

Abstract
A series of stereochemically defined cyclic ethers as P2-ligands were incorporated in an allophenylnorstatine-based isostere to provide a new series of HIV-1 protease inhibitors. Inhibitors 3b and 3c, containing conformationally constrained cyclic ethers, displayed impressive enzymatic and antiviral properties and represent promising lead compounds for further optimization.
Keywords: HIV protease, Inhibitors, Darunavir, Allophenylnorstatine, Design, Synthesis
The introduction of protease inhibitors into highly active antiretroviral treatment (HAART) regimens with reverse transcriptase inhibitors represented a major breakthrough in AIDS chemotherapy.1 This combination therapy has significantly increased life expectancy, and greatly improved the course of HIV management. Therapeutic inhibition of HIV-1 protease leads to morphologically immature and noninfectious viral particles.2 However, under the selective pressure of chemotherapeutics, rapid adaptation of viral enzymes generates strains resistant to one or more antiviral agents.3 As a consequence, a growing number of HIV/AIDS patients harbor multi-drug-resistant HIV strains. There is ample evidence that such strains can be readily transmitted.4 Therefore, one of the major current therapeutic objectives has been to develop novel protease inhibitors (PIs) with broad-spectrum activity against multidrug-resistant HIV-1 variants. In our continuing interest in developing concepts and strategies to combat drug-resistance, we have reported a series of novel PIs including Darunavir, TMC-126, GRL-06579 and GRL-02031.5–8 These inhibitors have shown exceedingly potent enzyme inhibitory and antiviral activity as well as exceptional broad spectrum activity against highly cross-resistant mutants. Darunavir, which incorporates (R)-(hydroxymethyl)-sulfonamide isostere and a stereochemically defined bis-tetrahydrofuran (bis-THF) as the P2-ligand, was initially approved for the treatment of patients with drug-resistant HIV and more recently, it has been approved for all HIV/AIDS patients including pediatrics.9
Darunavir was designed based upon the ‘backbone binding’ concept developed in our laboratories. Darunavir-bound X-ray structure revealed extensive hydrogen bonding with the protease backbone throughout the enzyme active site.10 The P2-bis-THF ligand is responsible for its superior drug-resistance properties. The bis-THF ligand has been documented as a privileged ligand for the S2-subsite. Incorporation of this ligand into other transition-state isosteres also resulted in significant potency enhancement.11 Besides 3(S)-THF, [3aS,5S,6R]-bis-THF, we have designed a number of other novel cyclic ether-based high affinity ligands. Incorporation of these ligands in (R)-(hydroxyethyl)-sulfonamide isosteres provided PIs with excellent potency and drug-resistance properties.6–8 We have then investigated the potential of these structure-based designed P2-ligands in KNI-764-derived isostere designed by Mimoto and co-workers.12 This PI incorporates an allophenylnorstatine isostere. Interestingly, KNI-764 has maintained good activity against HIV-1 clinical strains resistant to several FDA-approved PIs. The flexible N-(2-methyl benzyl) amide P2′-ligand may have been responsible for its activity against drug-resistant HIV-1 strains as the flexible chain allows better adaptability to mutations.12,13 The bis-THF and other structure-based designed P2-ligands, make several critical hydrogen bonds with the protein backbone, particularly with Asp-29 and Asp-30 NH’s.11 Therefore, incorporation of these ligands into the KNI-764-derived isostere, may lead to novel PIs with improved potency and efficacy against multidrug-resistant HIV-1 variants. Furthermore, substitution of P2-phenolic derivative in KNI-764 with a cyclic ether-based ligand could result in improved metabolic stability and pharmacological properties since phenol glucuronide is readily formed when KNI-764 is exposed to human hepatocytes in vitro.12
The synthesis of target compounds 3a–e was accomplished as described in Scheme 1. Our synthetic plan for the synthesis of carboxylic acid 7 (Scheme 1) involved the preparation of the key intermediate 5 which was prepared through two different synthetic pathways. In the first approach, known optically active azidodiol 414 was first hydrogenated in the presence of Boc2O. The resulting diol was converted to 5 by selective acylation of the primary alcohol with acetic anhydride in the presence of pyridine and a catalytic amount of DMAP at 0 °C for 4 h to provide 5 in 77% overall yield. As an alternative approach, commercially available optically active epoxide 6 was exposed to lithium acetate, formed in situ from lithium carbonate and acetic acid in DMF. This resulted in the regioselective opening15 of the epoxide ring and afforded compound 5 in 62% yield. The alcohol 5 thus obtained was protected as the corresponding acetonide by treatment with 2-methoxypropene in the presence of a catalytic amount of CSA. The acetate group was subsequently hydrolyzed in the presence of potassium carbonate in methanol to afford the corresponding alcohol. This latter was subjected to an oxidation reaction using ruthenium chloride hydrate and sodium periodate in a mixture of aqueous acetonitrile and CCl4 at 23 °C for 10 h. This resulted in the formation of target carboxylic acid 7 in 61% yield. Amine 9a was prepared by activation of carboxylic acid 7 into the corresponding mixed anhydride by treatment with iso-butylchloroformate followed by reaction with amine 8a.16,17
Scheme 1.

Reagents and conditions: (a) H2, Pd/C, Boc2O, EtOAc; (b) Ac2O, Pyr, DMAP; (c) LiCO3, AcOH, DMF; (d) 2- methoxypropene, CSA, DCM; (e) K2CO3, MeOH; (f) RuCl3, NaIO4, CCl4-MeCN-H2O (2:2:3); g) N-methylmorpholine, iBuOCOCl, 8a, THF.
Synthesis of various inhibitors was carried out as shown in Scheme 2. Deprotection of Boc and acetonide groups was carried out by exposure of 9 to 1 M solution of hydrochloric acid in methanol at 23 °C for 8 h. This provided amine 10 in quantitative yield. Reaction of 11a with amine 10 in CH2Cl2 in the presence of Et3N at 23 °C for 6 h, provided inhibitor 3a in 62% yield. The 3(S)-tetrahydrofuranyl carbonate 11a was prepared as described previously.18 Similarly, allophenylnorstatine-based inhibitors 3b–e were synthesized. As shown, carbonates 11b19, 11c7, and 11d–e19 were prepared as previously described. Reaction of these carbonates with amine 10 furnished the desired inhibitors 3b–e in 45–62% yield.
Scheme 2.
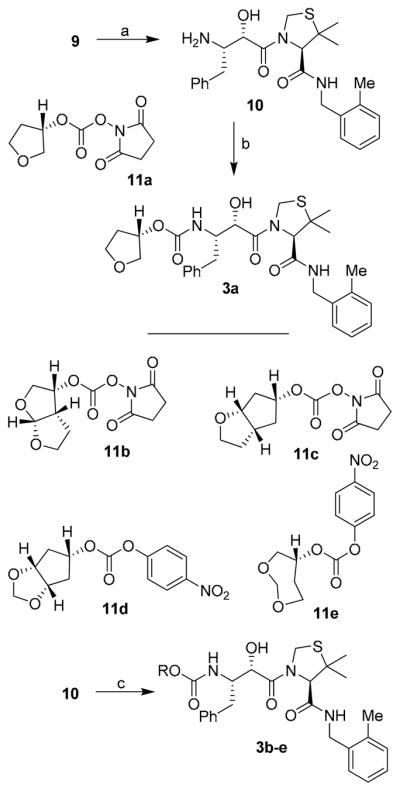
(a) 1 M HCl, MeOH; (b) 11a, Et3N, CH2Cl2; (c) 11b,c Et3N, CH2Cl2; or, 11d,e, DIPEA, THF.
The syntheses of inhibitors 14a,b and 16a–c were carried out as shown in Scheme 3. Inhibitors 14a,b, containing hydroxyethylamine isostere were prepared by opening of epoxide 6 with amine 8a in the presence of lithium perchlorate in diethyl ether at 23 °C for 5 h to provide amino alcohol 12 in 64% yield. Removal of Boc-group by exposure to 1M HCl in MeOH at 23 °C for 12 h afforded amine 13. Reactions of amine 13 with activated carbonates 11a and 11b afforded urethane 14a and 14b in 44% and 59% yields, respectively. For the synthesis of inhibitors 16a–c, commercially available (R)-5,5-dimethyl-thiazolidine-4-carboxylic acid was protected as its Boc-derivative. The resulting acid was coupled with amines 15a–c in the presence of DCC and DMAP in CH2Cl2 to provide the corresponding amides. Removal of Boc-group by exposure to 30% trifluoroacetic acid afforded 8b–d. Coupling of these amines with acid 7 as described in Scheme 1, provided the corresponding products 9b–d. Removal of Boc group and reactions of the resulting amine with activated carbonate 11b furnished inhibitors 16a–c in good yields (55–60%).
Scheme 3.

Reagents and conditions: (a) 8a, Li(ClO4), Et2O; (b) CF3CO2H, CH2Cl2; (c) 11a or, 11b, Et3N, CH2Cl2; (d) N-methylmorpholine, iso-butylchloroformate, 8b–d, THF; (e) CF3CO2H, CH2Cl2, then 11b, Et3N, CH2Cl2.
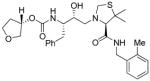
Inhibitors 3a–e were first evaluated in enzyme inhibitory assay utilizing protocol described by Toth and Marshall.20 Compounds that showed potent enzymatic Ki values were then further evaluated in antiviral assay. The inhibitor structure and potency are shown in Table 1. As shown, incorporation of a stereochemically defined 3(S)-tetrahydrofuran ring as the P2-ligand provided inhibitor 3a, which displayed an enzyme inhibitory potency of 0.2 nM and antiviral IC50 value of 20 nM. The corresponding derivative 14a with a hydroxyethylamine isostere exhibited over 400-fold reduction in enzyme inhibitory activity. Introduction of a stereochemically defined bis-THF as the P2-ligand, resulted in inhibitor 3b, which displayed over 40-fold potency enhancement with respect to 3a. Inhibitor 3b displayed a Ki of 5.2 pM in the enzyme inhibitory assay. Furthermore, compound 3b has shown an impressive antiviral activity with an IC50 value of 9 nM. Inhibitor 14b with hydroxyethylamine isostere is significantly less potent than the corresponding norstatine-derived inhibitor 3b. Inhibitor 3c with a (3aS, 5R, 6aR)-5-hydroxy-hexahydrocyclopenta[b]furan as the P2-ligand has displayed excellent inhibitory activity, and particularly, antiviral activity, showing an IC50 value of 13 nM. Other structure-based designed ligands in inhibitors 3d and 3e have shown subnanomolar enzyme inhibitory activity. However, inhibitor 3b with a bis-THF ligand has shown most impressive activity.
Table 1.
Enzymatic inhibitory and antiviral activity of allophenylnorstatine-derived inhibitors
| Entry | Inhibitor | Kj(nM) | IC50(μM)a,b |
|---|---|---|---|
| 1. |
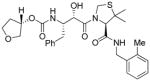 3a |
0.21 | 0.02 |
| 2. |
 14a |
86.2 | nt |
| 3. |
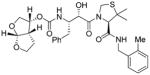 3b (GRL-0355) |
0.0052 | 0.009 |
| 4. |
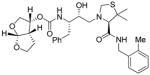 14b |
2.6 | nt |
| 5. |
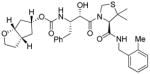 3c |
0.29 | 0.013 |
| 6. |
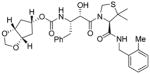 3d |
0.65 | nt |
| 7. |
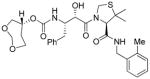 3e |
0.78 | nt |
| 8. |
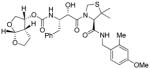 16a |
2.03 | 0.051 |
| 9. |
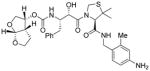 16b |
1.01 | 0.53 |
| 10. |
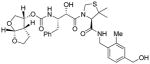 16c |
0.31 | 0.23 |
Values are means of at least three experiments.
Human T-lymphoid (MT-2) cells were exposed to 100 TCID50 values of HIV-1LAI and cultured in the presence of each PI, and IC50 values were determined using the MTT assay. Darunavir exhibited Kj = 16 pM, IC50 = 1.6 nM.
To obtain molecular insight into the possible ligand-binding site interactions, we have created energy-minimized models of a number of inhibitors based upon protein-ligand X-ray structure of KNI-764 (2).21 An overlayed model of 3b with X-ray structure of 2-bound HIV-1 protease is shown in Figure 2. This model for inhibitor 3b was created from the X-ray crystal structure of KNI-764 (2)-bound HIV-1 protease (KNI-764, pdb code 1MSM21) and the X-ray crystal structure of darunavir (pdb code 2IEN22), by combining the P2-end of the darunavir structure with the P2′-end of the KNI-764 structure, followed by 1000 cycles of energy minization. It appears that both oxygens of the bis-THF ligand are suitably located to form hydrogen bonds with the backbone atoms of Asp-29 and Asp-30 NH’s, similar to darunavir-bound HIV-1 protease.10 Furthermore, the KNI-764-X-ray structure-derived model of 3b suggested that the incorporation of appropriate substituents on the phenyl ring could interact with Asp-29′ and Asp-30′ in the S2′-subsite. In particular, it appears that a 4-hydroxymethyl substitutent on the P2′-phenyl ring could conceivably interact with backbone Asp-30′ NH in S2′-subsite. Other substituents such as a methoxy group or an amine functionality also appears to be within proximity to Asp-29′ and Asp-30′ backbone NHs. Based upon these speculations, we incorporated p-MeO, p-NH2 and p-CH2OH substituents on the P2′-phenyl ring of inhibitor 3b. As shown in Table 1, neither p-MeO nor p-NH2 groups improved enzyme inhibitory potency compared to inhibitor 3b. Of particular note, compound 16a, displayed a good antiviral potency, possibly suggesting a better penetration through the cell membrane. Inhibitor 16c with a hydroxymethyl substituent showed sub-nanomolar enzyme inhibitory potency but its antiviral activity was moderate compared to unsubstituted derivative 3b. As it turned out, inhibitor 3b is the most potent inhibitor in the series. We subsequently examined its activity against a clinical wild-type X4-HIV-1 isolate (HIV-1ERS104pre) along with various multidrug-resistant clinical X4- and R5- HIV-1 isolates using PBMCs as target cells.5b As can be seen in Table 2, the potency of 3b against HIV-1ER104pre (IC50 = 31 nM) was comparable to FDA approved PI amprenavir with IC50 value of 45 nM. Darunavir and atazanavir on the other hand, are significantly more potent with IC50 values of 5 nM and 3 nM respectively. Inhibitor 3b, while less potent than darunavir, maintained 5-fold or better potency over amprenavir against HIV-1MDR/C, HIV-1MDR/G, HIV-1MDR/TM and HIV-1MDR/MM. It maintained over 2-fold potency against HIV-1MDR/JSL. In fact, inhibitor 3b maintained comparable potency to atazanavir against all multidrug-resistant clinical isolates tested. The reason for its impressive potency against multidrug-resistant clinical isolates is possibly due to its ability to make extensive hydrogen-bonds with protease backbone in the S2 subsite and its ability to fill in the hydrophobic pockets in the S1′-S2′ subsites effectively.
Figure 2.
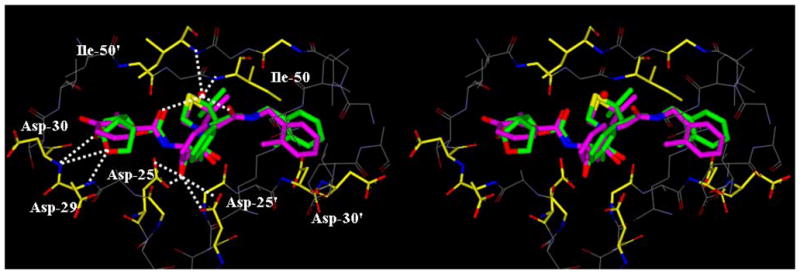
Structure of inhibitor 3b, modeled into the active site of HIV-1 protease, superimposed on the X-ray crystal structure of KNI-764. Inhibitor 3b carbons are shown in green and KNI-764 carbons are shown in magenta.
Table 2.
Antiviral activity of 3b (GRL-0355) against multi-drug resistant clinical isolates in PHA-PBMs
| Virus | IC50 (μM)
|
|||
|---|---|---|---|---|
| 3b (GRL-0355) | APV | ATV | DRV | |
| HIV-1ERS104pre (wild-type: X4) | 0.031 ± 0.002 | 0.045 ± 0.014 | 0.003 ± 0.003 | 0.005 ± 0.001 |
| HIV-1MDR/C (X4) | 0.061 ± 0.005 (2) | 0.346 ± 0.071 (8) | 0.045 ± 0.026 (15) | 0.010 ± 0.006 (2) |
| HIV-1MDR/G (X4) | 0.029 ± 0.002 (1) | 0.392 ± 0.037 (9) | 0.029 ± 0.020 (10) | 0.019 ± 0.005 (4) |
| HIV-1MDR/TM (X4) | 0.064 ± 0.032 (2) | 0.406 ± 0.082 (9) | 0.047 ± 0.009 (16) | 0.007 ± 0.003 (1) |
| HIV-1MDR/MM (R5) | 0.042 ± 0.001 (1) | 0.313 ± 0.022 (7) | 0.040 ± 0.002 (13) | 0.027 ± 0.008 (5) |
| HIV-1MDR/JSL (R5) | 0.235 ± 0.032 (8) | 0.531 ± 0.069 (12) | 0.635 ± 0.065 (212) | 0.028 ± 0.008 (6) |
The amino acid substitutions identified in the protease-encoding region of HIV-1ERS104pre, HIV-1C, HIV-1G, HIV-1MM, HIV-1JSL compared to the consensus type B sequence cited from the Los Alamos database include L63P; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q,V82A, L89M; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, L90M; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M; L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, Q92K; and L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, V82A, respectively. HIV-1ERS104pre served as a source of wild-type HIV-1. The IC50 values were determined by using PHA-PBMs as target cells and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of IC50 values for each isolate compared to the IC50 values for wild-type HIV-1ERS104pre. All assays were conducted in duplicate, and the data shown represent mean values (± 1 standard deviations) derived from the results of two or three independent experiments. Amprenavir = APV; Atazanavir = ATV; Darunavir = DRV.
In conclusion, incorporation of stereochemically defined and conformationally constrained cyclic ethers into the allophenylnorstatine resulted in a series of potent protease inhibitors. The promising inhibitors 3b and 3c are currently being subjected to further in-depth biological studies. Design and synthesis of new classes of inhibitors based upon above molecular insight are currently ongoing in our laboratories.
Figure 1.

Structures of inhibitors1, 2, and 3b
Acknowledgments
The financial support of this work is provided by the National Institute of Health (GM 83356).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sepkowitz KA. N Engl J Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 2.Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RAF, Scolnick EM, Sigal IS. Proc Natl Acad Sci, USA. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Pillay D, Bhaskaran K, Jurriaans S, Prins M, Masquelier B, Dabis F, Gifford R, Nielsen C, Pedersen C, Balotta C, Rezza G, Ortiz M, de Mendoza C, Kücherer C, Poggensee G, Gill J, Porter K. AIDS. 2006;20:21–28. doi: 10.1097/01.aids.0000196172.35056.b7. [DOI] [PubMed] [Google Scholar]; (b) Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. AIDS. 2000;14:141–149. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- 4.Wainberg MA, Friedland G. JAMA. 1998;279:1977–1983. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- 5.(a) Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J Bioorg Med Chem Lett. 1998;8:687–690. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]; (b) Koh Y, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Louis JM, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK, Pretzer E, Cho H, Hussain KA, Duzgunes N. Antiviral Res. 2002;54:29–36. doi: 10.1016/s0166-3542(01)00209-1. [DOI] [PubMed] [Google Scholar]
- 6.Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. J Virol. 2002;76:1349–1358. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JK, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 8.Koh Y, Das D, Leshchenko S, Nakata H, Ogata-Aoki H, Amano M, Nakayama M, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2009;53:997–1006. doi: 10.1128/AAC.00689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) FDA approved Darunavir on June 23, 2006: FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108676.htm (b) On October 21, 2008, FDA granted traditional approval to Prezista (darunavir), co-administered with ritonavir and with other antiretroviral agents, for the treatment of HIV-1 infection in treatment-experienced adult patients. In addition to the traditional approval, a new dosing regimen for treatment-naïve patients was approved
- 10.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh AK, Ramu Sridhar P, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 12.Mimoto T, Terashima K, Nojima S, Takaku H, Nakayama M, Shintani M, Yamaoka T, Hayashi H. Bioorg Med Chem. 2004;12:281–293. doi: 10.1016/j.bmc.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura K, Kato R, Yusa K, Kavlick MF, Maroun V, Nguyen A, Mimoto T, Ueno T, Shintani M, Falloon J, Masur H, Hayashi H, Erickson J, Mitsuya H. Proc Natl Acad Sci U S A. 1999;96:8675–8680. doi: 10.1073/pnas.96.15.8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Thompson WJ, Holloway MK, McKee SP, Duong TT, Lee HY, Munson PM, Smith AM, Wai JM, Darke PL, Zugay J, Emini EA, Schleif WA, Huff JR, Anderson PS. J Med Chem. 1993;36:2300–10. doi: 10.1021/jm00068a006. [DOI] [PubMed] [Google Scholar]
- 15.Ohmoto K, Okuma M, Yamamoto T, Kijima H, Sekioka T, Kitagawa K, Yamamoto S, Tanaka K, Kawabata K, Sakata A, et al. Bioorg Med Chem Lett. 2001;9:1307–1323. doi: 10.1016/s0968-0896(01)00007-4. [DOI] [PubMed] [Google Scholar]
- 16.Ikunaka M, Matsumoto J, Nishimoto Y. Tetrahedron: Asymmetry. 2002;13:1201–1208. [Google Scholar]
- 17.Iwona Kudyba I, Raczko J, Jurczak J. J Org Chem. 2004;69:2844–2850. doi: 10.1021/jo0358269. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh AK, Duong TT, McKee SP. Tetrahedron Lett. 1992;33:2781–2784. doi: 10.1016/S0040-4039(00)78856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Ghosh AK, Leshchenko S, Noetzel M. J Org Chem. 2004;69:7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]; (b) Ghosh AK, Gemma S, Takayama J, Baldridge A, Leshchenko-Yashchuk S, Miller HB, Wang YF, Kovalevsky AY, Koh Y, Weber IT, Mitsuya H. Org Biomol Chem. 2008;6:3703–3713. doi: 10.1039/b809178a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK, Gemma S, Baldridge A, Wang YF, Kovalevsky AY, Koh Y, Weber IT, Mitsuya H. J Med Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toth MV, Marshall GRA. Int J Pep Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 21.Vega S, Kang LW, Velazquez-Campoy A, Kiso Y, Amzel LM, Freire E. Proteins. 2004;55:594–602. doi: 10.1002/prot.20069. [DOI] [PubMed] [Google Scholar]
- 22.Kovalevski AY, Louis JM, Aniana A, Ghosh AK, Weber IT. J Mol Biol. 2008;384:178–192. doi: 10.1016/j.jmb.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]