

Abstract
Development of novel therapeutic approaches to repair fracture non-unions remains a critical clinical necessity. We evaluated the capacity of human embryonic stem cell (hESC)-derived mesenchymal stem/stromal cells (MSCs) to induce healing in a fracture non-union model in rats. In addition, we placed these findings in the context of parallel studies using human bone marrow MSCs (hBM-MSCs) or a no cell control group (n = 10 to 12 per group). Preliminary studies demonstrated that both for hESC-derived MSCs and hBM-MSCs, optimal induction of fracture healing required in vitro osteogenic differentiation of these cells. Based on biomechanical testing of fractured femurs, maximum torque and stiffness were significantly greater in the hBM-MSC as compared to the control group that received no cells; values for these parameters in the hESC-derived MSC group were intermediate between the hBM-MSC and control groups, and not significantly different from the control group. However, some evidence of fracture healing was evident by X-ray in the hESC-derived MSC group. Our results thus indicate that while hESC-derived MSCs may have potential to induce fracture healing in non-unions, hBM-MSCs function more efficiently in this process. Additional studies are needed to further modify hESCs to achieve optimal fracture healing by these cells.
Keywords: embryonic stem cells, mesenchymal stem/stromal cells, osteogenic, bone repair, fracture non-union
INTRODUCTION
Five to ten percent of all fractures are complicated by delayed union or non-union (1). Thus, development of novel therapeutic approaches to treat and repair non-union fractures remains a critical clinical necessity. Human bone marrow mesenchymal stem/stromal cells (hBM-MSCs) are considered a promising candidate for clinical applications. The availability of autologous hBM-MSCs from patients is advantageous for therapeutic applications. hBM-MSCs are multipotent and possess the ability to regenerate cell types specific for different tissues, including adipose tissue, bone, and cartilage. Implantation of autologous hBM-MSCs using different scaffolds resulted in bone regeneration in several animal models (2–5). However, long-term cultures of hBM-MSCs have limitations because, over time, the cells exhibit a reduced proliferation rate attributable to telomere shortening and senescence (6).
In recent years, significant advances have been made in examining the potential of human embryonic stem cells (hESCs) in regenerative medicine. ESCs are undifferentiated, pluripotent cells derived from mammalian pre-implantation blastocysts. Unlike hBM-MSCs, hESCs can be maintained in culture indefinitely in an undifferentiated state, thus offering potential advantages over the use of adult hBM-MSCs. Since the initial report describing the derivation of hESCs in 1998 (7), these cells have been widely used for studies of human developmental biology and to define methods that support the differentiation of hESCS into specific lineages (8–10).
Recently, several groups reported the development of MSCs from hESCs (11–14). These MSCs have the surface phenotype typical of hBM-MSCs isolated from human bone marrow, including expression of CD73, CD105, Stro-1, and CD44. Molecular studies show expression of transcription factors and matrix proteins also typically produced by hBM-MSCs. A stromal cell co-culture system demonstrated that hESC-derived CD73+ cells function as MSCs with robust formation of adipocytes, chondrocytes, and osteoblasts (13,14).
The goal of our study was to define the optimal conditions for using hESC-derived populations to repair fractures in a fracture non-union model (15,16). We initially identified optimal culture conditions for CD73+ hESC-derived MSCs to induce fracture healing. Next, we evaluated the capacity of these CD73+ hESC-derived MSCs to induce healing in a fracture non-union model in rats. In addition, we placed these findings in the context of parallel studies using hBM-MSCs or a control group that received no cells.
METHODS
Animals
Male athymic nude rats (Charles River Labs), age 8 to 12 wks, weighing 270 to 370 g were used. Following pilot studies, the rats were divided into 3 groups for the main study: (1) control, atelocollagen matrix infused with saline (n = 10); (2) atelocollagen infused with CD73+ hESC-derived MSCs differentiated under osteogenic conditions (see below) for 7 days (n = 10); and (3) atelocollagen infused with hBM-MSCs differentiated under osteogenic conditions (see below) for 7 days (n = 12). All rats were housed, treated, and handled in accordance with the guidelines set forth by the Mayo Clinic Institutional Animal Care and Use Committee.
Cell culture
The hESC line H9 (obtained from WiCell) was maintained as undifferentiated cells as previously described (17,18) by coculture with irradiated mouse embryonic fibroblast (MEF) cells in DMEM/F12 supplemented with 15% Knockout Serum Replacer (KOSR) (Invitrogen), 1% MEM nonessential amino acids (Invitrogen), 0.5% penicillin-streptomycin (P/S), 2 mM L-Glutamine, 0.1 mM β-mercaptoethanol (Sigma), and 4 ng/mL human bFGF (Invitrogen). hBM-MSCs (Lonza) were cultured in phenol red-free αMEM (Invitrogen) supplemented with 10% FBS and 1% P/S. Media were changed every 2 to 3 days, and cells were passaged upon reaching 80 to 90% confluency. Sorted CD73+ hESC-derived MSCs and hBM-MSCs were differentiated on plastic for 7 days in the presence osteogenic differentiation medium containing phenol red-free αMEM, 10% FBS, 50 μM ascorbic acid, and 10−8 dexamethasone.
Mesenchymal differentiation by stromal co-culture
The mouse bone marrow stromal cell line M2-10B4 (ATCC) was grown in DMEM (Invitrogen) containing 10% fetal bovine serum (FBS) (Hyclone), 1% P/S, 1% MEM-nonessential amino acids, and 0.1 mM β-mercaptoethanol. M2-10B4 cells were inactivated with 10 μg/mL mitomycin C in M2-10B4-conditioned medium for 3 hrs at 37°C with 5% CO2 prior to culture on gelatin-coated plates (Sigma). Mesenchymal differentiation of hESCs occurred during co-culture on stromal cell layers. hESCs were passaged onto M2-10B4 mouse stromal cells with differentiation medium consisting of RPMI supplemented with 15% defined FBS (Hyclone), 2 mML-glutamine, 0.1 mM β-mercaptoethanol, 1% MEM-nonessential amino acids, and 1% P/S. Media were changed every 2 to 3 days. After 17 to 21 days, differentiated hESCs were made into a single cell suspension by treatment with collagenase IV (1 mg/mL) (Invitrogen), followed by 0.05% trypsin/ EDTA (Gibco/Invitrogen) supplemented with 2% chick serum (Sigma). Cells were treated with an antibody against human CD73 conjugated with PE (Pharmingen). Antibodies for PE conjugated with magnetic beads were used to select CD73+ cells from single cell suspension using the EasySep PE selection kit (Stemcell Technologies). Post-sorted CD73+ hESCs were plated onto gelatin-coated plates and grown in mesenchymal stem cell medium consisting of αMEM (Invitrogen) supplemented with 10% FBS, 1% P/S, 1% MEM-nonessential amino acids, 2 mM L-Glutamine, and 0.1 mM β-mercaptoethanol. Media were changed on sorted cells every 2 to 3 days. For flow cytometric analysis, post-sorted cell populations were washed with Mg2+ and Ca2+-free phosphate buffered saline (PBS) (Hyclone) and removed from culture plates using 0.25% Trypsin/EDTA. After washing with PBS supplemented with 2% FBS and 0.1% sodium azide, the cells were incubated with antibodies for CD34-APC, CD73-PE, CD90-PE, CD105-APC, and CD146-FITC (all IgG1, all from Pharmingen) and their corresponding IgG1 controls. Live cell populations identified using 7AAD exclusion were analyzed on FACS Calibur (Becton Dickenson) for different surface antigen expression using Flow-Jo Software (TreeStar).
Induction of fracture non-union
We modified a fracture non-union protocol described by Matsumoto and colleagues (15,16). Briefly, a lateral parapatellar knee incision was made on the right limb to expose the distal femoral condyle. To avoid displacement of the fracture, a K-wire was inserted from the trochlear groove into the femoral canal in a retrograde manner. The tendons were attached over the patellofemoral joint with absorbable sutures. To avoid complex fractures, we modified the previously published protocol (15,16), and did not cut the femur with a thin saw cut but still used 3-point bending directly to fracture the femur. Thus, a transverse femoral shaft fracture was produced in the right femur of each rat using a C-shaped instrument applying 3-point bending. To achieve non-union, the periosteum was cauterized circumferentially at a distance of 2 mm on each side of the fracture. The rats immediately received a local implantation of saline or cells mixed with atelocollagen (105 cells of each type in 100 μl of the solution) at the fracture site (16). The wound was closed, and postoperative pain was managed by administration of subcutaneous injection of buprenorphine hydrochloride after surgery. Normal weight bearing activities were allowed following the operation.
Implantation of cells
Immediately after the creation of the non-union fracture, the rats received the appropriate local implantation of saline or cells mixed with atelocollagen (105 cells of each type in 100 μl of the solution) at the fracture site. We used atelocollagen as a scaffold based on an established model of fracture repair (16).
Radiographical assessment of fracture healing
Radiographs of the fractured femurs were taken under anesthesia immediately after surgery and every 2 wks with the animal in the supine position and both limbs fully extended. Radiographs were evaluated by 2 blinded readers and scored for fracture healing using a modification of a previously describe scale (19): 0 – no change from post-fracture appearance; 1 – trace of radiodense material in the fracture defect; 2 – flocculent radiodensity with flecks of calcification; 3 – defect bridged at least at one point with material of non-uniform density; 4 – at least one of four cortices obscured by new bone, or the fracture was bridged on dorsal and ventral sides with uniform callus; and 5 – defect bridged by uniform new bone, cut ends of cortex no longer distinguishable. For each animal and time point, we used the mean of the scores of the two readers.
Tissue harvesting
8 wks after fracture, rats were euthanized by CO2inhalation. The fractured femurs used for biomechanical testing were dissected free from the surrounding muscle, and testing was performed as described below. As part of the pilot studies, femurs were also used in histological analyses; bones were harvested and embedded in methyl methacrylate. Serial sections were mounted on silane-coated glass slides and stained immediately as described below.
Biomechanical assessment of fracture union
Without removal of the intramedullary K-wires, standardized torsional testing was performed on the fractured site. The torsional load was applied at a speed of 5°/sec for a maximum of 36 secs. Maximum torque (N-cm) and stiffness (N-cm/degree) were used as the primary endpoints (20). Maximum torque was the highest load that the bone sustained before fracture, and stiffness was calculated from the linear portion of the loading curve (higher values for both are indicative of stronger bone). The area under the curve represents the energy to failure (N-cm*degrees/cm), which was also assessed.
Detection of human cells in the fracture callus
To identify human cells in the fracture callus, immunohistochemistry with anti-human nuclear monoclonal antibody (Chemicon), which stains nuclei of all human cell types, was performed on sections of femurs embedded in methyl methacrylate. A human iliac crest biopsy specimen was used as a positive control. Serial sections were mounted on silane-coated glass slides and stained immediately. Unstained deplasticized sections were rehydrated, and antigen retrieval was performed (Target Retrieval Solution, BioGenex). Sections were blocked with 10% goat serum and 10% mouse serum. The sections were incubated for 1 hr at 37°C with or without 1:100 dilution of mouse anti-human nuclei monoclonal antibody (Chemicon), followed by incubation for 30 min at room temperature with 1:100 dilution of goat anti-mouse IgG FITC antibody (Jackson). Sections were subsequently dehydrated and mounted with Prolong Gold plus DAPI (Invitrogen/Molecular Probes). Immunofluorescent cells were imaged on a Zeiss Axiovert 200M fluorescent microscope (Zeiss) with AxioVision Ver. 4.6 imaging software.
Statistical analysis
All data are reported as the mean ± SEM, and P < 0.05 was considered significant. Maximum torque and stiffness were compared in the CD73+ hESC-derived MSC and the hBM-MSC groups to the control (no cell) group using two-sample t-tests. In each group, complete non-union of fractures was manifested by the inability to perform the biomechanical testing as the bones tended to come apart during the course of processing; in these animals, the lowest value for maximum torque, stiffness, and energy to failure was assigned (rather than values of 0, thus representing a conservative bias).
Results
Our modification of the published protocol (15,16) with circumferential cauterization of the periosteum reproducibly resulted in fracture non-union as compared to mid-shaft fractures that were not cauterized (Fig. 1). In preliminary studies, we tested the ability of CD73+ hESC-derived MSCs to induce fracture healing in this non-union model. As previously described (14), we used hESC-derived MSCs that express CD73, CD90, CD105, and CD146 following 12 days of in vitro co-culture with the murine bone marrow stromal cell line M2-10B4 (Fig. 2). Under the appropriate culture conditions in vitro, these cells (CD73+ hESC-derived MSCs) could differentiate into osteoblasts capable of mineral deposition, adipocytes, and chondrocytes (14). When we used these CD73+ hESC-derived MSCs that were co-cultured for 12 days with the murine bone marrow stromal cell line M2-10B4 and then seeded on a collagen-based scaffold and implanted into the fracture site, a bone repair response was present as early as 2 wks following surgery (Fig. 3A). However, rats receiving these cells required euthanasia at 6 wks following surgery due to the appearance of a palpable mass in the right thigh (Fig. 3A, B). Histological examination of the fracture site implanted with the CD73+ hESC-derived MSCs revealed a marked excess of bone formation (Fig. 3C), but no tumor or teratoma formation. By contrast, when CD73+ hESC-derived MSCs were further differentiated in vitro under osteogenic conditions for an additional 7 days prior to implantation, we did not see aberrant bone formation (Fig. 4). These osteogenically differentiated CD73+ hESC-derived MSCs were used in the subsequent studies.
Figure 1.

Fracture healing response 6 wks following surgery by X-ray of non-cauterized and cauterized femurs following fracture induction.
Figure 2.

Phenotypic identification of hESC-derived MSCs. (A) Phase contrast image of hESC-derived MSCs in vitro (total magnification = 100×). Flow cytometric analysis of hESC-derived MSCs showing negative CD34 expression (B) (green = isotype control), with positive CD73 (C), CD90 (D), CD105 (E) and CD146 (F) expression typically associated with the mesenchymal phenotype.
Figure 3.

(A) Fracture healing assessed by x-ray in rat femurs that received CD73+ hESC-derived MSCs 2 wks and 6 wks following surgery; (B) Gross examination of a femur that received undifferentiated CD73+ hESC-derived MSCs or a control femur that was fractured but did not undergo cautery (both at 6 wks); and (C) histology of the femur that received undifferentiated CD73+ hESC-derived MSCs.
Figure 4.
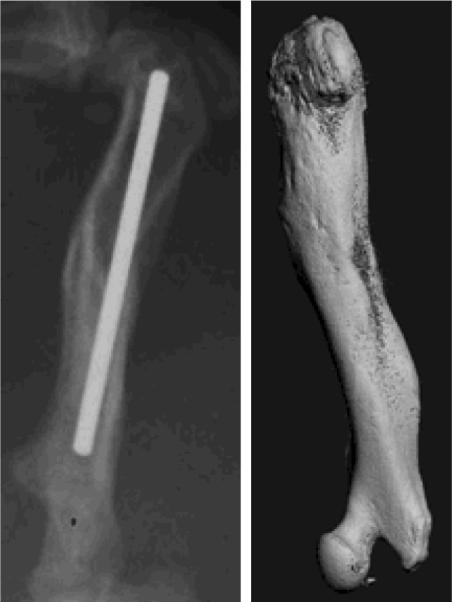
Optimal fracture healing induced by CD73+ hESC-derived MSCs differentiated along an osteoblastic phenotype based on radiographic and μCT analysis 8 wks following surgery.
We next performed a similar analysis comparing hBM-MSCs cultured either under osteogenic conditions or in growth medium without osteogenic supplements in vitro for 7 days. We generally observed better fracture healing using osteogenically pre-differentiated hBM-MSCs (Fig. 5B) as compared to undifferentiated hBM-MSCs (Fig. 5A). Based on these results, we sought to compare the ability of CD73+ hESC-derived MSCs and hBM-MSCs (both first cultured in vitro under osteogenic conditions for 7 days) to induce fracture healing in our non-union model.
Figure 5.

Comparison of the fracture healing response by hBM-MSCs cultured in the (A) absence or (B) presence of osteogenic supplements, as shown by x-ray and μCT scans taken 8 wks following surgery.
Radiographs from representative rats that underwent femoral fractures followed by cauterization of the periosteum and treatment with atelocollagen scaffolds containing saline (no cells), differentiated hESC-derived MSCs, and differentiated hBM-MSCs are shown in Figure 6. There was fracture healing in both the hESC-derived MSC and hBM-MSC groups as compared to the control (no cell) group. The hBM-MSC group showed significantly improved fracture healing as compared to the no cell group (Fig. 7), with the hESC-derived MSC group having intermediate scores. To objectively quantify the degree of fracture healing, we performed torsional testing of the femurs. Consistent with the radiological scores, maximum torque (Fig. 8A) and stiffness (Fig. 8B) were significantly greater in the hBM-MSC as compared to the control group that received no cells. Values for these parameters in the animals receiving the hESC-derived MSCs were intermediate between the hBM-MSC and control groups, and not different from the control group. Energy to failure (N-cm*degrees/cm) was 405 ± 49 in the no cell group, 412 ± 105 in the hESC-derived MSC group (P = 0.954 versus no cell), and somewhat higher in the hBM-MSC group (617 ± 159, P = 0.254 versus no cell).
Figure 6.

Comparative radiographic assessment of fracture healing in rat femurs that received no cells, CD73+ hESC-derived MSCs, or hBM-MSCs differentiated under similar osteogenic conditions for 7 days.
Figure 7.

Fracture healing grades in the rat femurs that received no cells, CD73+ hESC-derived MSCs, or hBM-MSCs differentiated under similar osteogenic conditions for 7 days.
Figure 8.

(A) Maximum torque and (B) Stiffness based on biomechanical testing of fractured femurs that received no cells, CD73+ hESC-derived MSCs, or hBM-MSCs differentiated under similar osteogenic conditions for 7 days.
Finally, to test for the presence of donor human cells at the fracture site, we performed immunological analysis with a human nuclear antibody in rats that had received CD73+ hESC-derived MSCs following fracture. This revealed the presence of human donor cells along the lining of trabeculae in the newly formed fracture callus at 6 wks following fracture (Fig. 9, panel C); donor cells were also detected in the bone marrow of newly formed bone.
Figure 9.

Identification of human CD73+ hESC-derived MSCs at the fracture site 6 wks following fracture based on immunostaining for a human anti-nuclear antibody. Shown are a fractured femur that were not implanted with human cells (A), a femur implanted with CD73+ hESC-derived MSCs but without the primary antibody (B), and a femur implanted with CD73+ hESC-derived MSCs and stained with both the primary and secondary (FITC-labeled) antibody (C). Panel D shows a positive control using human bone.
DISCUSSION
In these studies, we initially assessed how CD73+ hESC-derived MSCs and hBM-MSCs induced bone formation upon implantation in vivo. Initial studies with local implantation of CD73+ hESC-derived MSCs resulted in excess bone formation at the fracture site. However, in vitro osteogenic differentiation of these CD73+ hESC-derived MSCs prior to implantation induced more normal bone formation. Recent studies demonstrated the in vivo potential of hESC-derived MSCs to form bone (14,21–23), and hBM-MSCs have been used successfully in vivo for bone repair in animal models (2–5). But little data exist directly comparing the ability of hESCs versus hBM-MSCs to induce fracture healing. Our results show that CD73+ hESC-derived MSCs could induce callus formation at the fracture site similar to hBM-MSCs; however, these cells needed pre-differentiation towards osteoblastic lineage in vitro for efficient fracture repair in vivo. We went a step further to address the functionality of the repaired fractures by conducting biomechanical testing on the fractured femurs 8 wks post-surgery, which constituted the first demonstration of the ability of hESC-derived MSCs to form functional bone in vivo, especially under weight bearing conditions. The femurs that received osteogenically differentiated CD73+ hESC-derived MSCs showed a trend towards an increase in maximum torque and stiffness compared to control group receiving no cells, whereas the femurs receiving the osteogenically differentiated hBM-MSCs had clearly superior biomechanical properties as compared to the control group.
Bone formation is a well coordinated process that involves interactions between the osteoprogenitor cells, local cells, and extracellular matrix. Local signals might influence the degree of differentiation that multipotent stem cells can undergo under pathological conditions. The limited ability of osteogenically differentiated CD73+ hESC-derived MSCs to form functional bone in vivo compared to hBM-MSCs could be influenced by the environment in which they are differentiating and their interactions with local cells. Also, local signals might favor the hBM-MSCs, which are already at an intermediate state of osteoblastic differentiation as compared to the pluripotent hESCs, even though the latter were differentiated towards an osteogenic phenotype. Along these lines, we recently demonstrated (14) that hESC-derived MSCs and hBM-MSCs exhibit important differences in gene expression, at least prior to osteogenic differentiation. Specifically, the hESC-derived MSCs have increased expression of pluripotent and multipotent stem cell and endothelial cell-associated genes. However, further studies comparing gene expression and other properties of osteogenically differentiated hESC-derived MSCs and hBM-MSCs are needed to assess why the latter functioned better in our fracture model.
A key step towards the clinical success of cellular therapeutics is to define culture conditions that drive hESC differentiation to osteoblasts in an efficient and stable manner. More important for in vivo differentiation of hESCs is to avoid development of unwanted cell phenotypes, since implantation of undifferentiated hESCs might result in teratoma formation (7). Our results show that there was no teratoma or tumor formation in vivo, which suggests that these hESCs were further differentiated into a mesenchymal lineage. A plausible explanation for the excess bone formation we observed by undifferentiated CD73+ hESC-derived MSCs could be differentiation heterogeneity of these cells in vivo that led to an uncontrolled differentiation into bone. hESC-derived MSCs have more vascular characteristics than hBM-MSCs (14), which might have led to excess bone formation by CD73+ hESC-derived MSCs at the fracture site.
hBM-MSC populations express high levels of CD73 (24). The CD73+ hESC-derived MSC population used in our study was phenotypically and functionally similar to hBM-MSCs. Kopher etal (14) showed that the majority of hESC-derived MSCs express CD73 (>98%) after 12 days of in vitro culture. These cells could form mineralized nodules as shown by positive von Kossa staining after additional culture in osteogenic medium and were able to differentiate into adipocytes and chondrocytes with appropriate stimulation (14).
To verify the presence of donor cells at the fracture site, we also performed analyses with a human nuclear antibody that detected the presence of donor cells along the lining of trabeculae in the newly formed fracture callus. Thus, while further studies are needed to quantify the relative contributions of host versus donor cells towards bone formation in this fracture model, our data do demonstrate the presence of the donor cells at the fracture site.
In conclusion, our results indicate that while hESC-derived MSCs may have the potential to induce fracture healing in non-unions, hBM-MSCs function more efficiently in this process, at least under the conditions used in our study. Additional studies are needed to further modulate the phenotype of hESCs and potentially enhance their ability to induce fracture healing or other types of bone regeneration.
ACKNOWLEDGEMENTS
This work was supported by a grant from Mayo Foundation and the University of Minnesota, NIH AG004875 (SK), AR48147 (JJW), HL077923 (DSK). We would like to thank Mr. James Bronk for help with the rat surgeries.
REFERENCES
- 1.Einhorn TA. Enhancement of fracture-healing. J Bone Joint Surg Am. 1995;77:940–956. doi: 10.2106/00004623-199506000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Arinzeh TL, Peter SJ, Archambault MP, et al. Allogeneic mesenchymal stem cells regenerate bone in a critical-sized canine segmental defect. J Bone Joint Surg Am. 2003;85-A:1927–1935. doi: 10.2106/00004623-200310000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Bruder SP, Kraus KH, Goldberg VM, Kadiyala S. The effect of implants loaded with autologous mesenchymal stem cells on the healing of canine segmental bone defects. J Bone Joint Surg Am. 1998;80:985–996. doi: 10.2106/00004623-199807000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Kon E, Muraglia A, Corsi A, et al. Autologous bone marrow stromal cells loaded onto porous hydroxyapatite ceramic accelerate bone repair in critical-size defects of sheep long bones. J Biomed Mater Res. 2000;49:328–337. doi: 10.1002/(sici)1097-4636(20000305)49:3<328::aid-jbm5>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 5.Viateau V, Guillemin G, Bousson V, et al. Long-bone critical-size defects treated with tissue-engineered grafts: a study on sheep. J Orthop Res. 2007;25:741–749. doi: 10.1002/jor.20352. [DOI] [PubMed] [Google Scholar]
- 6.Abdallah BM, Kassem M. Human mesenchymal stem cells: from basic biology to clinical applications. Gene Ther. 2008;15:109–116. doi: 10.1038/sj.gt.3303067. [DOI] [PubMed] [Google Scholar]
- 7.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocytes. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 8.Kaufman DS, Hanson ET, R.L. L, et al. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci USA. 2001;98:10716–10721. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levenberg S, Huang NF, Lavik E, et al. Differentiation of human embryonic stem cells on three-dimensional polymer scaffolds. Proc Natl Acad Sci USA. 2003;100:12741–12746. doi: 10.1073/pnas.1735463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segev H, Fishman B, Ziskind A, et al. Differentiation of human embryonic stem cells into insulin-producing clusters. Stem Cells. 2004;22:265–274. doi: 10.1634/stemcells.22-3-265. [DOI] [PubMed] [Google Scholar]
- 11.Barberi T, Willis LM, Socci ND, Studer L. Derivation of multipotent mesenchymal preecursors from human embryonic stem cells. PloS Med. 2005;2:e161. doi: 10.1371/journal.pmed.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olivier EN, Rybicki AC, Bouhassira EE. Differentiation of human embryonic stem cells into bipotent mesenchymal stem cells. Stem Cells. 2006;24:1914–1922. doi: 10.1634/stemcells.2005-0648. [DOI] [PubMed] [Google Scholar]
- 13.Trivedi P, Hematti P. Simultaneous generation of CD34+ primitive hematopoietic cells and CD73+ mesenchymal stem cells from human embryonic stem cells cocultured with murine OP9 stromal cells. Exp Hematol. 2007;35:146–154. doi: 10.1016/j.exphem.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Kopher RA, Penchev VR, Islam MS, et al. Human embryonic stem cell-derived CD34+ cells function as MSC progenitor cells. Bone. 2010;447:718–728. doi: 10.1016/j.bone.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto T, Kawamoto A, Kuroda R, et al. Therapeutic potential of vasculogenesis and osteogenesis promoted by peripheral blood CD34-positive cells for functional bone healing. Am J Pathol. 2006;169:1440–1457. doi: 10.2353/ajpath.2006.060064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mifune Y, Matsumoto T, Kawamoto A, et al. Local delivery of granulocyte colony stimulating factor-mobilized CD34-positive progenitor cells using bioscaffold for modality of unhealing bone fracture. Stem Cells. 2008;26:1395–1405. doi: 10.1634/stemcells.2007-0820. [DOI] [PubMed] [Google Scholar]
- 17.Tian X, Kaufman DS. Hematopoietic development of humon embryonic stem cells in culture. Meth Mol Biol. 2008;430:119–133. doi: 10.1007/978-1-59745-182-6_8. [DOI] [PubMed] [Google Scholar]
- 18.Hill KL, Kaufman DS. Hematopoietic differentiation of human embryonic stem cells by cocultivation with stromal layers. Curr Protoc Stem Cell Biol Chapter. 2008;1:1–12. doi: 10.1002/9780470151808.sc01f06s6. [DOI] [PubMed] [Google Scholar]
- 19.Cook SD, Salkeld SL, Patron LP. Bone defect healing with an osteogenic protein-1 device combined with carboxymethylcellulose. J Biomed Mater Res B Appl Biomater. 2005;75:137–145. doi: 10.1002/jbm.b.30271. [DOI] [PubMed] [Google Scholar]
- 20.Hak DJ, Makino T, Niikura T, et al. Recombinant human BMP-7 effectively prevents non-union in both young and old rats. J Othop Res. 2006;24:11–20. doi: 10.1002/jor.20022. [DOI] [PubMed] [Google Scholar]
- 21.Arpornmaeklong P, Brown SE, Wang Z, Krebsbach PH. Phenotypic characterization, osteoblastic differentiation and bone regeneration capacity of human embryonic stem cell-derived mesenchymal stem cells. Stem Cells Dev. 2009;18:955–968. doi: 10.1089/scd.2008.0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harkness L, Mahmood A, Ditzel N, et al. Selective isolation and differentiation of a stromal population of human embryonic stem cells with osteogenic potential. Bone. doi: 10.1016/j.bone.2010.09.023. in press doi:10.1016/j/bone.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 23.Both SK, van Apeldoorn AA, Jukes JM, et al. Differential bone-forming capacity of osteogenic cells from either embryonic stem cells or bone marrow-derived mesenchymal stem cells. J Tissue Eng Regen Med. doi: 10.1002/term.303. in press doi:10.1002/term.303. [DOI] [PubMed] [Google Scholar]
- 24.Werstat K, Meckbach D, Weis-Klemm M, et al. TGF-beta enhances the integrin alpha2beta1-mediated attachment of mesenchymal stem cells to type I collagen. Stem Cells Dev. 2010;19:645–656. doi: 10.1089/scd.2009.0208. [DOI] [PubMed] [Google Scholar]