

Abstract
Alterations in nuclear factor kappa B (NF-κB) essential modulator (NEMO; HUGO-approved symbol IKBKG) underlie most cases of ectodermal dysplasia with immune deficiency (EDI), a human disorder characterized by anhidrosis with diminished immunity. EDI has also been associated with a single heterozygous mutation at position Ser32 of the NF-κB inhibitor IκBα, one of two phosphorylation sites that are essential for targeting IκBα for proteasomal degradation and hence for activation of NF-κB. We report a novel heterozygous nonsense mutation in the IKBA (HUGO-approved symbol, NFKBIA) gene of a 1-year-old male child with EDI that introduces a premature termination codon at position Glu14. An in-frame methionine downstream of the nonsense mutation allows for reinitiation of translation. The resulting N-terminally truncated protein lacks both serine phosphorylation sites and inhibits NF-κB signaling by functioning as a dominant negative on NF-κB activity in lymphocytes and monocytes. These findings support the scanning model for translation initiation in eukaryotes and confirm the critical role of the NF-κB in the human immune response.
Keywords: primary immunodeficiency, ectodermal dysplasia, EDI, lymphocyte activation, antigen presenting cell, NEMO, NF-κB, IKBKG, IκBα, IKBA, NFKBIA
INTRODUCTION
The nuclear factor kappa B (NF-κB) family of transcription factors regulates the expression of genes essential for host defense and inflammation. In the resting cell, NF-κB factors are retained in the cytoplasm by interaction with members of the inhibitor of the NF-κB (IκB) family that includes IκBα, IκBβ, and IκBε[Ghosh et al., 1998; Hayden and Ghosh, 2004]. In the canonical pathway of NF-κB activation, IκB components are phosphorylated on two amino-terminal serine residues by the IκB kinase (IKK) complex composed of two catalytic proteins, IKKα and IKKβ and the regulatory subunit IKKγ (NEMO, NF-κB essential modulator; HUGO-approved symbol, IKBKG). This modification targets IκBs for degradation by the proteasome, permitting the release and nuclear translocation of NF-κB [Li and Verma, 2002]. IκBα degradation is induced in response to stimulation by the tumor necrosis factor (TNF) receptor superfamily that includes ectodysplasin receptor (EDAR) and CD40, Toll-like receptors (TLRs), and antigen receptors on T and B cells [Li and Verma, 2002; Ruland and Mak, 2003]. Because of its central role in NF-κB signaling, hypomorphic mutations in the NEMO gene (MIM# 300248) can cause ectodermal dysplasia, a disorder characterized by anhidrosis and absent or misshapen teeth with severe immune deficiency (EDI) [Jain et al., 2001; Puel et al., 2004; Temmerman et al., 2006; Zonana et al., 2000]. EDI has also been associated with a single hypermorphic mutation in IKBA (HUGO-approved symbol, NFKBIA; MIM# 164008) [Courtois et al., 2003; Janssen et al., 2004]. Herein, we describe an 1-year-old male with EDI associated with a novel heterozygous nonsense mutation in IKBA. This mutation conditions an early termination of translation that is compensated by a reinitiation of translation at downstream methionine. The resultant N-terminal truncated protein is resistant to degradation and exerts a dominant negative effect on NF-κB activity.
MATERIALS AND METHODS
Case Report
A male Caucasian infant was born after an uneventful 37-week pregnancy, as the first child to nonconsanguineous parents. His neonatal course was uncomplicated, and he was fed initially with breast milk alone and then in combination with artificial milk. He received standard immunizations at birth and at 2 months without complications. He did not receive any live virus vaccines. At the age of 2 months, he started suffering from gastrointestinal problems with feeding intolerance, frequent episodes of diarrhea, and episodes of fever with leukocytosis and neutrophilia. By that time, failure to thrive was evident.
At 7 months of age he was referred for evaluation and admitted to the intensive care unit with respiratory impairment; extensive interstitial lung disease was evident by a chest computerized tomography scan, which also identified a normal-sized thymus. Bronchoscopy revealed parainfluenza virus and Pneumocystis carinii, which was successfully treated with trimethoprim-sulfa-methoxazole. In his first months of life he also suffered from recurrent episodes of bacteremia. Testing excluded HIV infection. A skin biopsy from the antecubital region revealed an absence of sweat glands, and the results of two sweat tests supported the diagnosis of anhidrosis. The patient underwent an allogeneic cord blood transplant and expired in the peritransplant period due to gram-negative sepsis.
Study Subjects
The patient, family members, and unaffected normal controls were studied at the National Jewish Medical and Research Center and the Clinical Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIAID/NIH; protocol 89-I-006). Immunologically normal adult individuals served as controls. The Institutional Review Board of the NIAID approved the open protocol, and informed consent was obtained from the parents before the enrollment in the study.
Immunophenotyping
Lymphocyte subpopulations were studied by flow cytometry on at least three occasions prior to transplant with the whole-blood lysis technique and analyzed with a FACScan (Becton Dickinson, San Jose, CA) with CellQuest software. The monoclonal antibodies used included anti-CD3, anti-CD4, anti-CD8, anti-CD19, anti-CD16, anti-CD56, anti-CD45RA, anti-CD45RO, anti-CD20, anti-CD5, anti-IgG, anti-IgM, anti-IgA, and anti-CD27 (all from Becton Dickinson). Irrelevant antibodies to the different subclasses were used to ascertain background staining. To calculate the absolute number of each subpopulation, the percentage was multiplied by the absolute count of peripheral blood lymphocytes as determined by Coulter counter (Celdyne, San Jose, CA) followed by a differential cell leukocyte count of a blood sample that had been obtained simultaneously.
Cell Preparation and Culture Conditions
Peripheral blood mononuclear cells (PBMCs) were obtained from patients and healthy adult volunteers by centrifugation of heparinized blood over Ficoll-Hypaque density gradient lymphocyte separation medium (Pharmacia Biotech, Piscataway, NJ), with standard techniques. To measure interleukin IL-12 production, 2 × 106 PBMCs were cultured in RPMI 1640 complete medium (1 ml) for 36 hr. The following reagents were also used: human interferon (IFN)-γ (1,000 U/ml; Peprotech, Rocky Hill, NJ), SAC (Pansorbin cells, 1:10,000 w/v, Calbiochem, La Jolla, CA), LPS from E. coli 01127:B8 (1 μg/ml; Sigma, St. Louis, MO), OspA (a gift of Alan Sher, Bethesda, MD), and CD40L trimer (2.5 μg/ml; a gift of Immunex Corp., Seattle, WA). For anti-CD3 stimulation, OKT3 antibody (gift of Ortho Biotech, Somerset, NJ) was first dissolved in carbonate buffer at a concentration of 10 μg/ml and aliquotted into 24-well plates at 250 μl/well. After overnight incubation at 4°C, the plates were washed twice in sterile PBS. Cell suspensions were then added. The cytokine IL-12 was used (10 μg/ml; R&D Systems, Minneapolis, MN). After 36 hr, supernatants were removed. IFN-γ, IL-12, and TNF-α concentrations were determined by specific ELISA (R&D Systems), according to the manufacturer’s instructions.
Molecular Diagnosis
DNA and RNA were extracted from lymphocytes by standard methods. A NEMO gene study was performed as previously described [Jain et al., 2001]. For IKBA gene, exons 1–6 were amplified by PCR from genomic DNA with primers flanking each exon as described [Courtois et al., 2003]. The PCR products were purified (Qiagen, Valencia, CA) and cycle-sequenced at both the 5′ and the 3′ ends with the dye-terminator dideoxy nucleotides. Sequencing results were compared to the reference sequence GenBank NM_020529.1 and positions numbered as position 1 being the A of the first ATG initiation codon.
Western Blot Studies
Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines cultured in RPMI with 10% fetal calf serum (FCS) were stimulated with CD40L trimer (2.5 μg/ml) in the presence of cycloheximide (CHX, 50 μg/ml; Sigma), an inhibitor of protein translation, to prevent de novo IκBα protein synthesis. Cell lysates were prepared by resuspending cell pellet in 100 mM NaCl, 50 mM Tris-Cl at pH 8.0, 0.5% NP-40, 50 mM NaF, 30 mM sodium pyrophosphate, 1 mM Na3VO4, 0.6 % diisopropyl fluorophosphates (Sigma), and 1 × complete TM protease inhibitor mixture (Boehringer Mannheim, Indianapolis, IN) for 15 min on ice followed by centrifugation at 38,000 rpm for 30 min. The supernatants were collected as cell lysates and the protein concentration determined by the Bio-Rad protein assay method (Bio-Rad, Hercules, CA) with bovine albumin as a standard. Samples were then applied to SDS-PAGE followed by transfer onto nitrocellulose membranes. The membranes were probed with either anti-IκBα N-terminal specific, or anti-IκBα C-terminal specific antibody (catalog numbers SC203 and SC271; Santa Cruz Biotech, Santa Cruz, CA). This was followed by the addition of horseradish peroxidase-conjugated donkey anti-rabbit IgG (Amersham, Piscataway, NJ). Films were developed by the enhanced chemiluminescence (ECL) method (Amersham). For protein loading control, membranes were stripped and reblotted with anti-β-actin specific antibody (Sigma).
T Cell Proliferation Assay
PBMCs were suspended in complete medium (RPMI containing 1% penicillin/streptomycin with 10% FCS) at 3 × 106 cells/ml. In triplicate wells, the PBMC cultures were either unstimulated or stimulated with phytohemagglutinin (PHA) diluted 1:100 (In-vitrogen, Carlsbad, CA) or tetanus toxoid (1:400 concentration; Connaught Laboratories, Philadelphia, PA), or Conclavin A (Con-A) 5 μg/ml (Invitrogen), or Pokweed mitogen (PWM) 1 μg/ml (Sigma-Aldrich, St. Louis, MO). Tetanus toxoid cultures were pulsed after 4 days and all other culture conditions were pulsed at 2 days with 1 μCi of [3H]thymidine per well (ICN Biomedicals, Costa Mesa, CA) and incubated an additional 18 hr before harvesting onto printed filter mats (Wallac/Perkins Elmer, Waltham, MA) using a 96-well cell harvester (TOMTEC, Orange, CA). Incorporation of 3H on filter mats was measured as counts per minute (cpm) by a BetaPlate reader (Wallac/Perkins Elmer).
Luciferase Reporter Gene Assay
Human embryonic kidney 293 (HEK293) cells were seeded into 24-well plates (1 × 105 cells/well). Following an 8-hr incubation, the cells were transfected with 1.1 μg of a luciferase expressing reporter plasmid under an NF-κB promoter (pNF-κB-Luc) (BD Biosciences) with 0.2 μg β-galactosidase control vector as a transfection control to normalize luciferase activity. A total of 2.2 μg of either an IκBα–wild-type-expressing plasmid (pCMV-IKBA-WT), an IκBα–mutant-expressing plasmid (pCMV-IKBA-40G>T), or empty vector (pCMV-empty). Transfections were carried out using a Calcium Phosphate Transfection Kit (Invitrogen). Approximately 36 hr after transfection, the cells were stimulated with TNF-α (10 ng/ml) for 6 hr. The cells were then harvested and luciferase activity was measured with a Bright-GloTM luciferase assay system (Promega, Madison, WI). The presented data represents one of three independent transfection experiments. Transfections with pCMV-IKBA-WT and pCMV-IKBA-40G>T were carried out in triplet. Luciferase activity of each sample was normalized to an internal reference standard of β-galactosidase activity.
RESULTS
Clinical Characteristics and Immunologic Evaluation of the Patient
The clinical presentation of this male infant with failure to thrive, recurrent pyogenic bacterial infections, oral candidiasis, and PCP pneumonia suggested a severe immunodeficiency with compromised T-cell function. Immunologic evaluation revealed normal serum immunoglobulin levels on at least two occasions prior to the institution of intravenous immunoglobulin (IVIG) therapy: Serum levels at 7 months of age were: IgG 515 mg/dl, IgA 38 mg/dl, and IgM 33 mg/dl. Lymphocyte phenotyping showed normal percentages and absolute numbers of T-cells (CD4+ and CD8+), B-cells (CD19+), and major histocompatibility complex (MHC) class II (human leukocyte antigen [HLA]-DR)-expressing cells but reduced natural killer (NK) cells (CD16+/56+, 0.06%) and γ/δ T-cells (0.1%; normal range 3–28%). At the 5-month evaluation, CD4+T cells with exclusive CD45RO was 19.5%, while cells with exclusive expression of CD45RA was 57.4%. Mitogen-induced proliferation to PHA, pokeweed mitogen (PWM), and Concanavalin-A (Con-A) were normal. Tetanus-induced proliferation was also normal. Taken together, these finding suggests that the patient’s clinical predisposition to pyogenic and opportunistic infections was not a result of obvious defects in B and T cell development or activation.
Abnormalities of Antigen-Presenting Cell Function
The absence of sweat glands in the patient’s skin biopsy was indicative of ectodermal dysplasia and this in combination with the clinical history severe infections strongly suggested impaired NF-κB signaling. We further delineated the immunologic characteristics of the patient by assessing the capacity of patient monocytes in PBMC to produce the NF-κB regulated cytokines TNF-α and IL-12 following stimulation with mediators of innate and adaptive immunity. We stimulated patient and control cells with the TLR4 agonist LPS plus IFNγ, the B. burgdorferi–derived TLR2 agonist OspA plus IFNγ, SAC plus IFNγ, or CD40L plus IFNγ and measured TNF-α and IL-12 secretion by ELISA. In comparison to controls, patient monocytes in PBMCs synthesized markedly reduced amounts of TNFα and IL-12 (Fig. 1a). These findings suggest that the patient’s monocytes were defective in the production of NF-κB regulated cytokines in response to diverse stimuli and could explain the patient’s predisposition to microbial infections.
FIGURE 1.

Monocytes and T cells in PBMCs have impaired production of cytokines in response to different stimuli. A total of 2 × 106 patient and control PBMCs were stimulated with LPS (1 μg/ml) +IFNγ (1,000 U/ml), SAC (Pansorbin cells, 1:10,000 w/v)+IFNγ, OspA (1 μg/ml) +IFNγ, or CD40L (3.5 μg/ml)+IFNγ. TNFα and IL-12 production were measured at 36 hr by ELISA. TNFα and IL-12 production from unstimulated (control) conditions were <50 pg/ml and <10 pg/ml, respectively, for both patient and control. Data are representative of two independent experiments. b: Patient and control T cells in 2 × 106 PBMCs were stimulated with plate coated OKT3 or OKT3+IL-12 (10 μg/ml). Production of IFNγ and TNFα was analyzed by ELISA after 36 hr. IFNγ and TNFα production from unstimulated conditions was<50 pg/ml for both patient and control. Data are representative of two independent experiments.
Abnormalities of T-Cell Function
Although the patient was shown to have normal T-cell numbers and cellular subsets, the clinical history of Pneumocystis pneumonia suggested impairment in cellular immunity. Furthermore, the deficiency of IL-12 production from patient monocytes upon stimulation with various mediators of innate and adaptive immunity suggested that cognate interactions between APCs and T cells would result in reduced IFNγ production. To explore this possibility, we cultured PBMCs with anti-CD3 for 36 hr and then measured IFNγ secretion into the culture fluid by specific ELISA. As shown in Figure 1b, the patient produced markedly reduced amounts of IFNγ compared with controls. To demonstrate that the reduced IFNγ production was due to the lack of the NF-κB regulated cytokine IL-12, we stimulated PBMCs from controls and the patient with anti-CD3 as before, but this time in the presence of recombinant IL-12. Recombinant IL-12 restored the deficient IFNγ production in the patient (Fig. 1b).
Molecular Studies
Several aspects of the phenotype in this patient resembled that associated with hypomorphic mutations in NEMO [Courtois et al., 2003; Doffinger et al., 2001; Jain et al., 2001; Temmerman et al., 2006]. However, NEMO sequence and protein expression were normal. Other candidate genes involved in the NF-κB signaling pathway were then considered as possible disease susceptibility genes. In particular, we looked at IKBA because a single heterozygous mutation at position Ser32 of IκBα has previously been associated with EDI [Courtois et al., 2003; Janssen et al., 2004]. Genomic sequencing of the IKBA gene revealed a novel, and previously undescribed, heterozygous nonsense mutation c.40G>T in exon 1 that introduces a premature stop codon p.Glu14X (Fig. 2a). This mutation was confirmed by an independent PCR and sequencing reaction. RT-PCR and cDNA sequencing indicated that both IKBA alleles were transcriptionally active in the patient’s PBMCs (data not shown). The mutation was not present in either parent and therefore constituted a de novo mutation in the patient (Fig. 2a).
FIGURE 2.

Molecular analysis of the IKBA gene, and its effect on NF-κB signailing and IκBα protein expression. a: Chromatogram of the IKBA gene showing the heterozygous c.40G>T (p.Glu14X) mutation in the patient. Protein sequence for the healthy and mutated allele are represented above and below the DNA sequence respectively. b: Western blot analysis shows aberrant expression of an N-truncated IκBα protein in EBV-transformed patient B cells when a C-terminal specific antibody is used. The degradation of the aberrant N-truncated IκBα protein in response to CD40L stimulation is impaired. c: Impaired RelA and cRel binding in patient cells following CD40 stimulation. EBV transformed B cells were isolated from the patients and a normal control. Cells were stimulated in the presence of CD40L for 30 min. Cellular extracts were prepared and analyzed on the same gel by electrophoretic mobility shift assay for NF-κB binding activity. For supershift, all samples were incubated with anti c-Rel antibody prior to incubation with the labeled probe. One representative experiment of two is shown. d: In comparison to the control, resynthesis of wild-type IκBα protein level was notably reduced in the EBV-transformed B cell lines following stimulation with CD40L and in the absence of CHX. Cytoplasmic extracts were prepared from cells from a normal control and the patients and subjected to immunoblotting. Blots were probed with anti C-terminal IκBα antibody.
The c.40G>T IKBA Mutation Inhibits NF-κB Activation
The IκBα protein is a key regulator of the canonical activation pathway that leads to the nuclear translocation of NF-κB. The identification in this patient of a potentially disease-causing mutation in the IKBA gene prompted us to explore the consequences for IκBα expression and NF-κB activation. EBV-transformed lymphoblastoid cell lines were generated from the patient and normal PBMC. The presence and transcriptional activity of the IKBA mutation was confirmed by direct sequencing of PCR and RT-PCR products in the EBV-transformed cell line (data not shown). Translocation of NF-κB to the nucleus results from phosphorylation and degradation of IκBα. Thus, the fate of IκBα was measured by the immunoblotting of cellular extracts of EBV-transformed B cells from the patient and controls following stimulation in vitro with CD40L in the presence of CHX. IκBα expression was measured at several intervals. An N-terminal specific IκBα antibody showed a band of the expected size in both control and patient cells, which rapidly disappeared with CD40L stimulation (Fig. 2b). However, the IκBα band in patient cells was reduced in intensity and this prompted us to analyze the protein extracts with a C-terminal specific IκBα antibody. This revealed a second, lower molecular weight band only in patient cells that remained stable during CD40L stimulation. Activation with TNF-α induced delayed degradation of the wild-type protein in patient cells, but did not affect the level of the mutant protein in the patient’s lymphoblastoid cell line (our unpublished observation). In further studies, we more precisely defined the effect of the c.40G>T IKBA mutation on NF-κB activation. In particular, we measured p65 and c-Rel activation by electrophoretic mobility shift assay in cellular lysates prepared from lymphoblastoid B cells from the patient and the normal control. In contrast to the normal control, p65 and c-Rel activity in cells from the patient was impaired after stimulation with CD40L (Fig. 2c). These results indicate that the mutation, rather than generating a null allele, is inducing the translation of an alternative IκBα product that lacks the N-terminal part of the protein (ΔN-IκBα). The mutant IκBα is resistant to the degradation by the proteasome and is associated with reduced downstream NF-κB activity.
IKBA is an NF-κB target gene and in the absence of CHX, resynthesis of IκBα can be normally detected in mammalian in vitro systems following proinflammatory stimulation. To determine the effect of the c.40G>T IKBA mutation on the resynthesis of IκBα, we stimulated an EBV-transformed B cell line from the patient and controls with CD40L and in the absence of CHX. IκBα expression was measured at several intervals with the C-terminal IκBα antibody (Fig. 2d). Resynthesis of IκBα was detectable in the control at 60 min and was nearly complete at 2 hr. In contrast, resynthesis of wild-type IκBα was notably reduced, while the level of the mutant protein in the patient’s lymphoblastoid cell line was unchanged. It is interesting to note that the vast majority of IκBα species in the patient’s EBV-transformed cell line was the mutant protein. The mutant species likely accumulate because they are not subject to degradation and contribute to the patient’s phenotype by sequestering NF-κB.
c.40G>T IKBA Mutation Results in an N-Terminal Truncated IκBα That Exerts a Dominant Negative Effect on NF-κB Signaling
To validate the effect of the c.40G>T IKBA mutation on NF-κB activation, 293 cells were transfected with an NF-κB reporter construct in combination with either a wild-type IKBA plasmid, c.40G>T IKBA mutant plasmid, or with empty vector as a control. Cells were stimulated with TNF-α, and luciferase activity was measured. The c.40G>T IKBA mutant plasmid suppressed activation of NF-κB at higher levels in comparison to the vector control or the wild-type IKBA plasmid (Fig. 3). In addition, we transfected 293 cells with C-terminal V5-His tagged expression vectors with the type wild-type IKBA or the c.40G>T IKBA mutant allele. Protein expression was analyzed by immunoprecipitation with an anti-His antibody in cellular lysates followed by immunoblotting with a C-terminal IKBA antibody. A protein of ~40 kDa was detected in wild type IKBA-transfected cells and a lower molecular weight protein was detected in c.40G>T IKBA mutant-transfected cells (Fig. 3B). Taken together, these findings demonstrate that the c.40G>T IKBA mutation results in the translation of a mutant protein that inhibits NF-κB and supports the cellular and clinical findings in our patient.
FIGURE 3.
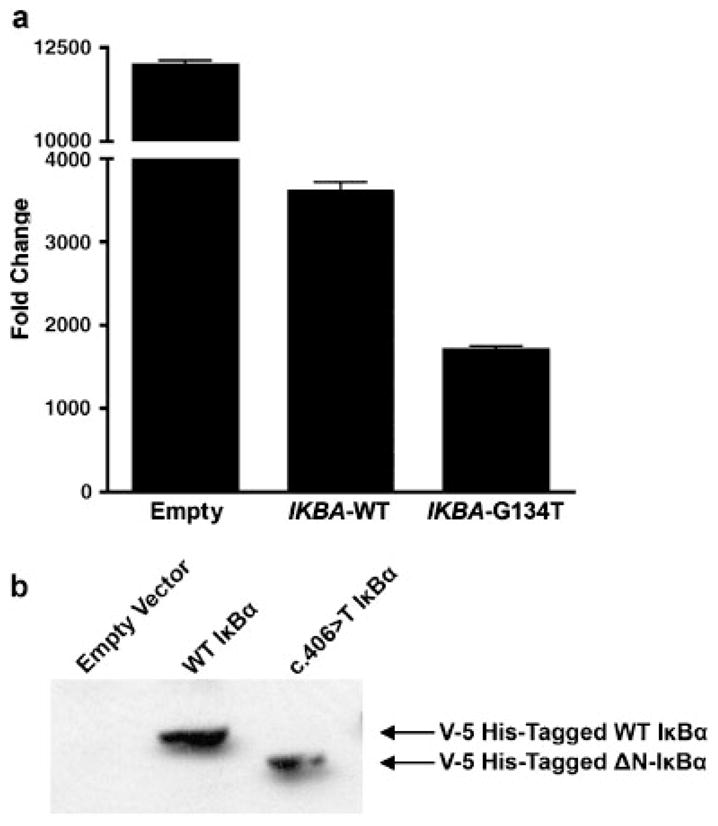
c.40G>T IKBA mutation results in an N-terminal truncated IκBα that exerts a dominant negative effect on NF-κB signaling. a: Cotransfection experiment in HEK293 cells with pNF-kB-Luc reporter plasmid and either pCMV-IKBA-WT or pCMV-IKBA-40G>T. Luciferase activity was measured in response to TNFα stimulation. The luciferase activity of each sample was normalized to an internal reference standard of β-galactosidase activity. Data represent one of three independent cotransfection experiments. b: c.40G>T IKBA encodes a truncated IκBα protein .293 cells were transfected with C-terminal V5-His tagged expression vectors with the wild-type IKBA or the c.40G>T IKBA mutant allele. Protein expression was analyzed by immunoprecipitation with an anti-His antibody in cellular lysates followed by immunoblotting with a C-terminal IKBA antibody.
DISCUSSION
In this work we describe a novel IκBα mutation in a 1-year-old patient with EDI. The c.40G>T IKBA mutation creates a premature termination codon but an in-frame methionine downstream of the mutation leads to reinitiation of translation. The amino-truncated IκBα protein does not affect fetal development but is not sufficient for the development of protective immune responses.
According to the linear scanning model, the polyribosome scans the mRNA from the 5′ region and starts translation at the first favorable AUG codon. If this is followed by a short open reading frame (ORF), some ribosomes may resume scanning and reinitiate translation at the next downstream AUG [Kozak, 2001, 2002a, 2002b]. Premature termination of translation in bacteria often leads to reinitiation at nearby start codons [Kozak, 2001; Poyry et al., 2004]. The analysis of the IKBA cDNA sequence (Fig. 4a) revealed a second in-frame AUG codon (Met37) downstream of the p.Glu14X mutation. Several computational tools have been developed to assist in the identification of translation initiation sites (TIS). We analyzed the patient’s IKBA gene sequence with DNA functional sites finder (DNAFS), a program that estimates a score for the likelihood of an AUG codon to initiate translation according to its context and similarity to the Kozakian sequence [Liu et al., 2005]. The score of the first AUG in the mutated allele decreases from 0.96 (observed in the wild type) to 0.844 and is comparable to the 0.78 score for translation initiation at Met37. These predictions further reinforce the hypothesis that the nonsense mutation at position 14 allows the use of this downstream start codon through a reinitiation of translation mechanism. As a result, an aberrant N-terminal truncated IκBα protein (ΔN-IκBα) lacking both serines (Ser32 and Ser36) required for degradation by the proteasome is expressed in patient cells (Fig. 4b). The molecular weight of the truncated protein and the fact that it was only recognized by C-terminal specific IκBα antibody is in agreement with this model. With complementation experiments, we show that the c.40G>T IKBA mutant construct inhibits NF-κB activity and results in the translation of a mutant protein. This is in keeping previously published results in which IκBα mutant constructs lacking both serine phosphorylation sites retained the capacity to bind and sequester NF-κB heterodimers, but could not release them because the mutant IκBα protein was no longer subjected to degradation by the ubiquitin-proteasome pathway [Boothby et al., 1997; Brockman et al., 1995]. Examples supporting the reinitiation of translation are rare in human genetics but have been described in other inherited blood disorders [Puel et al., 2006; Santagata et al., 2000; Yamashita et al., 1996].
FIGURE 4.

Model for the translation of the mutant IκBα allele E 14 X. a: N-terminal sequence of the IKBA cDNA and likelihood scores for the use of different TIS in the WT and the mutated allele. b: Proposed model for the translation of the δN-IκBα protein. The proximity of the new stop codon induced by the nonsense mutation to a second in-frame methionine residue would allow the translation of the majority of the IκBα ORF by a reinitiation of translation mechanism.
The IKBA mutation affecting Ser32 and the new mutation in our patient were associated with normal T and B cell numbers, proliferation to nonspecific mitogens and to tetanus, and a clinical history indicative of a defective T cell function [Courtois et al., 2003; Janssen et al., 2004]. However, the reduced numbers of NK cells, the normal serum immunoglobulin levels, as well as the normal numbers of CD4+ T cells expressing CD45RO and CD45RA in our patient differ from the Ser32 phenotype. These findings indicate that the immunologic impairments may vary with different IKBA mutations and expand the possible phenotypes associated with IκBα mutations. The in vitro production of T cell cytokines such as TNF-α and INF-γ was severely impaired in our patient, thus dissociating requirements for T cell proliferation from those for cytokine production. Surprisingly, the deficient responses could be restored by coculture with IL-12, suggesting that effector T cell responses were not completely dependant on IκBα degradation.
A child with the Ser 32 IKBA mutation was successfully transplanted with T-cell-depleted bone marrow from a haploid identical parent and was healthy at follow-up 7 years posttransplant [Dupuis-Girod et al., 2006]. Our patient received a matched unrelated cord blood transplant following myeloablative conditioning with busulfan and cyclophosphamide, but expired from sepsis prior to engraftment. The differences in clinical outcomes highlights the fact that bone marrow transplantation, while an effective treatment for patients with primary immune deficiency, is also associated with morbidity and mortality that limits its utility. Administration of recombinant IL-12 could be an important therapeutic intervention in patients with alterations in IKBA through the restoration of deficient Th-1 T cell immune responses.
While this work was under review, McDonald et al. [2007] reported an IKBA mutation in a patients exhibiting EDI. The mutation leads to a reinitiation of translation in IKBA and confirm our observations.
Acknowledgments
We thank the patient’s family for their willingness to participate in this study; Ron Hornung and Margaret Brown for immunologic evaluations; Mary Derry for editorial assistance; and Steve Holland and Harry Malech for helpful discussions.
Footnotes
This article is a US Government work, and, as such, is in the public domain in the United States of America
References
- Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, Dupuis-Girod S, Bodemer C, Livadiotti S, Novelli F, Rossi P, Fischer A, Israel A, Munnich A, Le Deist F, Casanova JL. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. 2003;112:1108–1115. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- Dupuis-Girod S, Cancrini C, Le Deist F, Palma P, Bodemer C, Puel A, Livadiotti S, Picard C, Bossuyt X, Rossi P, Fischer A, Casanova JL. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics. 2006;118:e205–e211. doi: 10.1542/peds.2005-2661. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–228. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J, van de Vosse E, van Tol M, Bredius R, Ottenhoff TH, Weemaes C, van Dissel JT, Lankester A. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. J Exp Med. 2004;200:559–568. doi: 10.1084/jem.20040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Constraints on reinitiation of translation in mammals. Nucleic Acids Res. 2001;29:5226–5232. doi: 10.1093/nar/29.24.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Emerging links between initiation of translation and human diseases. Mamm Genome. 2002a;13:401–410. doi: 10.1007/s00335-002-4002-5. [DOI] [PubMed] [Google Scholar]
- Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002b;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Liu H, Han H, Li J, Wong L. DNAFSMiner: a web-based software toolbox to recognize two types of functional sites in DNA sequences. Bioinformatics. 2005;21:671–673. doi: 10.1093/bioinformatics/bth437. [DOI] [PubMed] [Google Scholar]
- McDonald DR, Mooster JL, Reddy M, Bawle E, Secord E, Geha RS. Heterozygous N-terminal deletion of IkappaBalpha results in functional nuclear factor kappaB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J Allergy Clin Immunol. 2007;120:900–907. doi: 10.1016/j.jaci.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Poyry TA, Kaminski A, Jackson RJ. What determines whether mammalian ribosomes resume scanning after translation of a short upstream open reading frame? Genes Dev. 2004;18:62–75. doi: 10.1101/gad.276504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol. 2004;16:34–41. doi: 10.1016/j.coi.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Puel A, Reichenbach J, Bustamante J, Ku CL, Feinberg J, Doffinger R, Bonnet M, Filipe-Santos O, Beaucoudrey L, Durandy A, Horneff G, Novelli F, Wahn V, Smahi A, Israel A, Niehues T, Casanova JL. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet. 2006;78:691–701. doi: 10.1086/501532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruland J, Mak TW. Transducing signals from antigen receptors to nuclear factor kappaB. Immunol Rev. 2003;193:93–100. doi: 10.1034/j.1600-065x.2003.00049.x. [DOI] [PubMed] [Google Scholar]
- Santagata S, Gomez CA, Sobacchi C, Bozzi F, Abinun M, Pasic S, Cortes P, Vezzoni P, Villa A. N-terminal RAG1 frameshift mutations in Omenn’s syndrome: internal methionine usage leads to partial V(D)J recombination activity and reveals a fundamental role in vivo for the N-terminal domains. Proc Natl Acad Sci USA. 2000;97:14572–14577. doi: 10.1073/pnas.97.26.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temmerman ST, Ma CA, Borges L, Kubin M, Liu S, Derry JM, Jain A. Impaired dendritic-cell function in ectodermal dysplasia with immune deficiency is linked to defective NEMO ubiquitination. Blood. 2006;108:2324–2331. doi: 10.1182/blood-2006-04-017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Wu N, Kupfer G, Corless C, Joenje H, Grompe M, D’Andrea AD. Clinical variability of Fanconi anemia (type C) results from expression of an amino terminal truncated Fanconi anemia complementation group C polypeptide with partial activity. Blood. 1996;87:4424–4432. [PubMed] [Google Scholar]
- Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, Emmal SA, Ferguson BM. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:1555–1562. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]