

Abstract
To date, natural products containing 2-benzyl-4H-pyran-4-one and 2-benzylpyridin-4(1H)-one substructures have been encountered in relatively few fungi outside of the black aspergilli clade. While exploring the occurrence of these compounds among Aspergillus spp., it was determined that the structures of the unusual furopyrrols tensidols A and B (5 and 6) and JBIR-86 and JBIR-87 (9 and 10) were incorrect and should be reassigned as 2-benzyl-4H-pyran-4-ones (7, 8, 11e, and 12, respectively). The origin of the unique N-phenyl groups in the 2-benzylpyridin-4(1H)-ones nygerones A and B (1 and 2) was also examined and it was established that N-phenylamides added to the culture medium were suitable substrates for generating these metabolites; however, this phenomenon remained limited to a single fungus in our collection (Aspergillus niger ATCC 1015). A variety of 2-benzyl-4H-pyran-4-ones and 2-benzylpyridin-4(1H)-ones were detected among the black aspergilli, but only pestalamide B (13) was found in all eleven of the tested strains. These metabolites, as well as a group of synthetic analogues demonstrated weak antifungal activity against several Candida strains, Aspergillus flavus, and Aspergillus fumigatus.
Previously, we reported the discovery of the 2-benzylpyridin-4(1H)-one-containing metabolites nygerones A and B (1-2) from Aspergillus niger.1-4 Subsequent 1H NMR-guided efforts by our group to purify additional 2-benzylpyridin-4(1H)-one and related 2-benzyl-4H-pyran-4-one metabolites from A. niger yielded several compounds that we presumed were structurally similar to 1 and 2. However, during the course of dereplication studies, a variety of natural products possessing α-pyridone,5γ-pyrone,6 and furopyrrol7 skeletons were found in the literature and all of these compounds were reported to possess 1H NMR chemical shifts that were virtually identical to the key diagnostic resonances in 1 and 2. Fortunately, the α-pyridone-containing variant had been previously examined by Ye and colleagues who successfully revised the proposed structure of aspernigrin A (3)5 as the more plausible 2-benzylpyridin-4(1H)-one shown for compound 4.8 Our analysis of the furopyrrol-containing molecules, which included tensidols A and B (5-6),7 led to the observation that these structures had arisen from a strikingly different interpretation of 1H and 13C NMR data that were indistinguishable from the values reported for the 2-benzyl-4H-pyran-4-ones carbonarone A (7)6 and pestalamide A (8),9 respectively. In this report, we address the structure revision of the tensidols using a combination of NMR spectroscopy, ab initio carbon chemical shift calculations, and total synthesis. We have also analyzed the distribution of the 2-benzylpyridin-4(1H)-one and 2-benzyl-4H-pyran-4-one metabolites among black aspergilli, and explored the origins of the N-phenyl groups in 1 and 2, as well as investigated the biological activities of these compounds.
Results and Discussion
Structure Revision of the Furopyrrols
The tensidol family of metabolites, which includes 5-6, as well as JBIR-86 (9) and JBIR-87 (10),10 were reported from the fermentation broths of a marine isolate of A. niger and an unidentified Aspergillus sp., respectively. We were initially prompted to examine the spectroscopic data for 5 and 6 since the unusual furopyrrol skeleton in these structures was reportedly unprecedented.7 Focusing on 5, we noted several unusual chemical shift assignments that we could not reconcile. Namely, the 13C NMR data presented for C-2 (162.0), C-3 (178.0), C-3a (119.1), C-4 (164.3), C-5 (116.0), and C-6a (168.8) were inconsistent with our expectations for carbon resonances in isolated or fused furan and pyrrole systems (Supporting Information, Figure S1). At the same time, we purified a small molecule from A. niger whose MS and NMR data matched those reported for 5 (Supporting Information, Table S1); however, de novo structure characterization studies carried out by our group suggested that γ-pyrone 7 was a more reasonable alternative to consider. The molecular formula for 7 exhibited a low H:C ratio (0.85) which, along with the presence of 4JH-C couplings in the HMBC data, might have played confounding roles in previous attempts to unambiguously assign the correct structure for this metabolite.11, 12 Further inspection of the literature demonstrated that nearly one year after the disclosure of 5 in 2006, Zhang and colleagues reported the alternative structure 7 from a marine isolate of Aspergillus carbonarius, although no mention of 5 was provided in their account.6 We propose that 2-benzyl-4H-pyran-4-one 7 represents the correct structure of this metabolite.
A similar set of circumstances surrounded compound 6, which was originally described as a methyl succinate derivative of 5.7 In this case, a 2008 report by Ding et al. unknowingly provided structure 89 as a reasonable revision for compound 6. Our evaluation of both data sets confirmed that the 1H and 13C NMR chemical shifts reported for the two compounds were identical and that 8 should be regarded as the correct structure of this metabolite.
In 2010, we encountered a new opportunity to further test our assertion that the furopyrrol metabolites should be revised as 2-benzyl-4H-pyran-4-ones. We noted that Takagi et al. had obtained what appeared to be two new furopyrrol-containing metabolites, JBIR-86 (9) and JBIR-87 (10);10 however, both compounds exhibited some problematic features. Among our initial concerns were the incongruous 13C NMR chemical shifts (Supporting Information, Figure S1) and presence of suspect 4JH-C couplings within the furopyrrol ring system.10 Additionally, the spectrometric analysis of 9 and 10 was reported to have yielded HRESIMS data containing even-mass quasi-molecular ions at m/z 244.0986 [M + H]+ and 358.1286 [M + H]+, respectively. This would presumably necessitate the incorporation of an odd number of nitrogens in each metabolite. However, the published 13C NMR data for 9 clearly supported the presence of a γ-pyrone, thus eliminating candidate 11a from further consideration (Figure 1A). In addition, the observed chemical shifts for the methyl group in 9 (δ 52.8) suggested that it was attached to an oxygen atom, not the N-methyl amide shown for candidate 11b (Figure 1A). These restrictions left open the possibilities that the revised structure contained a primary ketimine (11c) or methyl imidate (11d) (Figure 1A), but both candidate compounds were also rejected based on biosynthetic considerations and evaluations of their respective predicted 13C NMR chemical shift data (Supporting Information, Figure S2).
Figure 1.

A) Candidate structures (11a-11e) evaluated as alternatives during the revision of 9. B) Mean average errors (MAE) were caluculated using Σ(δcalc − δexp)/#. A smaller MAE value represents better overall agreement between the computational and published 13C-NMR data. C) 1H→13C HMBC correlations observed for synthetic 11e.
With no plausible nitrogen-containing candidate structures left to consider for 9, we focused on formulating a reasonable alternative structure from the NMR data. This approach led us to propose 11e as a candidate structure (Figure 1A). We proceeded to test this hypothesis using gauge-including atomic orbital based 13C NMR chemical shift calculations on both 9 and 11e. Density functional theory calculations performed at the b3lyp/6-31+g(d,p) level yielded a poor mean absolute error value of 16.0 ppm for 9, but a reasonable value of 3.8 ppm for 11e (Figure 1B). Figure 2 illustrates that structure 9 exhibited several pronounced deviations in the calculated carbon NMR values for positions C-2, C-3, C-3a, C-4, C-5, and C-6a from the published data (Δδcalc-δexp −46.5, −20.0, −27.0, −41.1, −25.8, and −20.6 ppm, respectively). In contrast, 11e showed promise as a revised candidate structure with much less deviation in the predicted versus observed 13C NMR data for each of the corresponding carbon atoms (Δδcalc-δexp −5.8, −2.3, 6.6, −7.1, −2.7 and −1.7 ppm, respectively).
Figure 2.
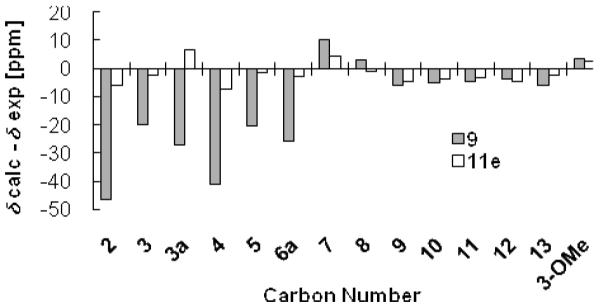
Deviations in the calculated versus observed 13C NMR chemical shifts for 9 and 11e. Differences approaching zero demonstrate good agreement between calculated versus reported 13C NMR data, whereas large differences indicate poor agreement between calculated and reported 13C NMR experimental data.
We further tested the prospect of revising 9 to candidate structure 11e by engaging in its total synthesis. We prepared 11e using a modification of the procedure reported by McCombie et al. (Scheme 1).13 Upon purification of the product, HRESIMS yielded a quasi-molecular ion at m/z 245.0815 [M + H]+ (calcd for C14H13O4, 245.0814). The 1H and 13C NMR data of synthetic 11e was remarkably consistent with each of the chemical shift values reported for 9 (Table 1). Moreover, both the previously published and new 1H→13C HMBC data that we examined showed that all but one of the observed heteronuclear couplings (H-5→C-3) were now accounted for as two-to-three bond correlations (Figure 1C). We are still unable to provide an explanation for the disagreement between the ESIMS data published by Takagi et al. and our own results. However, in light of the evidence, we are quite confident that 11e represents the correct structure for JBIR-86. Based on this finding, we also reevaluated compound 10. Upon inspection of its 1H, 13C, and 1H→13C HMBC NMR data (Supporting Information, Table S1 and Figure S3), reconsideration of its probable shared biosynthetic origins with 11e, and co-isolation of compound 8 from the same crude extract, we have concluded that 10 should be revised to the 2-benzyl-4H-pyran-4-one-containing structure 12.
Scheme 1.
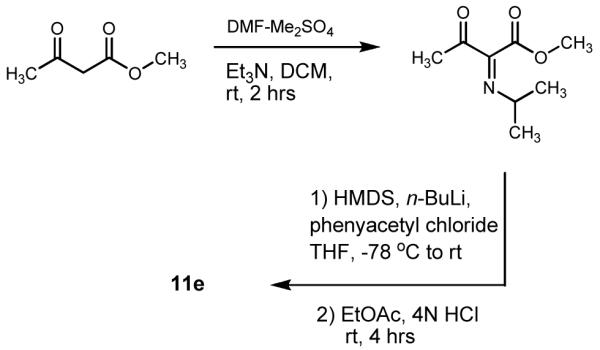
Synthesis of 11e.
Table 1.
Comparisons of the observed 1H and 13C NMR data for 11e versus the values reported for 9.
| Synthetic 11e | 9a | |||
|---|---|---|---|---|
| Position | δ H | δ C | δ H | δ C |
| 2 | 8.41, s | 162.0 | 8.43, s | 162.0 |
| 3a | 119.3 | 119.1 | ||
| 4 | 175.0 | 175.0 | ||
| 5 | 6.19, s | 116.9 | 6.21, s | 117.3 |
| 6a | 167.4 | 167.6 | ||
| 7 | 3.81, s | 39.7 | 3.83, s | 40.0 |
| 8 | 133.9 | 134.1 | ||
| 9,13 | 7.22, m | 129.1 | 7.24, m | 129.4 |
| 10,12 | 7.35, m | 129.1 | 7.32, m | 129.4 |
| 11 | 7.32, m | 127.8 | 7.32, m | 128.0 |
| 3 | 163.6 | 163.9 | ||
| 3-OCH3 | 3.85, s | 52.7 | 3.87, s | 52.8 |
Data reproduced from Takagi, M.; Motohashi, K.; Hwang, J.-H.; Nagai, A.; Kazuo, S. J. Antibiotics. 2010, 63, 371-373.
Distribution of 2-Benzylpyridin-4(1H)-Ones and 2-Benzyl-4H-Pyran-4-Ones among Black Aspergilli
To date, the majority of secondary metabolites bearing 2-benzylpyridin-4(1H)-one and 2-benzyl-4H-pyran-4-one skeletons have been reported from black aspergilli.14, 15 Given our previous encounter with this family of metabolites from A. niger, as well as our desire to better understand its distribution among the members of this clade, we prepared a focused library of 11 Aspergillus strains for further investigation. The fungi were grown with and without epigenetic modifiers2-4 and the extracts examined by LC-ESIMS. By probing extracted-ion chromatograms for each of the target compounds (1, 2, 4, 7, 8, and 13), we were able to determine which black aspergilli were producers of the 2-benzylpyridin-4(1H)-one and 2-benzyl-4H-pyran-4-one metabolites under our culture conditions (Supporting Information, Table S2). Interestingly, the 2-benzylpyridin-4(1H)-one-containing compound pestalamide B (13)9 was the only metabolite detected in all 11 of the black aspergilli strains (note – the 13C NMR data provided for 13 by Ding et al. have been revised16). In contrast, the 2-benzyl-4H-pyran-4-one-containing analog of 13, compound 8, was observed in a much more restricted group of five fungal strains and in some cases it was detected only under chemical epigenetic induction conditions. The unsubstituted amide analogs of 13 and 8, metabolites 4 and 7, respectively, were also found in relatively few fungi (two and four strains, respectively). At this time, the role of methyl succinate conjugation of these metabolites is unknown.
We were struck by the limited distribution of compound 1, which was detected only in A. niger ATCC 1015 treated with suberoylanilide hydroxamic acid (SAHA) (Supporting Information, Table S2). The origin of the N-phenyl group in 1 is an intriguing feature of this metabolite and its biosynthetic source was not readily apparent to us. Our original investigation of 1,1 including follow-up feeding studies involving the incorporation of aniline in the fungal growth medium did not support the idea that the phenyl ring of SAHA was the N-phenyl group precursor. To further verify the role SAHA plays in the production of nygerones, a subsequent test employing p-fluoro SAHA (Supporting Information, Scheme S1A) was used as the eliciting agent. The addition of p-fluoro SAHA resulted in the production of p-fluoro nygerone B (14) from A. niger ATCC 1015 (Supporting Information, Table S1). This led us to use p-fluoro aniline for further feeding studies (Supporting Information, Scheme S2), but we did not observe any production of 14 when the substrate was administered alone or with SAHA present (Supporting Information, Table S1B and S1C). These data indicated that aniline was not a suitable substrate for generating the N-phenyl group in 1. We next considered the possibility that an acetanilide precursor might be utilized in establishing the N-phenyl group in compound 1. Accordingly, we prepared p-fluoro acetanilide (Supporting Information, Scheme S1D and S1E) from p-fluoro aniline and acetyl chloride and added it alone or with SAHA to the growth medium. Under both conditions, 14 was obtained, which allowed us to purify the compound and confirm its structure using ESIMS, 1H, 13C, and 19F 1D and 2D NMR (Supporting Information, Table S1). These data indicate that acetanilides and SAHA are suitable precursor substrates for generating the N-phenyl group of the nygerones.
Evaluation of Antimicrobial Activity
In light of the sparse biological activity data reported for this family of metabolites, we prepared a group of several naturally occurring and synthetic γ-pyrone. and γ-pyridone-containing compounds for investigation (1, 8, 11e, and 14). We also took this opportunity to make 4-oxo-6-phenyl-4H-pyran-3-carboxylic acid (15) and associated ester derivatives (16, 17) using the synthetic approach presented in Scheme 1 (Supporting Information, Scheme S2 and Table S1). None of the compounds exhibited any inhibitory effects against bacteria (Acinetobacter baumannii ATCC 19606, A. baumannii ATCC BAA-1605, Burkholderia cepacia ATCC 25608, Enterobacter cloacae ATCC 13047, Escherichia coli ATCC 10798, E. coli ATCC 11775, Pseudomonas aeruginosa ATCC 10145, Klebsiella pneumonia ATCC 33495, K. pneumonia ATCC 51503, Staphylococcus epidermidis ATCC 12228, Staphylococcus aureus ATCC 25923, and S. aureus ATCC 700787) at a concentration of 100 μg/mL.
Previously, Fukuda et al.7 and Takagi et al.10 had reported that tensidols (7, 8, 11e, and 12) were capable of potentiating the activity of a sub-inhibitory concentration of miconazole (0.06 μM) against Candida albicans in a disk diffusion assay. Our attempts to confirm this activity in a broth microdilution assay against C. albicans ATCC 38245 failed to detect any imidazole potentiation. However, we did observe that compounds 11e, 16, and 17 provided complete growth inhibition against five Candida spp. (C. albicans, ATCC 38245, Candida glabrata NRRL Y-65, Candida krusei ATCC 27803, Candida parapsilosis ATCC 12969, and Candida tropicalis ATCC 12968) and two non-black aspergilli (Aspergillus fumigatus NRRL A1100 and Aspergillus flavus NRRL A1120) at a concentration of 100 μg/mL. At 10 μg/mL, 11e was only capable of inhibiting A. flavus growth, whereas 16 and 17 exhibited broader ranges of activities against C. krusei, A. flavus, and A. fumigatus at this reduced concentration.
EXPERIMENTAL SECTION
General Experimental Procedures
All solvents were of ACS grade or better. NMR data were obtained on Varian 300, 400, and 500 spectrometers and processed using VNMRj software. LC-ESIMS data were collected using a Thermo-Finnigan Surveyor LC system and a Finnigan LCQ Deca mass analyzer. HRESIMS data were obtained by electrospray ionization employing an Agilent 6538 UHD Accurate-Mass Quadrupole TOF mass analyzer. Initial separations were performed by normal phase flash chromatography using a Biotage Isolera system. Further isolation and purification was performed using preparative and semi-preparative Shimadzu HPLC systems with Phenomenex C18 Gemini columns.
Density Functional Theory Calculations (DFT)
Full geometry optimization and carbon chemical shift calculations for the proposed 11e and JBIR-86 (9) were performed on eight nodes of the Pentium-4 Xeon54 Quad Core Linux Cluster (running Red Hat Linux) using Gaussian03. Geometry optimization with frequency check and GIAO methods for determining carbon chemical shifts were performed using the basis set b3lyp/6-31+g(d,p). DFT calculations of tetramethylsilane (TMS) were also performed to determine the carbon chemical shift to be used as a reference. Carbon chemical shifts were referenced by subtracting the calculated δC of each carbon from the calculated δC of TMS. Analysis of results was performed by comparing the calculated chemical shifts with the published chemical shifts of JBIR-86.
Synthesis of 3-carbomethoxy-6-benzyl-4-pyrone (11e)
The general scheme for syntheszing 11e followed the methodology developed by McCombie et al.13 The enaminone, methyl 2-[(dimethylamino) methylene]-3-oxobutanonte, was synthesized as described with the application of methyl acetoacetate as a starting material instead of ethyl acetoacetate. Methyl acetoacetate (5.4 mL, 50 mmol) was added to the DMF-Me2SO2 adduct in CH2Cl2 (75 mL) at 0 °C and triethylamine (10 mL) was added slowly over 3 min. The mixture was stirred for 2 h at room temperature, washed with aqueous tartaric acid (10% w/v, 100 mL) and H2O (100 mL × 2), and dried over anhydrous MgSO4. The sample was gravity filtered (Whatman #1 filter paper) and the solvent from the filtrate was evaporated under vacuum. The crude material was subjected to silica gel flash chromatography using a CHCl3-EtOAc gradient (100:0 to 0:100). The solvent was evaporated from the enaminone under vacuum and further purified by flash chromatography using a hexane/EtOAc gradient (90:10 to 0:100). The solvent was dried to give the white solid product (Z)-methyl 2-((dimethylamino)methylene)-3-oxobutanoate.
To construct the pyrone ring, a solution of hexamethldisilazine (1 mL) in THF (6 mL) was stirred at −78 °C and n-BuLi in hexane (3 mL of a 1.6 M solution) was added slowly over 2 min. After the mixture cooled, a solution of the enaminone ((Z)-methyl 2-((dimethylamino)methylene)-3-oxobutanoate) (2 mmol) and phenyl acetyl chloride (2.4 mmol) in THF (5 mL) was added over 1 min. The dry ice/acetone bath was removed and the solution was allowed to warm for 5 min. Diethyl ether (20 mL) was added to the reaction, followed by 3 N HCl (8 mL). The solution was stirred rapidly overnight. The following day, the organic phase was washed with saturated sodium bicarbonate (100 mL) and H2O (100 mL × 2) then dried over anhydrous MgSO4. The sample was gravity filtered (Whatman #1 filter paper) and the filtrate dried under vacuum. The residue was subjected to silica gel flash chromatography using a CH2Cl2/EtOAc gradient (100:0 to 0:100). Final purification was performed by preparative HPLC using a MeOH/H2O gradient (50:50 to 100:0). The resulting product appeared as a red-brown amorphous solid (16 mg, 6%); 1H (CD3OD, 400 MHz) δH 8.45 (1H, s, H-2), 7.32 (3H, m, H-10, H-11, H-12), 7.24 (2H, m, H-9, H-13), 6.22 (1H, s, H-5), 3.88 (3H, s, 14-OCH3), 3.84 (2H, s, H-7); 13C (CD3OD, 100 MHz) δC 175.1 (C-4), 167.7 (C-6), 163.9 (C-14), 161.8 (C-2), 134.0 (C-8), 129.4 (C-9, C-13), 129.3 (C-10, C-12), 128.0 (C-11), 119.7 (C-3), 117.2 (C-5), 52.8 (14-OCH3), 39.72 (C-7); HRESIMS m/z 245.0815 [M + H]+ (calcd for C14H13O4, 245.0814).
LC-ESIMS analysis of extracts from black aspergilli grown under chemical epigenetic induction conditions
Freeze-dried spores from 11 strains of black aspergilli (Aspergillus aculeatus NRRL 5094, Aspergillus basiliensis NRRL 35542, A. carbonarius NRRL 346, A. carbonarius NRRL 369, Aspergillus ellipticus NRRL 5120, Aspergillus ibericus NRRL 35644, Aspergillus japonicus NRRL 2053, A. niger NRRL 326, A. niger NRRL 1015, Aspergillus parasiticus NRRL 465, and Aspergillus tubingensis NRRL 4875) were obtained from the ARS mycology collection. All strains were cryopreserved in 15% (vol/vol) glycerol solution at −80 °C. Cryopreserved samples were removed and plated on potato-dextrose agar and grown for 14 d at 25 °C. Hyphae from each strain were suspended in sterile H2O and used to inoculate three 1 L Erlenmeyer flasks containing 250 mL of sterile potato-dextrose broth. Each culture flask was treated with either 50 μM suberoylanilide hydroxamic acid (dissolved in 50:50 DMSO/H2O), 50 μM 5-azacytidine (dissolved in 50:50 DMSO/H2O), or bolus of 50:50 DMSO/H2O. Cultures were grown for 21 d at room temperature on a rotary shaker at 150 rpm. After 21 d, 2% (wt/vol) Diaion HP-20 resin was added to the flasks, which were further shaken for 4 h, followed by filtration of the cell mass and resin. The cell mass and resin from each flask was extracted in 250 mL aliquots of MeOH over a 48 h period (2×). The MeOH extracts were combined and solvent removed by rotary evaporation.
Extracts were prepared for LC-ESIMS analysis by suspending samples in a minimal amount of MeOH and adsorbing the solution onto C18. The dried C18 stationary phases loaded with samples were then placed in SPE cartridges and rinsed with 5 mL of deionized H2O followed by 5 mL of MeOH (each collected separately on a vacuum manifold). The MeOH fractions were evaporated in pre-weighed vials. The vials which contained the dried material from the MeOH fractions were weighed and the dried material was resuspended in a 90:10 MeOH/H2O solution at a concentration of 20 mg/mL. A portion of each sample was then transferred to a 2 mL Eppendorf tube and centrifuged at 14,000 rpm for 5 min. The supernatants were placed in vials for LC-ESIMS analysis using a MeOH/H2O gradient (25:75 to 100:0). The mass detector was set to scan a range from m/z 120-1500 in positive mode.
Synthesis of p-fluoro SAHA
Suberic acid monomethyl ester (2.01 g, 10.7 mmol), p-fluroraniline (1.42 g, 12.8 mmol), and hydroxybenzotriazole (1.73 g, 12.8 mmol) were dissolved in 25 mL of DMF. To this solution, diisopropylcarbodiimide (1.62 g, 12.8 mmol) was added and the reaction mixture was stirred at room temperature for 3 h. The reaction was quenched by pouring the mixture over 450 mL of ice stirred in H2O. After the ice had melted, the resulting precipitate was filtered using vacuum assisted filtration, followed by purification via flash silica chromatography using a hexane/EtOAC gradient. The procedure provided the suber-p-fluoroanilic acid methyl ester in 82% yield (2.47 g). Fresh methanolic hydroxyl amine was prepared by dissolving hydroxylamine hydrochloride (9.43 g, 135.6 mmol) in MeOH (25 mL) followed by the addition of KOH (7.60 g, 135.6 mmol). The mixture was allowed to stir under an atmosphere of nitrogen in an ice bath to aid the precipitation of the resulting salt, which was subsequently removed by filtration. The filtrate was transferred to a round bottom flask and a solution of suber-p-fluoroanilic acid methyl ester (2.07 g, 7.36 mmol) in 10 mL of MeOH was added. This reaction mixture was stirred under nitrogen at 50 °C for 12 h. The reaction was quenched via dilution in MeOH with ice H2O (250 mL). The resulting aqueous solution was neutralized with acetic acid resulting in the precipitation of p-fluoro SAHA. The white precipitate was filtered and evaporated under vacuum to provide an overall product yield of 61%. Analysis of the product by 1H, 13C, 19F NMR revealed that the p-fluoro SAHA was obtained in high chemical purity without need for further purification. 1H ((CD3)2SO, 500 MHz) δH 10.35 (1H, br s, N-H-12), 9.91 (1H, br s, N-H-2), 7.60 (2H, m, H13, H-17), 7.10 (2H, m, H14, H-16), 2.27 (1H, t, J = 7.5 Hz, H-9), 1.93 (2H, t, J = 7.5 Hz, H-4), 1.55 (2H, m, H-8), 1.48 (2H, m, H-5), 1.26 (4H, m, H-6 and 7); 13C ((CD3)2SO, 125 MHz): δ 171.7 (C-10), 169.8 (C-3), 159.6 (C-15), 136.3 (C-12), 121.1 (C-13, C-17), 115.7 (C-14, C-16), 36.7 (C-9), 32.8 (C-4), 28.9 (C-6, C-7), 25.5 (C-5, C-8); 19F ((CD3)2SO, 282 MHz) δC −119.96 (F-15); HRESIMS m/z 305.1274 [M + Na]+ (calcd for C14H19FNaO3, 305.1277).
Isolation and purification of p-fluoro nygerone B (14)
A cryopreserved sample of A. niger ATCC 1015 was plated on potato-dextrose agar and grown for 14 d at 25 °C. Hyphae were suspended in sterile H2O and used to inoculate a set of 20 Erlenmeyer flasks (1 L) each containing 250 mL of sterile potato-dextrose broth. Each flask was treated with 50 μM p-fluoro suberoylanilide hydroxamic acid (p-F-SAHA) (dissolved in 50:50 DMSO/H2O). Controls were created by inoculating four 1 L Erlenmeyer flasks containing 250 mL of sterile potato-dextrose broth and 0.5% (vol/vol) of 50:50 DMSO/H2O. Cultures were grown on a rotary shaker (150 rpm) for 14 d at room temperature and the combined culture broth were partitioned (3×) against an equivalent volume of CH2Cl2. The organic phase was collected and evaporated under vacuum. Treated and control extracts were analyzed by LC-ESIMS as described above. Further fractionation was performed on the p-F-SAHA culture extract by preparative HPLC using a MeOH/H2O gradient (35:65 to 100:0) with final purification being performed by semi-preparative HPLC to yield 2 mg of light-yellow amorphous solid; 1H (CDCl3, 400 MHz) δH 9.89 (1H, br s, N-Hb-15), 8.49 (1H, s, H-2), 7.23 (1H, m, H-11), 7.21 (2H, m, H-10, H-12), 7.10 (2H, m, H-3′, H-5′), 7.05 (2H, m, H-2′, H-6′), 6.83 (2H, m, H-9, H-13), 6.61 (1H, s, H-5), 6.06 (1H, br s, N-Ha-15), 3.71 (2H, s, H-7); 13C (CDCl3, 100 MHz) δC 177.7 (C-4), 166.0 (C-14), 164.1 (C-4′), 162.0 (C-1′), 151.3 (C-6), 146.9 (C-2), 134.8 (C-8), 128.9 (C-2′, C-6′, C-10, C-12), 128.5, C-9, C-13), 127.7 (C-11), 121.9 (C-5), 117.7 (C-3), 116.9 (C-3′, C-5′), 39.74 (C-7); 19F (CDCl3, 376 MHz) δ −112.36 (F-11); HRESIMS m/z 323.1192 [M + H]+ (calcd for C19H16FN2O2, 323.1196).
3-carboethoxy-6-phenyl-4-pyrone (16)
Compound 16 was prepared and purified based on a previously reported method.13 The resulting product was a red crystalline solid; 1H-NMR was in agreement with the literature;13 HRESIMS m/z 245.0816 [M + H]+ (calcd for C14H13O4, 245.0814).
3-carbomethoxy-6-phenyl-4-pyrone (17)
Compound 17 was prepared and purified based on a previously reported method,13 with the exception that methyl acetoacetate was utilized instead of ethyl acetoacetate. The resulting product was a red crystalline solid; 1H (CD3OD, 500 MHz) δH 8.82 (1H, s, H-2), 7.89 (2H, m, H-8, H-12), 7.55 (2H, m H-10), 7.53 (2H, m, H-9, H-11), 6.94 (1H, s, H-5), 3.86 (3H, s, H-14); 13C (CD3OD, 125 MHz) δC 175.6 (C-4), 166.0 (C-2), 164.1 (C-6), 163.0 (C-13), 130.8 (C-9, C-10, C-11), 129.6 (C-7), 126.2 (C-8, C-12), 118.3 (C-3), 112.4 (C-5), 52.3 (C-14); HRESIMS m/z 231.0659 [M + H]+ (calcd C13H11O4, 231.0657).
3-carboxylic-6-phenyl-4-pyrone (15)
A 10 mg sample of 17 was reacted with 1 N NaOH in distilled THF for 3 h at room temperature then adjusted to pH 3 with 1 N HCl. The resulting solution was partitioned against EtOAc and the solvent removed under vacuum. Purification was performed by preparative HPLC yielding 6.2 mg of a yellow amorphous solid. 1H ((CD3)2CO, 500 MHz) δH 9.88 (1H, s, H-2), 8.04 (2H, m, H-8, H-12), 7.66 (1H, m, H-10), 7.60 (2H, m, H-9, H-11), 6.95 (1H, s, H-5); 13C ((CD3)2CO, 125 MHz) δC 193.2 (C-2), 178.5 (C-4), 167.1 (C-6), 161.7 (C-13), 132.8 (C-10), 130.4 (C-7), 129.2, (C-9, C-11), 126.7 (C-8, C-12), 100.6 (C-3), 97.6 (C-5); HRESIMS m/z 215.0358 [M + H]− (calcd for C12H7O4, 215.0350).
Pestalamide A (8)
1H- and 13C-NMR were in agreement with published values;9 HRESIMS m/z [M + Na]+ 366.0956 (calcd for C18H17NO6Na, 366.0954).
Biological Assays
Twelve bacteria and seven fungi were used in the antimicrobial assays. Stock cultures of strains were delivered into presterilized 96-well plates, and compounds prepared in DMSO were added to each well of the 96-well plate (final DMSO concentration of 1%). Plates were shaken orbitally for 5 s and an initial OD600 value recorded. Bacteria and fungi in 96-well plates were incubated for 24 h and 48 h, respectively, in a humidified incubator at 37 °C. Plates were removed, orbitally shaken, and a final OD600 value recorded. The final OD600 reading was subtracted from the initial OD600 reading to obtain the change in OD600 (antimicrobial activity was determined by the change in optical density). Chloramphenicol and ketoconazole were used as antibacterial and antifungal positive controls, respectively.
Supplementary Material
Acknowledgment
We wish to acknowledge the National Institutes of Health (1RO1GM092219-01 and 1RO1AI085161-01) for financial support. DFT calculations was performed at the OU Supercomputing Center for Education & Research (OSCER) at the University of Oklahoma. We also thank J. Hayle for his assistance and training with Gaussian03.
Footnotes
Supporting Information. Predicted (ACD) 13C NMR of furan and pyrrol ring systems (Figure S1) and partial structures of 11c and 11d (Figure S2), comparison of HMBC correlations of JBIR-87 (10) and (12) (Figure S3), tables of 1H and 13C NMR chemical shifts of 1, 2, 5/7, 6/8, 9/11e, 10/12, 13, 14, 15, and 17 (Table S1), LC-ESIMS analysis of black aspergilli (Table S2), antimicrobial assay results (Table S3), schemes of feeding studies for the production of 14 (Scheme S1A-E), and synthesis of 15, 16, and 17 (Scheme S2) are available free of charge via the internet at http://pubs.acs.org.
References and Notes
- 1.Henrikson JC, Hoover AR, Joyner PM, Cichewicz RH. Org. Biomole. Chem. 2009;7:435–438. doi: 10.1039/b819208a. [DOI] [PubMed] [Google Scholar]
- 2.Fisch KM, Gillaspy AF, Gipson M, Henrikson JC, Hoover AR, Jackson L, Najar FZ, Wagele H, Cichewicz RH. J. Ind. Microbiol. Biotech. 2009;36:1199–1213. doi: 10.1007/s10295-009-0601-4. [DOI] [PubMed] [Google Scholar]
- 3.Williams RB, Henrikson JC, Hoover AR, Lee AE, Cichewicz RH. Org. Biomole. Chem. 2008;6:1895–1897. doi: 10.1039/b804701d. [DOI] [PubMed] [Google Scholar]
- 4.Cichewicz RH, Henrikson JC, Wang X, Branscum KM. Manual of Industrial Microbiology and Biotechnology. 3rd ed American Society for Microbiology; Washington DC: 2009. pp. 78–95. [Google Scholar]
- 5.Hiort J, Maksimenka K, Reichert M, Perović-Ottstadt S, Lin WH, Wray V, Steube K, Schaumann K, Weber H, Proksch P, Ebel R, Müller WEG, Bringmann G. J. Nat. Prod. 2004;67:1532–1543. doi: 10.1021/np030551d. [DOI] [PubMed] [Google Scholar]
- 6.Zhang YP, Zhu TJ, Fang YC, Liu HB, Gu QQ, Zhu WM. J. Anitbiot. 2007;60:153–157. [Google Scholar]
- 7.Fukuda T, Hasegawa Y, Hagimori K, Yamaguchi Y, Masuma R, Tomoda H, Omura S. J. Antibiot. 2006;59:480–485. doi: 10.1038/ja.2006.67. [DOI] [PubMed] [Google Scholar]
- 8.Ye YH, Zhu HL, Song YC, Liu JY, Tan RX. J. Nat. Prod. 2005;68:1106–1108. doi: 10.1021/np050059p. [DOI] [PubMed] [Google Scholar]
- 9.Ding G, Jiang LH, Guo LD, Chen XL, Zhang H, Che YS. J. Nat. Prod. 2008;71:1861–1865. doi: 10.1021/np800357g. [DOI] [PubMed] [Google Scholar]
- 10.Takagi M, Motohashi K, Hwang J-H, Nagai A, Shin-ya K. J. Antibiot. 2010;63:371–373. doi: 10.1038/ja.2010.45. [DOI] [PubMed] [Google Scholar]
- 11.White KN, Amagata T, Oliver AG, Tenney K, Wenzel PJ, Crews P. J. Org. Chem. 2008;73:8719–8722. doi: 10.1021/jo800960w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi H, Engene N, Smith JE, Preskitt LB, Gerwick WH. J. Nat. Prod. 2010;73:517–522. doi: 10.1021/np900661g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCombie SW, Metz WA, Nazareno D, Shankar BB, Tagat J. J. Org. Chem. 1991;56:4963–4967. [Google Scholar]
- 14.Nielsen KF, Mogensen JM, Johansen M, Larsen TO, Frisvad JC. Anal. Bioanal. Chem. 2009;395:1225–1242. doi: 10.1007/s00216-009-3081-5. [DOI] [PubMed] [Google Scholar]
- 15.Abarca ML, Accensi F, Cano J, Cabañes FJ. Antonie Leeuwenhoek. 2004;86:33–49. doi: 10.1023/B:ANTO.0000024907.85688.05. [DOI] [PubMed] [Google Scholar]
- 16.The 13C NMR chemical shift data published by Ding et. al.9 in Table 4 for pestalamide B were found to be incorrect. It appears as though the 13C NMR shift data for pestalamide A in Table 3 were superimposed onto Table 4. A list of the corrected chemical shifts is found in Table S1 of the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.