397. doi: 10.1093/carcin/23.8.1387. [DOI] [PubMed] [Google Scholar]
- Wei M, Wanibuchi H, Yamamoto S, Li W, Fukushima S. Urinary bladder carcinogenicity of dimethylarsinic acid in male F344 rats. Carcinogenesis. 1999;20:1873–2187. doi: 10.1093/carcin/20.9.1873. [DOI] [PubMed] [Google Scholar]
- Welch K, Higgins I, Oh M, Burchfiel C. Arsenic exposure, smoking and respiratory cancer in copper smelter workers. Arch. Environ. Health. 1982;37:325–335. doi: 10.1080/00039896.1982.10667586. [DOI] [PubMed] [Google Scholar]
- Winski SL, Carter DE. Arsenate toxicity in human erythrocytes: characterization of morphologic changes and determination of the mechanism of damage. J. Toxicol. Environ. Health. 1998;53:345–355. doi: 10.1080/009841098159213. [DOI] [PubMed] [Google Scholar]
- Wijeweera JB, Gandolfi AJ, Parrish A, Lantz RC. Sodium arsenite enhances AP-1 and NFkappaβ and DNA binding and induces stress protein expression in precision-cut rat lung slices. Toxicol. Sci. 2001;61:283–294. doi: 10.1093/toxsci/61.2.283. [DOI] [PubMed] [Google Scholar]
- Wood ES. Arsenic as a Domestic Poison. Boston, MA: Wright & Potter Printing Co.; 1885. [Google Scholar]
- Wood JM. Biological cycles for toxic elements in the environment. Science. 1974;183:1049–1052. doi: 10.1126/science.183.4129.1049. [DOI] [PubMed] [Google Scholar]
- Wood TC, Salavagionne OE, Mukherjee B, Wang L, Klumpp AF, Thomae BA, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. Human arsenic methyltransferase (AS3MT) pharmacogenetics: gene resequencing and functional genomics studies. J. Biol. Chem. 2006;281:7364–7373. doi: 10.1074/jbc.M512227200. [DOI] [PubMed] [Google Scholar]
- WPSC. The Safety of Chromated Copper Arsenate (CCA)-Treated Wood. Wood Preservative Science Council; 2008. p. 2. pp. 2.Wood Preservative Science Council. Available at: http://www.woodpreservativescience.org/safety.shtml. Accessed December 3, 2010. [Google Scholar]
- Wu F, Burns FJ, Zhang R, Uddin AN, Rossman TG. Arsenite-induced alterations of DNA photodamage repair and apoptosis after solar-simulation UVR in mouse keratinocytes in vitro. Environ. Health Perspect. 2005;113:983–986. doi: 10.1289/ehp.7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager JW, Hicks JB, Fabianova E. Airborne arsenic and urinary excretion of arsenic metabolites during boiler cleaning operations in a Slovak coal-fired power plant. Environ. Health Perspect. 1997;105:836–842. doi: 10.1289/ehp.97105836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Hisanaga A, Ishinishi N. Tumorigenicity of inorganic arsenic compounds following intratracheal instillations to the lungs of hamsters. Int. J. Cancer. 1987;40:220–223. doi: 10.1002/ijc.2910400216. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Konishi Y, Matsuda T, Murai T, Shibata M-A, Matsui-Yuasa I, Otani S, Kuroda K, Endo G, Fukushima S. Cancer induction by an organic arsenic compound, dimethylarsinic acid (cacodylic acid), in F344/DuCrj rats after pretreatment with five carcinogens. Cancer Res. 1995;55:1271–1276. [PubMed] [Google Scholar]
- Yamanaka K, Hasegawa A, Sawamura R, Okada S. Dimethylated arsenics induce DNA strand breaks in lung via the production of active oxygen in mice. Biochem. Biophys. Res. Commun. 1989;165:43–50. doi: 10.1016/0006-291x(89)91031-0. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Hoshino M, Okamoto M, Sawamura R, Hasegawa A, Okada S. Induction of DNA damage by dimethylarsine, a metabolite of arsenics, is for the major part likely due to its peroxyl radical. Biochem. Biophys. Res. Commun. 1990;168:58–64. doi: 10.1016/0006-291x(90)91674-h. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Ohtsubo K, Hasegawa A, Hayashi H, Ohgi H, Kanisawa M, Okada S. Exposure to dimethylarsinic acid, a main metabolite of inorganic arsenics, strongly promotes tumorigenesis initiated by 4-nitroquinoline 1-oxide in the lungs of mice. Carcinogenesis. 1996;17:767–770. doi: 10.1093/carcin/17.4.767. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Okada S. Induction of lung-specific DNA damage by metabolically methylated arsenics via the production of free radicals. Environ. Health Perspect. 1994;102(Suppl. 3):37–40. doi: 10.1289/ehp.94102s337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan-Chu H. Arsenic distribution in soils. In: Nriagu JO, editor. Arsenic in the Environment, Part I; Cycling and Characterization. Hoboken, NJ: John Wiley and Sons, Inc.; 1994. pp. 17–47. [Google Scholar]
- Yang C, Frenkel K. Arsenic-mediated cellular signal transduction, transcription factor activation and aberrant gene expression: implications in carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 2002;21:331–342. [PubMed] [Google Scholar]
- Yarnell A. Salvarsan. Chem. Eng. News. 2005;83:116. [Google Scholar]
- Yokohira M, Arnold LL, Pennington KL, Suzuki S, Kakiuchi-Kiyota S, Herbin-Davis K, Thomas DJ, Cohen SM. Severe systemic toxicity and urinary bladder cytotoxicity and regenerative hyperplasia induced by arsenite in arsenic (+3 oxidation state) methyltransferase knockout mice. A preliminary report. Toxicol. Appl. Pharmacol. 2010;246:1–7. doi: 10.1016/j.taap.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Yorifuji T, Tsuda T, Grandjean P. Unusual cancer excess after neonatal arsenic exposure from contaminated milk powder. J. Natl. Cancer Inst. 2010;102:360–361. doi: 10.1093/jnci/djp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost LJ, Tao SH, Egan SK, Barraj LM, Smith KM, Tsuji JS, Lowney YW, Schoof RA, Rachman NJ. Estimation of dietary intake of inorganic arsenic in U.S. children. Hum. Ecol. Risk Assess. 2004;10:473–483. [Google Scholar]
- Yu D. A physiologically based pharmacokinetic model of inorganic arsenic. Regul. Toxicol. Pharmacol. 1999;29:128–141. doi: 10.1006/rtph.1999.1282. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Liaw J, Bates M, Smith AH. Kidney cancer mortality: fifty-year latency patterns related to arsenic exposure. Epidemiology. 2010;21:103–108. doi: 10.1097/EDE.0b013e3181c21e46. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Selvin S, Liaw J, Bates MN, Smith AH. Acute myocardial infarction mortality in comparison with lung and bladder cancer mortality in arsenic-exposed region II of Chile from 1950 to 2000. Am. J. Epidemiol. 2007;166:1382–1391. doi: 10.1093/aje/kwm238. [DOI] [PubMed] [Google Scholar]
- Zakharyan RA, Ayala-Fierro F, Cullen WR, Carter DM, Aposhian HV. Enzymatic methylation of arsenic compounds. VII. Monomethylarsonous acid (MMAIII) is the substrate for MMA methyltransferase of rabbit liver and human hepatocytes. Toxicol. Appl. Pharmacol. 1999;158:9–15. doi: 10.1006/taap.1999.8687. [DOI] [PubMed] [Google Scholar]
- Zakharyan RA, Sampayo-Reyes A, Healy SM, Tsaprailis G, Board PG, Liebler DC, Aposhian HV. Human monomethylarsonic acid (MMA(V)) reductase is a member of the glutathione-S-transferase superfamily. Chem. Res. Toxicol. 2001;14:1051–1057. doi: 10.1021/tx010052h. [DOI] [PubMed] [Google Scholar]
- Zernike K. Arsenic Case Is Considered Homicide, Maine Police Say. New York, NY: The New York Times; 2003. [Google Scholar]
- Zhang TD, Chen CQ, Wang ZG, Wang ZY, Chen SJ, Chen Z. Arsenic trioxide, a therapeutic agent for APL. Oncogene. 2001;20:7146–7153. doi: 10.1038/sj.onc.1204762. [DOI] [PubMed] [Google Scholar]
- Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong CX, Mass MJ. Both hypomethylation and hypermethylation of DNA associated with arsenite exposure in cultures of human cells identified by methylation-sensitive arbitrarily-primed PCR. Toxicol. Lett. 2001;122:223–234. doi: 10.1016/s0378-4274(01)00365-4. [DOI] [PubMed] [Google Scholar]
- Zhu B, Wang X, Li L. Human gut microbiome: the second genome of human body. Protein Cell. 2010;1:718–725. doi: 10.1007/s13238-010-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]