

Abstract
The total synthesis of the proposed structure of anticancer agent, stereocalpin A is described. The synthesis features a diastereoselective synthesis of a 5-hydroxy-2,4-dimethyl-3-oxo-octanoic acid unit with asymmetric anti- and syn-aldol reactions as the key steps. Initial cycloamidation led to complete epimerization at the C-11 stereocenter due to unique steric constraints in the 12-membered depsipeptide ring. A late stage methylation strategy led to the synthesis of the proposed structure of stereocalpin A.
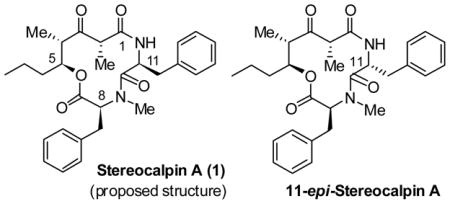
A variety of cyanobacterial metabolites have shown diverse biological properties including antitumor, antibiotic, antiviral, antimycobacterial, analgesic, and antipyretic properties.1 Stereocalpin A (1), a new cyclic depsipeptide, was isolated from the dry lichen, Ramalina terebrata, of Antarctica in 2008, by Oh et al. 2 Initial testing for cytotoxicity against three human solid tumor cell lines has shown good activity against human colon carcinoma cell lines (HT-29, IC50 = 6.5 μM), human skin carcinoma cell lines (B16F10, IC50 = 11.9 μM), and human liver carcinoma cell lines (HepG2, IC50 = 13.4 μM). In addition, stereocalpin A displayed a protein tyrosine phosphatase 1B (PTP1B) inhibitory activity in a dose-dependent manner with an IC50 value of 40 μM. Further biological investigations could not be carried out due to the lack of material.
The structure of stereocalpin A (1, Figure 1) was elucidated by extensive use of NMR and HPLC analyses of derivatives. Acid degradation (6 N HCl, 120 °C, 24 h) of 1 followed by derivatization with Marfey’s reagent3 and subsequent HPLC analysis revealed that stereocalpin A contains a L-Phe, a L-N-Me-Phe, and a 5-hydroxy-2,4-dimethyl-3-oxo-octanoic acid unit, which has not been previously reported as a component of any natural product. The absolute configuration of the octanoate derivative was resolved by a thorough analysis of NOESY data. The unique structure, interesting biological activity, and our interest in cyclic depsipeptides as antitumor agents4 led us to explore the chemistry and biology of stereocalpin A. Herein, we report our preliminary investigation leading to the stereoselective synthesis of the proposed structure of stereocalpin A and 11-epi-stereocalpin A. The present work suggested an incorrect assignment of the reported structure for stereocalpin A.2 Furthermore, we observed that the proposed structure has an unusual propensity to epimerize at the L-phenylalanine stereocenter.
Figure 1.

Retrosynthesis of Stereocalpin A.
As illustrated in Figure 1, our initial retrosynthetic analysis of stereocalpin A led us to disconnect the peptide bond between Phe and N-Me-Phe residues to provide amino acid derivative 2. The C2 chiral center is located between the C1 amide and C3 ketone, making this chiral center prone to epimerization. Therefore, we planned to keep the C3 ketone as a protected alcohol that will be oxidized into a ketone at the end of the synthesis. The acid 2 can be further disconnected to provide the octanoic acid subunit 5, Cbz-N-Me-Phe 4, and Phe-OMe 3. We planned to synthesize subunit 5 using Ghosh’s TiCl4 promoted anti-aldol reaction and Evans’ syn-aldol reaction as the key steps.
As depicted in Scheme 1, we utilized an ester-derived titanium enolate-based highly diastereoselective anti-aldol reaction to install the C4 and C5 stereocenters of stereocalpin A. 5 In a slightly modified protocol, tosylaminoindanol ester 6 was treated with TiCl4 (1.1 equiv) and diisopropylethylamine (3.8 equiv) in CH2Cl2 at 23 °C for 2 h. Addition of the resulting titanium enolate to the premixed n-butyraldehyde (2 equiv), TiCl4 (2 equiv) and MeCN (2 equiv) at −78 °C afforded the anti-aldol product 7 in 65% yield. The 1H-NMR and 13C-NMR analysis determined the anti-aldol diastereoselectivity to be 37:1 for aldol adduct 7. Protection of the resulting alcohol as a benzyl ether followed by reductive cleavage of the chiral auxiliary with LAH afforded alcohol 8 in 85% yield over two steps.6 We then employed Evans’ syn-aldol reaction to install the C2 and C3-stereocenters of 5. Thus, Swern oxidation of 8 followed by syn-aldol7 reaction with chiral imide 9 generated aldol product 10 in 89% yield (7:1 dr). Protection of alcohol 10 as its MOM ether using MOMCl, DIPEA and DMAP afforded the corresponding MOM ether in 90% yield. The removal of chiral imide with LiOOH at 0 °C to 23 °C for 12 h provided the acid 11 in 65% yield. It was coupled with L-phenylalanine methyl ester to provide the corresponding amide in 90% yield.8 The benzyl protecting group was removed by catalytic hydrogenation over Pearlman’s catalyst in a mixture (1:1) of ethyl acetate and methanol. The resulting alcohol was esterified with Cbz-N-Me-phenylalanine 4 using DCC and DMAP to afford the key amino acid derivative 12 in 85% yield.
Scheme 1.

Synthesis of Cyclization Precursor 12
As shown in Scheme 2, saponification of amine ester 12 with lithium hydroxide in t-butanol/H2O at 0 °C for 2 h followed by exposure of the resulting acid to catalytic hydrogenation over Pd(OH)2 afforded the corresponding amino acid precursor for cycloamidation. Treatment of the resulting amino acid with HATU (1.2 equiv) and HOAt (2 equiv) in a mixture (5:1) of CH2Cl2 and DMF at 23 °C for 24 h resulted in the cycloamide 13 as a single product in 95% yield.9 To complete the synthesis, the MOM group in 13 was deprotected using dimethylboron bromide.10 Oxidation of the resulting alcohol with Dess-Martin periodinane11 afforded 14, the presumed structure of stereocalpin A. However, the 1H-NMR and 13C-NMR of 14 did not match the reported data for the natural stereocalpin A.2 To our surprise, the X-ray crystal structure of 14 (Figure 2) 12 , revealed that the C11-stereocenter of L-phenylalanine had completely epimerized to D-phenylalanine, as shown in Scheme 2. For further confirmation of the structure, cycloamide 14 was hydrolyzed with 6N HCl at 110 °C for 24 h. The resulting amino acids were exposed to Marfey’s reagent3 and the resulting products were analyzed by HPLC which confirmed the presence of D-phenylalanine and N-Me-L-phenylalanine. To avoid this epimerization, other coupling reagents, such as TBTU and BOP reagents, have been examined. However, in each case, the same epimerization product was obtained. Although partial epimerization of amino acid during coupling reactions has been reported, 13 a complete epimerization during a macrolactamization reaction is unprecendented. The observed facile epimerization is presumably due to developing steric compression between the C2 methyl, C3 MOM group, and the C11 benzyl group in the 12-membered cycloamide.
Scheme 2.

Synthesis of 11-epi-Stereocalpin A
Figure 2.
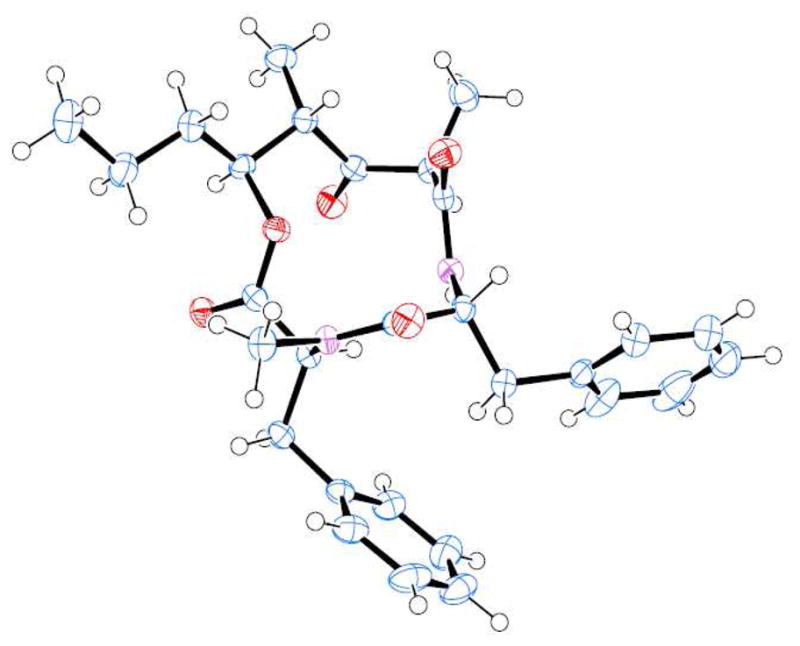
ORTEP drawing of Compound 14
Assuming that the C2 methyl and C3 MOM groups may be involved in facilitating the epimerization of L-phenylalanine during cyclization, we decided to assemble the methyl group at the C2 position at a later stage after the formation of the 12-membered ring. As outlined in Scheme 3, Swern oxidation of alcohol 8 followed by Mukaiyama aldol reaction of the resulting aldehyde with ketene acetal 15 afforded the corresponding aldol product14, the β-hydroxy ester 16 in 72% yield (5:1 dr). Saponification of methyl ester 16 with aqueous LiOH and 30% H2O2 afforded the corresponding acid which was coupled with L-phenylalanine methyl ester to provide the β-hydroxyamide derivative. Dess-Martin oxidation11 of the resulting β-hydroxyamide furnished amide 17 in 71% yield over two steps. Removal of the benzyl protecting group, followed by esterification of the resulting alcohol with N-Cbz-N-Me-phenylalanine afforded amino acid derivative 18 in 89% yield. Saponification of ester 18 with LiOH in aqueous t-butanol, and removal of the Cbz group by catalytic hydrogenation over Pd(OH)2 in a mixture (1:1) of EtOAc and MeOH, afforded the corresponding amino acid. The resulting amino acid was exposed to macrolactamization with HATU and HOAt in a mixture of CH2Cl2 and DMF (5:1) at 23 °C for 36 h as described above. The desired cycloamide 19 was formed in 47% yield, along with 10% of the C11-epimer 20. Methylation of 19 was carried out stereoselectively in the presence of cesium carbonate and methyl iodide in DMF for 24 h to afford the proposed stereocalpin A (1) in 50% yield (90% based on recovered starting material) as a single isomer. It appears that the alkylation presumably, proceeded from the less hindered face away from both benzyl and propyl substituents on the 12-membered ring. Interestingly, however, the spectral data of synthetic stereocalpin A (1) did not match with the data reported for the natural stereocalpin A.2 Our detailed structural analysis using 2D-NMR and NOESY of synthetic 1 fully supported our assignment of 1 as the proposed structure of stereocalpin A. Therefore, our stereocontrolled synthesis of the stereocalpin structure (1) now suggested that the structure of natural stereocalpin A had been assigned incorrectly. The comparison of NMR data is shown in the suporting information 1.
Scheme 3.

Synthesis of Stereocalpin A (proposed structure)
In conclusion, we have developed a short synthesis of the proposed structure of stereocalpin A. Our synthetic studies now ascertain that the original assignment of the stereocalpin A structure is incorrect. Interestingly, a combination of substituents, their stereochemistry and developing steric strain during the formation of a 12-membered depsipeptide ring led to an unprecedented complete epimerization at the C-11 amino acid center. Further investigation leading to the assignment of stereocalpin A’s structure and structure-activity studies are in progress.
Supplementary Material
Acknowledgments
Financial support by the National Institutes of Health is gratefully acknowledged. The authors thank Dr. Phillip E. Fanwick (Purdue University) for assistance with the X-ray crystal structure analysis.
Footnotes
Supporting Information Available: Experimental procedures and 1H- and 13C-NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Gupta RR. In: Marine Natural Products. Kiyota H, editor. Vol. 5. Springer; 2006. [Google Scholar]; (b) Burja AM, Banigs B, Abou-Mansour E, Burgess JG, Wright PC. Tetrahedron. 2001;57:9347. [Google Scholar]
- 2.Seo C, Kim JH, Lee HK, Park SM, Sohn JH, Oh H. Tetrahedron Lett. 2008;49:29. [Google Scholar]
- 3.Marfey P. Carlsberg Res Communi. 1984;49:51. [Google Scholar]
- 4.(a) Ghosh AK, Kulkarni S. Org Lett. 2008;10:3907. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Moon DK. Org Lett. 2007;9:2425. doi: 10.1021/ol070855h. [DOI] [PubMed] [Google Scholar]; (c) Ghosh AK, Xu X. Org Lett. 2004;6:2055. doi: 10.1021/ol049292p. [DOI] [PubMed] [Google Scholar]; (d) Bai R, Covell DG, Ghosh AK, Liu C, Hamel E. J Biol Chem. 2002;277:32165. doi: 10.1074/jbc.M205076200. [DOI] [PubMed] [Google Scholar]; (e) Ghosh AK, Liu C. Org Lett. 2001;3:635. doi: 10.1021/ol0100069. [DOI] [PubMed] [Google Scholar]; (f) Ghosh AK, Liu W, Xu Y, Chen Z. Angew Chem Int Ed. 1996;35:74. doi: 10.1002/anie.199600741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh AK, Onishi M. J Am Chem Soc. 1996;118:2527. doi: 10.1021/ja9539148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh AK, Liu C. J Am Chem Soc. 2003;125:2374. doi: 10.1021/ja021385j. [DOI] [PubMed] [Google Scholar]
- 7.Evans DA, Fitch DM. J Org Chem. 1997;62:454. doi: 10.1021/jo9621884. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Bilban M, Foster CA, Boger DL. J Am Chem Soc. 2002;124:5431. doi: 10.1021/ja020166v. [DOI] [PubMed] [Google Scholar]
- 9.Carpino LA. J Am Chem Soc. 1993;115:4397. [Google Scholar]
- 10.Olindon Y, Morton HE, Yoakim C. Tetrahedron Lett. 1983;24:3969. [Google Scholar]; Olindon Y, Yoakim C, Morton HE. J Org Chem. 1984;49:3912. [Google Scholar]
- 11.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277. [Google Scholar]
- 12.CCDC 724928 contains the supplementary crystallographic data for Compound 14. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 13.(a) Reszka P, Methling K, Lalk M, Xiao Z, Weiszc K, Bednarskia PJ. Tetrahedron:Asymmetry. 2008;19:49. [Google Scholar]; (b) Ehrlich A, Heyne HU, Winter R, Beyermann M, Haber H, Carpino LA, Bienert M. J Org Chem. 1996;61:8831. doi: 10.1021/jo951108d. [DOI] [PubMed] [Google Scholar]
- 14.(a) Mukaiyama T, Banno K, Narasaka K. J Am Chem Soc. 1974;96:7503. [Google Scholar]; (b) Paterson I, Smith JD. J Org Chem. 1992;57:3261. [Google Scholar]; (c) Paterson I, Smith JD, Ward RA. Tetrahedron. 1995;51:9413. [Google Scholar]; (d) Mutou T, Suenaga K, Fujita T, Itoh T, Takada N, Hayamizu K, Kigoshi H, Yamada K. Synlett. 1997:199. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.