

Abstract
The pathogenesis of multiple sclerosis (MS) remains elusive. Recent reports advocate greater involvement of B cells and immunoglobulins in the initiation and propagation of MS lesions at different stages of their ontogeny. The key role of B cells and immunoglobulins in pathogenesis was initially identified by studies in which patients whose fulminant attacks of demyelination did not respond to steroids experienced remarkable functional improvement following plasma exchange. The positive response to Rituximab in Phase II clinical trials of relapsing-remitting MS confirms the role of B cells. The critical question is how B cells contribute to MS. In this paper, we discuss both the deleterious and the beneficial roles of B cells and immunoglobulins in MS lesions. We provide alternative hypotheses to explain both damaging and protective antibody responses.
1. Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS) with varied clinical presentations and heterogeneous histopathological features. According to the National MS Society, approximately 400,000 people have been diagnosed with MS in the United States, with approximately 200 new cases every week. The main pathologic hallmark of MS is the demyelinated plaque, which has specific histological and immunocytological characteristics depending on the activity of the disease [1–4]. Another important immunopathological feature of MS is continuous synthesis of immunoglobulins (oligoclonal IgG's) in cerebrospinal fluid (CSF). The evidence associating antibodies with MS derives from a study by Kabat et al., who first described increased levels of immunoglobulin (Ig) in the cerebrospinal fluid (CSF) [5]. For many years, self-reactive antibodies have been associated with the pathogenesis of MS, and their presence was usually linked to demyelination based on studies done in experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of MS [6].
EAE is induced in animals by intradermal injection with whole or parts of various proteins that make up myelin. It is primordial to understand that EAE is not MS; it only resembles some forms and stages of MS. Hence, a number of significant assumptions made when proposing EAE as MS may only be partially true. Evidence from EAE studies favoring the role of antibodies in demyelination derived from three studies, (i) serum from animals with EAE produced periventricular demyelinating lesions when injected directly into the ventricles of normal animals, [7, 8], (ii) gamma-globulin was deposited in the CNS before the appearance of cells [9]; and (iii) serum from animals with EAE produced by sensitization with whole CNS-induced demyelination of CNS tissue cultures [10]. A feature that distinguishes species is the structural and functional heterogeneity of antibodies in higher species such as mammals. EAE is commonly studied in rodents and is mostly dependent on cellular immune responses even if antibodies worsen the disease, with the exception of models where pathogenic Th2 responses play a pivotal role. The reader should understand that the heterogeneity of antibody response and the diversity in their pathogenicity was mostly suggested and best studied in primate models of EAE [11] that closely reflect adequate complexity found in humans. Two other animal models of MS are virus-induced or toxin-induced demyelination [6]. Using a mouse model of Theiler's virus-induced demyelination, our research group showed that immunoglobulins and complement are involved in the pathogenesis of demyelination [12]. In a later study, in humans, we reported on four different pathologic subtypes of active MS lesions among 83 biopsies and autopsies of MS patients [1]. One of the subtypes, Type II, demonstrated a prominent presence of antibodies and complement. Antibodies directed against self-antigens are frequently considered deleterious and may play a pathogenic role in certain autoimmune diseases. A conventional method to remove circulating autoantibodies in patients is termed plasma exchange (PLEX). PLEX is a procedure that involves separating the blood from plasma and exchanging the plasma with albumin and returning the cellular components back to the patient. The large molecular weight substances that are removed from plasma following PLEX remain a question of open debate although it is certain that immunoglobulins and immune complexes are a major component of the substances that are removed. Few studies described evidence that PLEX does more than just remove antibodies; PLEX (i) can alter immune system by affecting T helper type-1/T helper type-II balance of circulating peripheral lymphocytes [13], (ii) can shift Th2-dominant status to Th1-dominant status [14], (iii) can decrease cytokine levels [15], and (iv) can improve cell function when administered along with prednisone [16].
2. Effect of Plasma Exchange in Patients with Demyelinating Disorders
Our group demonstrated conclusively for the first time the detrimental effect of plasma components in inflammatory demyelinating diseases of the CNS; PLEX in acute episodes of fulminant CNS inflammatory demyelination, which did not respond to high-dose methylprednisolone, led to a marked neurologic improvement in 6 patients [18]. These results were later confirmed in a double-blind placebo-controlled (sham procedure) trial performed at the Mayo Clinic, which demonstrated that approximately 40% of patients with acute, severe neurological deficits caused by MS or other CNS inflammatory demyelinating diseases who failed to recover after treatment with high-dose corticosteroids have dramatic recovery following PLEX [19]. In other well-designed, controlled studies, the beneficial effect of PLEX was demonstrated in patients with MS [20–22]. A more recent study [23] from our group performed in 153 patients with acute, steroid-refractory CNS inflammatory demyelinating disease (CNS-IDD) demonstrated moderate to marked functional neurological improvement in 59% of the cases within 6 months following PLEX. However, PLEX was less effective for patients with multiple sclerosis who subsequently developed a progressive disease course (P = .046). Accordingly, a recently published evidence-based guideline update concluded that PLEX is (i) probably effective and should be considered as a second-line treatment of steroid-resistant exacerbations in relapsing forms of MS, (ii) possibly effective and may be considered for acute fulminant demyelinating CNS disease, and (iii) is ineffective and should not be considered for chronic or secondary progressive MS [24]. Treatment in relapsing-remitting MS includes azathioprine, intravenous immunoglobulin (IVIg), interferon β-1a, glatiramer acetate, mitoxantrone hydrochloride, natalizumab, and cyclophosphamide, depending on the disease severity. A randomized, double-blinded, placebo-controlled trial of IVIg in patients with MS who had persistent muscle weakness concluded that IVIg does not reverse established weakness in MS [25]. PLEX has not been specifically studied in relapsing-remitting MS. A traditional hampering factor remains to be the excessive cost burden on the patient to undergo PLEX procedure. Nevertheless, a recent estimate established that a direct cost of five IVIg infusion sessions totaling 2.0 grams per kilogram (g/kg) body weight was $10,329.85 compared to a series of five TPE procedures, which had direct costs of $4,638.16 [26]. Taking this into consideration, further studies to understand the beneficial effects of PLEX are warranted given that the optimal protocol and the duration of benefit remains un-established.
3. The Presence of B Cells in CNS
An MS plaque is histologically characterized by inflammation, demyelination, and gliosis. Infiltration by mononuclear cells, particularly macrophages and T cells, is typical of the acute MS lesion. Esiri showed that autopsy material from the brains and spinal cords of 23 MS patients showed that Ig-containing cells were more numerous within the plaques than outside and were more common in recent than in old plaques [27]. Subsequently, Cepok et al. showed that B cells account for up to 25% of the CSF-infiltrating leukocytes during CNS inflammatory responses [28]. To better understand the potential mechanism of B cells in MS pathology, Baranzini et al. had PCR amplified and analyzed by size, spectratyping, and sequencing the Ig heavy chain CDR3 repertoire in 10 MS brain samples. Their work showed a higher level of rearranged transcripts in MS brains when compared to the noninflammatory brain tissues, suggesting that oligoclonal bands found by spectratyping are the result of antibody synthesis within the plaques [29]. The above findings supported the hypothesis that a germinal center- (GC-) like reaction takes place during the immune attack against CNS structures. This concept was further strengthened by Colombo et al. [30] who detected oligoclonal B cell accumulations in 10 of 10 MS patients. Upon analyses of the Ig V(D)J sequences from the CSF of patients, it was found that variable heavy chain domains, VH3 and VH4 genes, were extensively mutated as compared to the germline sequences. Their data suggested a compartmentalized clonal expansion in MS. This, however, raised the question on the capability of the brain tissue to host a GC-like reaction. The works of Knopf et al. provided the answers: they used a rodent model with an intact blood-brain barrier and demonstrated the trafficking of activated antigen-specific B cells into the brain, retention, and antibody production [31]. Their data proved that the brain microenvironment supports the development of antigen-directed humoral immunity. To confirm this concept, Corcione et al., showed that each B-cell subset participating in the GC reaction could be detected in the CSF of MS patients [32].
4. Autoimmune Hypothesis and the Presence of Immunoglobulins in CNS
Autoimmunity is the most accepted theory for MS pathogenesis, as self-reactive antibodies have been implicated in the pathogenesis of MS. Previous studies suggested intrathecal production of antibodies occurred after clonal expansion in patients as seen by the consistent identification of oligoclonal bands after CSF electrophoresis [33]. We reported on the heterogeneity of MS lesions in CNS tissue and their implications for the pathogenesis of demyelination. We performed a detailed immunohistochemical study of active MS lesions from 83 biopsies and autopsies of MS patients, following which we identified four different pathologic subtypes of active MS lesions. One of the subtypes, Type II, demonstrated the presence of macrophages and T-cells and, in addition, a prominent display of antibodies and complement [1]. Numerous publications during the last few decades supported the idea that CSF oligoclonal bands correlate to the level of B-cell involvement in MS [34]. In addition, evidence indicates that oligoclonal bands may have a prognostic value. One prospective study of patients with acute isolated demyelinating episode demonstrated intrathecal immunoglobulin synthesis to be a better predictor of MS progression than MRI [35]. Another prospective study showed that the presence of CSF oligoclonal bands in early MS generally correlated to a worse outcome [36]. A recent study showed strong correlation between levels of oligoclonal bands (OCBs) and prognosis for MS disability [37].
Our work [1, 18, 19] and others' results [38, 39] favor antibody-mediated demyelination in MS. On the contrary, in a recent study comparing the immunoglobulins and activated complement in MS and other neurologic diseases (ONDs) patients, the authors did not find IgG in myelin or ramified microglia from tissue remote to focal lesions [40]. Interestingly, C3d and C9neo stained positively for all IgG from MS and ONDs. The only MS-specific IgG deposition they found consisted of unusual microglial nodules containing short, linear deposits of activated complement (C3d) on partially demyelinated axons located in normal-appearing periplaque white matter. This study thus argued that the IgG and complement immunostaining of disrupted myelin in MS lesions is a nonspecific feature that cannot be interpreted as evidence of a distinct pathogenesis or serve to define particular variants of the disease. However, it is questionable whether many of the lesions in the study were as acute as those in the studies performed at Mayo [1].
In the past, Avrameas proposed that autoreactive antibodies present in healthy humans have natural physiological roles [41]. In line with this, we have shown that immunoglobulins directed against antigens of the lipid rafts of oligodendrocytes promote remyelination in CNS. Systemic injection of serum molecules from donor mice hyperimmunized with homogenized spinal cord induced the remyelination of CNS axons. This was the first evidence that immunoglobulins secreted in demyelinating lesions may have the potential to promote myelin repair [42]. We have further demonstrated that human immunoglobulins, when used to treat animal models of disease, can promote remyelination [43] and neurite extension [44]. Protecting axons of the CNS promises to be an effective strategy to limit axon loss and prevent permanent disability. Thus, there is a delicate balance between the presence of beneficial and deleterious immunoglobulins at the site of active lesion.
5. Antigen Specificity of Autoantibodies Found in MS
After several years of research, confirmation of the antigen-specificity of autoreactive antibodies in MS is still lacking. Due to their broad reactivity, IgG in CSF of patients with MS may represent synthesis of “nonsense” antibodies irrelevant to pathogenesis [45–47]. However, other experiments found molecular uniformity and temporal persistence of the Ig response in MS, thus conflicting with the nonsense antibody proposal [48]. It is possible that relevant molecules are limited to the myelin sheath. Warren and Catz studied the specificities of autoantibodies from MS patients and reported that most patients' IgG bound to MBP. They also reported that the peptide MBP [49–63] strongly inhibited autoantibody binding to MBP in almost all cases tested [64]. Genain et al. offered the most convincing evidence. Using immunogold-labeled peptides of myelin antigens and high-resolution microscopy (techniques that can detect antigen-specific antibodies in situ), they identified autoantibodies specific for the CNS myelin antigen myelin/oligodendrocyte glycoprotein (MOG). These autoantibodies specifically bound to disintegrating myelin around axons in acute MS lesions and the marmoset model of EAE [65]. O'Connor et al. examined whether autoantibodies that bind properly folded MOG protein are present in the CNS parenchyma of MS patients. Their data demonstrated that MOG-recognizing autoantibodies are present in substantially higher concentrations in the CNS parenchyma of MS patients than in the CSF and serum. This indicated that local production/accumulation is an important aspect of autoantibody-mediated pathology in CNS demyelinating diseases [66]. Further studies demonstrated the serological and/or CSF presence of antibodies directed against MBP and/or MOG in patients with MS [67].
Conversely, myelin-specific antibodies are not limited to MS. Using an enzyme-linked immunosorbent assay, Karni et al. compared levels and frequencies of anti-MOG antibody between patients with MS, patients with ONDs and healthy control subjects. Interestingly, the authors found higher plasma levels of antibodies to MOG and to MBP in MS patients compared with ONDs patients; however, the frequency of antibodies to MOG and MBP was similar in MS, ONDs and healthy controls [68]. Another group presented analogous results. Lampasona et al. used a liquid-phase radiobinding assay to measure serum anti-MOG IgG among 87 MS patients with MS, 12 encephalomyelitis patients and 47 healthy subjects. Surprisingly, the frequency of positive samples with low titers of anti-MOG IgG was similar in all the groups and subgroups. Binding-competition experiments showed that low affinity in these antibodies. These results demonstrated that anti-MOG antibodies are not disease specific [69].
In a parallel line of research, some reports suggested lipids or carbohydrates as possible candidate antigens for the humoral immune response. Arnon et al. reported the presence of anti-ganglioside antibodies in MS [70]. Endo et al. reported the presence of antibodies to glycosphingolipids in MS patients [71]. Their results indicated that antibodies against ganglioside GM1 and asialo GM1 were found commonly in 34 of 46 patients with MS. However, antilipid antibodies can be found in other diseases, such as systemic lupus erythematosus and stroke [72, 73]. An interesting report identified anti-alpha-glucose-based glycan IgM antibodies as predictors of relapse activity in MS after the first neurological event [74]. Others suggested that serum anti-Glc(alpha1, 4)Glc(alpha) antibodies serve as biomarkers for relapsing—remitting MS [75]. Villar et al. reported that the intrathecal synthesis of oligoclonal IgM against myelin lipids may predict an aggressive disease course in MS [76]. The most frequently recognized lipid in their study was phosphatidylcholine. Autoantibodies to myelin proteins, lipids and carbohydrates can be extracted from the tissue and sera of some MS patients.
A major discovery in demyelinating disorders was when Hinson et al. [77–79] discovered a potential pathogenic immunoglobulin G binding to the extracellular domain of a water channel aquaporin-4 (AQP4) in patients with neuromyelitis optica (NMO). Potential pathogenic immunoglobulins appear in approximately 70% of patients with NMO. It is unknown whether a detection problem limits the assay in the remaining patients or if an autoantibody to a different antigen drives the response. However, no clinical differences exist between antibody-positive and antibody-negative patients with clinical NMO. A randomized trial to determine whether IVIg reverses chronic visual impairment in MS patients with optic neuritis (ON) demonstrated that IVIg administration does not reverse persistent visual loss from ON to a degree that merits general use [80]. Interestingly, PLEX is a highly successful treatment for NMO arguing in favor of an autoimmune-mediated pathogenesis of this disease. In line with the autoimmune-mediated hypothesis, humoral immunity-suppressing drugs such as Mitoxantrone hydrochloride [81], (a synthetic anthracenedione that was approved for the treatment of worsening relapsing-remitting and secondary progressive MS), Mycophenolate Mofetil [82], (an immunosuppressive therapy), and Rituximab [83], (a B cell depleting therapy) were demonstrated to be beneficial for treatment of NMO.
It is now considered that NMO is likely the first true demyelinating autoimmune disorder. Autoantibodies may contribute to the ongoing immune response, but after more than 50 years of investigation in the field, one still cannot substantiate all the criteria necessary to classify MS as an autoimmune disease [84]. In light of this, the autoimmunehypothesis requires a complementary hypothesis/reason for pathogenesis of MS.
6. Alternate Hypothesis of MS Pathogenesis
MS is a complex and heterogeneous disease; hence, in addition to the autoimmune hypothesis, it is assumed that certain infectious agents play an important role in the pathogenesis. Epstein-Barr virus (EBV), human herpes virus-6 (HHV-6), varicella zoster virus (VZV), and Chlamydia pneumonia are some of the proposed infectious agents. Many studies in this area have demonstrated the presence of antibody titers to a broad range of pathogens in MS patients; however, many of these findings remain solitary and unconfirmed by other groups. EBV is a B-lymphotropic human DNA herpes virus that infects most individuals asymptomatically but causes infectious mononucleosis (IM) in some [85, 86]. Cepok et al. identified EBV proteins as putative targets of the immune response in MS [87]. Another study demonstrated the increased risk of MS in individuals with a clinical history of IM [88, 89]. Recently, researchers from the United Kingdom studied the prevalence of MS and infectious mononucleosis (IM) and how they relate to ultraviolet B (UVB) exposure [90]. As previously shown in other studies, MS highly correlated with IM [91, 92]. As a control, the authors also examined correlations of MS with cytomegalovirus prevalence and varicella prevalence, respectively, both of which were nonsignificant. Of note, the authors found that UVB in any season correlated more closely with MS than with IM. These results fit well with the EBV hypothesis, because there may be a mechanism through which UVB radiation mediates MS risk.
It has been suggested that low vitamin D levels as a result of immunosuppression lead to an increase in EBV infection. It is also known that a low amount of UVB decreases vitamin D levels. The geographical variation in the MS prevalence, with a higher prevalence of the disease in northern latitudes and a lower prevalence at the equator, is well established [93–95]. This variation in MS prevalence correlates positively with changes in the serum concentrations of 25-hydroxyvitamin D [96–98]. Several, but not all, studies show an inverse correlation between serum 25-hydroxyvitamin D concentrations and the incidence of MS, the severity and progression of disease [49–61]. Vitamin D, and its biologically active metabolite 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), not only plays an important role in the regulation of calcium and phosphorus homeostasis, but also is an important modulator of immune function. 1,25(OH)2D3 functions by associating with the vitamin D receptor that is widely distributed in a number of calcium-transporting tissues, neural tissues, and immune cells (dendritic cells, T-lymphocytes, B-lymphocytes, and macrophages) [62, 63, 99–104]. 1,25(OH)2D3 increases macrophage activity, inhibits dendritic cell maturation, inhibits B-cell functions, and favors the production of T-helper2 cells, thereby shifting the ratio of Th1/Th2 cells in favor of Th2 helper cells (Figure 1) [17, 105–111]. The polarization of activated CD4 + T-cells to a Th-1 phenotype (IL-2, IFNγ, TNFα and secretion) or to a Th-2 phenotype (IL-4, 5, 13, and 10 secretion) represents a major determinant of the nature of subsequent cellular and humoral immune responses. It is a self-perpetuating process in that one subtype inhibits the generation of the other [108, 110]. The primary generation of Th-1-type T-cell responses is potently inhibited by 1,25(OH)2D3 both in vitro and in vivo. 1,25(OH)2D3 also induces production of human cathelicidin, LL-37, which is particularly effective against respiratory viruses such as influenza [112]. The lack of vitamin D may result in abnormal response to EBV infection causing IM, thereby leading to a higher risk for MS [113].
Figure 1.
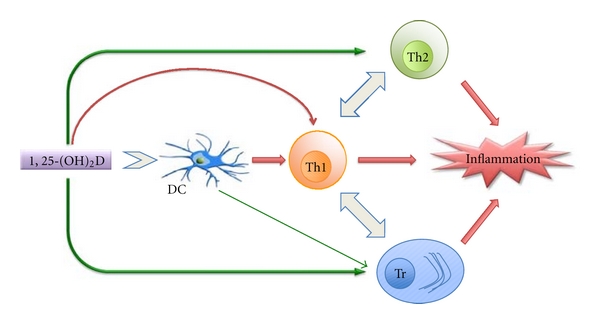
The in vitro effects of 1,25(OH)2D on the immune system. The effects of 1,25(OH)2D either directly or indirectly are depicted by arrows. While a green arrow represents positive influence, a red arrow represents the negative influence. The negative influence on inflammation indicates dampening of the inflammatory response. DC: dendritic cell, Th1: T helper type 1 lymphocyte, Th2: T helper type 2 lymphocyte, Tr: regulatory T lymphocyte [17].
7. Role of B Cells
7.1. As Antigen-Presenting Cells
The role of B cells in acute demyelination was discussed in the previous sections (accumulation of clonally expanded B cells, neuropathological studies, and presence of complement). Besides their role in acute demyelination, B cells can contribute to the disease progression in MS [114]. It was previously shown that epitope specificities generated in a mouse model of MS, EAE, overlap with encephalitogenic T cell epitopes, and human immune-dominant T and B cell epitopes [115–117]. Harp et al. studied the impact of myelin-specific antigen-presenting B cells on T cell activation in MS. Their results demonstrated a possible role of B cells as myelin-specific antigen-presenting cells (B-APCs) in MS [118].
Another treatment strategy used is Rituximab, an anti-CD20 monoclonal antibody. CD20 is a surface antigen with restricted expression on pre-B and mature B cells, but it is not expressed on stem cells and cells differentiating into plasma cells. Recent reports showed some favorable results of Rituximab in relapsing-remitting MS patients, as the administration of this drug reduced inflammatory brain MRI lesions and clinical relapses for 48 weeks in patients, after a single dose course [119, 120]. A study by Cross et al. showed that Rituximab reduces B and T cells in CSF of MS patients [121]. However, the rapid effect of Rituximab is not explained by the fact that it does not act on plasma cells or cells differentiating into plasma cells. Rituximab may target the processes such as antigen presentation by B cells and activation of T cells. This may affect the production of proinflammatory and regulatory cytokines in the CNS microenvironment. In line with this, a recent study in MS patients on Rituximab demonstrated that the episodic triggering of abnormal B-cell cytokine responses mediated “bystander activation” of disease-relevant proinflammatory T cells [122].
7.2. As Cytokine-Producing Cells
As described before, the study of Bar-Or et al. proposed that abnormal B-cell cytokine responses were responsible for new MS relapses through bystander activation of relevant proinflammatory T-cells [122]. This finding was important and may explain the established association between new MS relapses and infections. The importance of balanced proinflammatory and anti-inflammatory B-cell cytokines was demonstrated when selective deletion of IL-10 from B-cells aggravated clinical disease in EAE [123]. In addition, a recent study showed that IL-10 secretion by B-cells was deficient in MS patients as compared to healthy controls [124]. However, levels of IL-10 production were equivalent in patients with relapsing-remitting and secondary progressive MS.
8. Mechanisms of Action of MS Antibodies
Antibodies are the antigen-binding proteins present on the B-cell membrane and are secreted by the plasma cells. Membrane-bound antibody confers antigenic specificity on B cells; antigen-specific proliferation of B-cell clones is elicited by the interaction of membrane antibody with antigen. The versatility of secreted antibodies is demonstrated by the functions they mediate such as neutralization, agglutination, fixation with activation of complement, and activation of effector cells. Among the antibody functions is a novel property of the antibodies that fascinated scientists for several years. It is the ability of some antibodies to behave as enzymes with catalytic ability. Catalytic immunoglobulins, both of the IgM and IgG isotypes, have been detected in the serum of healthy donors, where they presumably participate in removing metabolic wastes and defend the organism against invading pathogens. Conversely, antigen-specific hydrolytic IgGs are seen in a number of inflammatory, autoimmune, and neoplastic disorders; their pathogenic effects have been demonstrated occasionally. Studies during the past decades have shown antibodies to be capable of participating in at least one of the functions presented below.
8.1. Functions of Immunoglobulins
(1) Neutralization —
The Fab region of the antibodies binds to the target (viruses, microbes [125], and/or toxins [126]) and blocks or neutralizes their action. There is some evidence that antibodies present in plaques bind to MBP and/or MOG and neutralize and cause deterioration of the tissue; however, the results remain controversial due to the presence of antibodies with similar specificities in healthy blood donors.
(2) Agglutination —
Antibodies are clonally generated for binding single specific antigens. The Fab regions of the antibodies link the antigens together, causing them to clump together, also known as agglutination [125]. Monoclonal antibodies derived from MS patients' agglutinated liposomes made from lipids of a chloroform/methanol extract of human myelin. Investigations by ELISA suggest that phospholipids are the reactive components, at least for some of these mAbs. Some antibodies reacted with liposomes containing galactocerebroside or sulfatide, others only with sulfatide-containing liposomes. However, there is no reported evidence for role of this binding for MS [127].
(3) Fixation and Activation of Complement —
The antibodies that bind to surface antigens on, for example, a macromolecular structure, will attract the first component of the complement cascade with their Fc region and initiate the activation of the “classical” complement system [128]. This results in the destruction of the molecule [125]. We previously described complement deposition in MS plaques [1]. Complement activation is known to occur in white-matter MS lesions. Storch et al. described a case of MS characterized by deposition of immunoglobulin and complement in the areas of active demyelination. In addition, macrophages in the lesions contained degradation products that were immunoreactive for myelin antigens and immunoglobulins. They observed that the destruction of myelin sheaths was associated with incomplete loss of oligodendrocytes in the active areas and reappearance of oligodendrocytes with remyelination in the inactive plaque center [129]. Finally, Piddlesden et al. showed that the demyelinating potential of antibodies to MOG was related to their ability to fix complement [130].
(4) Activation of Effector Cells —
Mast cells and phagocytes have Fc receptors that interact with the Fc region of IgA, IgG, and IgE antibodies. The engagement of a particular antibody with the Fc receptor on a particular cell triggers the effector function of that cell (e.g., phagocytes will phagocytose and mast cells degranulate) that will ultimately result in destruction of the invading microbe. One or more FcRs are expressed in microglia, astrocytes, oligodendrocytes, and neurons. Aberrant activation of FcRs in such neural cells may contribute to MS pathogenesis [131]. Antimyelin antibodies may mediate damage to myelin membranes through separate mechanisms such as receptor-mediated phagocytosis by macrophages and/or presentation of myelin autoantigens to specific T cells [65]. Autoantibodies may activate mast cells and cause their degranulation. The concept of mast cells in MS dates back to more than a century. Neumann in 1890 described the first association of mast cells to MS plaques [132]. Olsson identified mast cells in MS plaques, in 1974 [133]. Kruger et al. suggested in 1990 that mast cells contribute to the demyelinating process of MS [134]. A definite role for mast cells was finally demonstrated using the mast cell-deficient mice. These mice exhibited delayed onset and decreased EAE disease severity when compared to wild-type littermates [135].
(5) Effector Functions of Immunoglobulins —
The Fc region mediates effector functions, such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). In ADCC, the Fc region of an antibody binds to Fc receptors (FcγRs) on the surface of immune effector cells such as natural killers and macrophages, leading to the phagocytosis or lysis of the targeted cells. In CDC, antibodies kill the targeted cells by triggering the complement cascade at the cell surface. Frick and Stickl demonstrated that antibodies from sera of MS patients enable normal lymphocytes to exhibit a cytotoxic reaction against MBP [136, 137]. Later, Merrill et al. compared the peripheral blood lymphocytes (PBL) of MS patients to age- and gender-matched controls for ability to mediate ADCC. They demonstrated an enhanced ADCC of blood lymphocytes of MS patients [138].
(6) Catalytic Activity of Ig —
In most of the above-described functions of Ig, the antibodies tightly bind the antigen but does not specifically alter its chemical nature. Natural enzymes within the body bind biomolecules and subsequently catalyze their conversion to new products. Within the enzyme itself, the amino acid sequences that form the active site determine specificity and the efficiency. Considering the huge variability of the CDR regions, it is possible to find similar amino acid sequences in the hypervariable zone of the Igs, which could confer an enzymatic function to the antibody. Indeed, antibodies that catalyze a wide variety of reactions have been reported. The first description of catalytic antibodies to MBP was documented in SJL mice with EAE [139]. Since then, hydrolytic anti-MBP antibodies were isolated from MS patients [140]. Six preferential cleavage sites of the MBP molecule were identified by abzymes isolated from patients with MS [140, 141]. All the identified cleavage sites were located c-terminus to either arginine or lysine residues, thus providing an insight into the possible mechanism of action of abzymes (a serine protease-like activity). Aprotinin, a binding inhibitor of trypsin, could not block abzyme-mediated hydrolysis. The mechanism of action of these abzymes in comparison to conventional serine proteases would be of interest to protein biochemists studying structure-function relationship of proteins.
(7) Remyelinating Antibodies —
The discovery of the function of natural autoantibodies for CNS remyelination by our group was serendipitous. At the point of discovery, we were trying to induce more demyelination in an animal model of demyelinating disease (Theiler's virus infection) to test the hypothesis of virus-induced autoimmunity. However, when we immunized animals with myelin months after Theiler's virus infection, as opposed to extensive demyelination [142], we observed extensive remyelination [42]. As a result, we performed a classical passive transfer experiment in which we transferred antisera or purified immunoglobulins from uninfected animals immunized with myelin antigens into animals with extensive chronic demyelination following Theiler's infection [143, 144]. Of interest, the animals receiving hyperimmune sera or immunoglobulin directed against myelin showed extensive remyelination in contrast to animals that received the control antisera. Our research group then identified 8 different mouse mAbs that promoted remyelination [145]. Of note, each mAb bound unique antigens or to the surface of oligodendrocytes [146]. They all had relatively conserved germ line sequences [147, 148]. Based on this observation, we asked whether natural autoantibodies were present in the human population. We sought patients with disease processes that cause them to make their own mAbs, specifically patients with multiple myeloma, Waldenstrom's syndrome and monoclonal gammopathy of unknown significance. The serum samples from these patients were screened for binding to sliced live cerebellum and, specifically, to oligodendrocytes in culture. Two mAbs that we screened, sHIgM22 and sHIgM46, demonstrated very extensive remyelination [43] in both Theiler's virus and in lysolecithin-induced demyelination [149]. We also identified antibodies (sHIgM12 and sHIgM42) that support neurite extension in vitro as well as in the potent substrate laminin and override the neurite outgrowth inhibition of CNS myelin [44]. sHIgM22 [150] and sHIgM12 [151] are now available as recombinant IgM molecules. These results provide strong evidence for a beneficial response of antibodies in demyelinating diseases.
9. Role of Immunoglobulins
9.1. Deleterious Role
B cells also produce myelin-specific antibodies that bind to and destroy myelin within the plaque through an enzymatic mechanism. In this respect, catalytic antibodies have been reported to play a role in site-specific myelin destruction [139, 140]. The hydrolytic activity of the anti-MBP antibodies correlated with the expanded disability status [152]. Belogurov et al. [39] reported on the recognition and degradation of MBP peptides by serum autoantibodies. The authors used a constructed MBP-derived recombinant “epitope library” that spanned the entire MBP molecule to define the epitope-binding/ cleaving activities of autoantibodies isolated from the sera of 26 MS patients and 11 healthy individuals. The levels of autoantibodies to MBP fragments, as well as to whole MBP and myelin oligodendrocyte glycoprotein (MOG) molecules, were significantly higher in the sera of MS patients than in those of healthy donors. Patients with MS (77% of progressive and 85% of relapsing-remitting) were positive for catalysis, showing pronounced epitope specificity to the encephalitogenic MBP peptide 81–103. No healthy donors presented with these characteristics. Based on the results, anti-MBP binding and cleavage by abzymes may be regarded as a specific feature of MS as compared to healthy donors and may provide an additional marker of disease progression.
9.2. Beneficial Role
Our group reported on the identification of monoclonal IgM antibodies with remyelinating (sHIgM22 and sHIgM46) and neuroprotective (sHIgM12 and sHIgM42) properties. We have demonstrated that rHIgM22 binds to oligodendrocytes and myelin and promotes CNS remyelination in virus- and toxin-induced models of MS. Spinal cord remyelination is induced after a single low dose (25 μg/mL) of rHIgM22 [153]. It is remarkable that one peripheral (i.p.) treatment with a short-lived molecule (15 hr half life in mice) promotes maximal tissue repair within 5 weeks in a model of MS with little spontaneous repair. The properties of rHIgM22 are similar to a targetable growth factor. After peripheral injection, rHIgM22 crosses the blood brain barrier (BBB) and accumulates within brain and spinal cord lesions of mice with demyelination. Ferritin bead-labeled rHIgM22 has been detected in lesions in vivo by magnetic resonance imaging showing that the antibodies can readily cross the BBB [154]. In addition, repair-promoting antibodies induced distinct Ca2+ signals in both astrocytes and oligodendrocytes. The antibody's ability to induce Ca2+ signals is statistically correlated with promotion of myelin repair [155]. In addition, we showed that rHIgM22 strongly inhibits apoptotic signaling via reduction of caspase-3 and caspase-9 cleavage and reduces expression of differentiation markers MBP and MOG in oligodendrocyte cultures. We also documented that remyelination-promoting human IgMs protect oligodendrocytes in culture from peroxide-induced activation of caspase 3 [156], a marker of active apoptosis. Our group then identified Lyn kinase as a key player in rHIgM22-mediated effects in OLs. We isolated integrin αvβ3 and PDGFαR in a complex together with Lyn kinase, suggesting that rHIgM22 acts through a signaling complex containing Lyn, integrin αvβ3, and PDGFαR in oligodendrocytes. This implies that IgM-mediated remyelination is due to protection of oligodendrocyte progenitor cells and oligodendrocytes rather than promotion of OPC differentiation [157].
10. Conclusions
Over the past decades, accumulated evidence emphasized the role of immunoglobulins in MS. The antibodies are characterized with both detrimental as well as beneficial roles. A number of fundamental questions related to important problems of immunoglobulins in MS still have ambiguous answers. New experimental design strategies and technologies will help answer these questions more precisely. A pivotal question is why some immunoglobulins cause deleterious effects, whereas others are beneficial. Is antigen recognition a unique attribute that determines this role? This seems unlikely given that myelin-specific antibodies are reported from both MS patients and healthy controls [158].
We propose that their microenvironment determines whether immunoglobulins assume a pathogenic or reparative role. Antibodies play a deleterious role when bound to myelin and result in its degradation [140]. Alternatively, antimyelin antibodies clear myelin debris from sites of acute degradation to promote remyelination [159]. Another factor may be the intracellular events mediated in the target cell upon binding. We have shown that polyclonal Ig treatment strategies, such as IVIg or IVIgM, promote CNS repair via Fc-mediated activation of microglia and stimulation of IL-1β release. The interaction of Fcγ or Fcμ with receptors on microglia may initiate a cascade of events culminating in IL-1β-induced oligodendrocyte progenitor maturation and subsequent remyelination of demyelinated lesions [160]. These observations support the hypothesis that the oligoclonal immune response [27, 28, 32] found in the CSF and CNS parenchyma of MS patients with MS may represent a powerful endogenous repair system. The evidence of antimyelin antibodies in healthy donors again supports the idea that these responses are not pathogenic but may be part of an endogenous repair system.
All approved treatments for MS have targeted T-cells. Because of the clear role of B-cells in MS pathogenesis, there is a need to develop strategies to target this cell type. Treatment of MS patients with either B cell-depleting therapies, such as the administration of Rituximab or PLEX, has had very significant positive effects on a small percentage of the MS population. However, the majority of patients show minimal response to these treatments. In light of these findings, we consider it imperative to develop in vitro laboratory assays to distinguish, which patients respond to strategies to deplete B cells or remove immunoglobulins. Similarly, it will be critical to determine which patients respond to a human monoclonal antibody to promote remyelination. The results from these assays can be used to select or optimize patient's preventive or therapeutic care. This is especially important for patients with progressive forms of MS or patients who have not responded to any currently available treatments. For instance, PLEX could be considered as a first line of treatment for patients with injurious or deleterious immunoglobulins. Conversely, in patients with axonal preservation, an endogenous remyelination-enhancing approach may be appropriate. Ultimately, this supports the concept of “individualized medicine” (Figure 2). However, the paucity of available data from serology, genomics, proteomics, and metabolomics from large cohorts of MS patients remains a major limitation.
Figure 2.
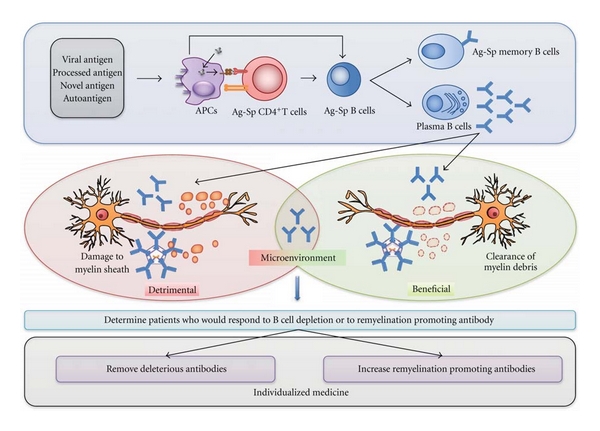
The role of B cells in MS pathogenesis. Antigens (viral, processed, novel, or auto) are internalized and presented via antigen presenting cells (APC's, such as dendritic cells, microglia and other mononuclear phagocyte system cells) to antigen-specific naïve T cells and/or B cells. B cells can sometimes act as APC's. Activated B cells undergo clonal expansion and mature into either antigen-specific memory B cells or into plasma cells that secrete antigen-specific antibodies that may have a detrimental (further damage to myelin sheaths of axons) or a beneficial effect (remyelination, clearing the metabolic wastes away, and neurite extension), depending on the microenvironment. The oligoclonal immune response observed in patients with MS thus represents an ambivalent role. It is of priority to determine clinical assays to delineate patients who would respond to B cell depletion therapies, or to remyelination promoting antibody therapy. This approach supports the concept of “individualized medicine”, where deleterious antibodies would be removed from circulation or in other cases endogenous remyelination would be promoted.
Disclosures
The technology for remyelination-promoting antibody rHIgM22 has been licensed to Acorda Therapeutics, Inc. No royalties have accrued to M. Rodriguez or Mayo Clinic to date, but both have rights to receive future royalties.
Acknowledgments
This work was supported by grants from the National Institutes of Health (no. R01s-GM092993, NS024180, NS032129, NS048357, and R21-NS073684), the National Multiple Sclerosis Society (CA1060A11), the Applebaum Foundation, the Hilton Foundation, the Peterson Foundation, and the Minnesota Partnership for Biotechnology and Medical Genomics. B. Wootla is the recipient of a fellowship from the Mayo-Applebaum funds. D. Astapenko is supported through an educational exchange program sponsored by Department of Histology and Embryology, Charles University in Prague. The authors thank Lea Dacy for editorial assistance.
References
- 1.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Annals of Neurology. 2000;47(6):707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 2.Frohman EM, Racke MK, Raine CS. Multiple sclerosis—the plaque and its pathogenesis. New England Journal of Medicine. 2006;354(9):942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 3.Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends in Molecular Medicine. 2001;7(3):115–121. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- 4.Wekerle H. Immune pathogenesis of multiple sclerosis. Neurological Sciences. 2005;26(supplement 1):S1–S2. doi: 10.1007/s10072-005-0386-9. [DOI] [PubMed] [Google Scholar]
- 5.Kabat EA, Moore DH, Landow H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. Journal of Clinical Investigation. 1942;21:571–577. doi: 10.1172/JCI101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denic A, Johnson AJ, Bieber AJ, Warrington AE, Rodriguez M, Pirko I. The relevance of animal models in multiple sclerosis research. Pathophysiology. 2011;18(1):21–29. doi: 10.1016/j.pathophys.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jankoovic BD, Rakic IM, Janjic M, Ivanus J, Mitrovic K. Effect of experimental allergic encephalomyelitis gamma-globulin upon the electrical activity of the brain. Experientia. 1966;22:459–460. doi: 10.1007/BF01900983. [DOI] [PubMed] [Google Scholar]
- 8.Jankoovic BD, Rakic IM, Janjic M, Mitrovic K, Ivanus J. Immuno-neurophysiological studies of the experimental allergic encephalomyelitis following the injection of antibrain antibodies and myelin protein into the cerebral cavity. Path Europ. 1966;2:p. 87. [Google Scholar]
- 9.Oldstone MB, Dixon FJ. Immunohistochemical study of allergic encephalomyelitis. American Journal of Pathology. 1968;52(2):251–263. [PMC free article] [PubMed] [Google Scholar]
- 10.Appel SH, Bornstein MB. The Application of tissue culture to the study of experimental allergic encephalomyelitis. Ii. Serum factors responsible for demyelination. Journal of Experimental Medicine. 1964;119:303–312. doi: 10.1084/jem.119.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raine CS, Cannella B, Hauser SL, Genain CP. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: a case for antigen-specific antibody mediation. Annals of Neurology. 1999;46(2):144–160. doi: 10.1002/1531-8249(199908)46:2<144::aid-ana3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez M, Lucchinetti CF, Clark RJ, Yaksh TL, Markowitz H, Lennon VA. Immunoglobulins and complement in demyelination induced in mice by Theiler’s virus. Journal of Immunology. 1988;140(3):800–806. [PubMed] [Google Scholar]
- 13.Goto H, Matsuo H, Nakane S, et al. Plasmapheresis affects T helper type-1/T helper type-2 balance of circulating peripheral lymphocytes. Therapeutic Apheresis. 2001;5(6):494–496. doi: 10.1046/j.1526-0968.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- 14.Kambara C, Matsuo H, Fukudome T, Goto H, Shibuya N. Miller Fisher syndrome and plasmapheresis. Therapeutic Apheresis. 2002;6(6):450–453. doi: 10.1046/j.1526-0968.2002.00466.x. [DOI] [PubMed] [Google Scholar]
- 15.Shariatmadar S, Nassiri M, Vincek V. Effect of plasma exchange on cytokines measured by multianalyte bead array in thrombotic thrombocytopenic purpura. American Journal of Hematology. 2005;79(2):83–88. doi: 10.1002/ajh.20342. [DOI] [PubMed] [Google Scholar]
- 16.De Luca G, Lugaresi A, Iarlori C, et al. Prednisone and plasma exchange improve suppressor cell function in chronic inflammatory demyelinating polyneuropathy. Journal of Neuroimmunology. 1999;95(1-2):190–194. doi: 10.1016/s0165-5728(98)00266-5. [DOI] [PubMed] [Google Scholar]
- 17.Griffin MD, Lutz WH, Phan VA, Bachman LA, McKean DJ, Kumar R. Potent inhibition of dendritic cell differentiation and maturation by vitamin D analogs. Biochemical and Biophysical Research Communications. 2000;270(3):701–708. doi: 10.1006/bbrc.2000.2490. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez M, Karnes WE, Bartleson JD, Pineda AA. Plasmapheresis in acute episodes of fulminant CNS inflammatory demyelination. Neurology. 1993;43(6):1100–1104. doi: 10.1212/wnl.43.6.1100. [DOI] [PubMed] [Google Scholar]
- 19.Weinshenker BG, O’Brien PC, Petterson TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Annals of Neurology. 1999;46(6):878–886. doi: 10.1002/1531-8249(199912)46:6<878::aid-ana10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 20.Keegan M, Konig F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. The Lancet. 2005;366(9485):579–582. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- 21.Keegan M, Pineda AA, McClelland RL, Darby CH, Rodriguez M, Weinshenker BG. Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology. 2002;58(1):143–146. doi: 10.1212/wnl.58.1.143. [DOI] [PubMed] [Google Scholar]
- 22.Moldenhauer A, Haas J, Wascher C, et al. Immunoadsorption patients with multiple sclerosis: an open-label pilot study. European Journal of Clinical Investigation. 2005;35(8):523–530. doi: 10.1111/j.1365-2362.2005.01518.x. [DOI] [PubMed] [Google Scholar]
- 23.Magana SM, Keegan BM, Weinshenker BG, et al. Beneficial plasma exchange response in central nervous system inflammatory demyelination. Archives of Neurology. 2011;68(7):870–878. doi: 10.1001/archneurol.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae-Grant A. Evidence-based guideline update: plasmapheresis in neurologic disorders: report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology. Neurology. 2011;76(3):294–300. doi: 10.1212/WNL.0b013e318207b1f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noseworthy JH, O’Brien PC, Weinshenker BG, et al. IV immunoglobulin does not reverse established weakness in MS. Neurology. 2000;55(8):1135–1143. doi: 10.1212/wnl.55.8.1135. [DOI] [PubMed] [Google Scholar]
- 26.Winters JL, Brown D, Hazard E, Chainani A, Andrzejewski C., Jr. Cost-minimization analysis of the direct costs of TPE and IVIg in the treatment of Guillain-Barre syndrome. BMC Health Services Research. 2011;11, article 101 doi: 10.1186/1472-6963-11-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esiri MM. Multiple sclerosis: a quantitative and qualitative study of immunoglobulin-containing cells in the central nervous system. Neuropathology and Applied Neurobiology. 1980;6(1):9–21. doi: 10.1111/j.1365-2990.1980.tb00199.x. [DOI] [PubMed] [Google Scholar]
- 28.Cepok S, Jacobsen M, Schock S, et al. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain. 2001;124(11):2169–2176. doi: 10.1093/brain/124.11.2169. [DOI] [PubMed] [Google Scholar]
- 29.Baranzini SE, Jeong MC, Butunoi C, Murray RS, Bernard CC, Oksenberg JR. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. Journal of Immunology. 1999;163(9):5133–5144. [PubMed] [Google Scholar]
- 30.Colombo M, Dono M, Gazzola P, et al. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. Journal of Immunology. 2000;164(5):2782–2789. doi: 10.4049/jimmunol.164.5.2782. [DOI] [PubMed] [Google Scholar]
- 31.Knopf PM, Harling-Berg CJ, Cserr HF, et al. Antigen-dependent intrathecal antibody synthesis in the normal rat brain: rissue entry and local retention of antigen-specific B cells. Journal of Immunology. 1998;161(2):692–701. [PubMed] [Google Scholar]
- 32.Corcione A, Casazza S, Ferretti E, et al. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(30):11064–11069. doi: 10.1073/pnas.0402455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. Journal of Clinical Investigation. 1998;102(5):1045–1050. doi: 10.1172/JCI3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cross AH, Wu GF. Multiple sclerosis: oligoclonal bands still yield clues about multiple sclerosis. Nature Reviews Neurology. 2010;6(11):588–589. doi: 10.1038/nrneurol.2010.142. [DOI] [PubMed] [Google Scholar]
- 35.Paolino E, Fainardi E, Ruppi P, et al. A prospective study on the predictive value of CSF oligoclonal bands and MRI in acute isolated neurological syndromes for subsequent progression to multiple sclerosis. Journal of Neurology Neurosurgery and Psychiatry. 1996;60(5):572–575. doi: 10.1136/jnnp.60.5.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amato MP, Ponziani G. A prospective study on the prognosis of multiple sclerosis. Neurological Sciences. 2000;21(4, supplement 2):S831–S838. doi: 10.1007/s100720070021. [DOI] [PubMed] [Google Scholar]
- 37.Joseph FG, Hirst CL, Pickersgill TP, Ben-Shlomo Y, Robertson NP, Scolding NJ. CSF oligoclonal band status informs prognosis in multiple sclerosis: a case control study of 100 patients. Journal of Neurology, Neurosurgery and Psychiatry. 2009;80(3):292–296. doi: 10.1136/jnnp.2008.150896. [DOI] [PubMed] [Google Scholar]
- 38.Bruck W, Neubert K, Berger T, Weber JR. Clinical, radiological, immunological and pathological findings in inflammatory CNS demyelination—possible markers for an antibody-mediated process. Multiple Sclerosis. 2001;7(3):173–177. doi: 10.1177/135245850100700307. [DOI] [PubMed] [Google Scholar]
- 39.Belogurov AA, Jr., Kurkova IN, Friboulet A, et al. Recognition and degradation of myelin basic protein peptides by serum autoantibodies: novel biomarker for multiple sclerosis. Journal of Immunology. 2008;180(2):1258–1267. doi: 10.4049/jimmunol.180.2.1258. [DOI] [PubMed] [Google Scholar]
- 40.Barnett MH, Parratt JD, Cho ES, Prineas JW. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Annals of Neurology. 2009;65(1):32–46. doi: 10.1002/ana.21524. [DOI] [PubMed] [Google Scholar]
- 41.Avrameas S. Natural autoantibodies: from ’horror autotoxicus’ to ’gnothi seauton’. Immunology Today. 1991;12(5):154–159. doi: 10.1016/S0167-5699(05)80045-3. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez M, Lennon VA, Benveniste EN, Merrill JE. Remyelination by oligodendrocytes stimulated by antiserum to spinal cord. Journal of Neuropathology and Experimental Neurology. 1987;46(1):84–95. doi: 10.1097/00005072-198701000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Warrington AE, Asakura K, Bieber AJ, et al. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warrington AE, Bieber AJ, Van Keulen V, Ciric B, Pease LR, Rodriguez M. Neuron-binding human monoclonal antibodies support central nervous system neurite extension. Journal of Neuropathology and Experimental Neurology. 2004;63(5):461–473. doi: 10.1093/jnen/63.5.461. [DOI] [PubMed] [Google Scholar]
- 45.Mattson DH, Roos RP, Arnason BG. Isoelectric focusing of IgG eluted from multiple sclerosis and subacute sclerosing panencephalitis brains. Nature. 1980;287(5780):335–337. doi: 10.1038/287335a0. [DOI] [PubMed] [Google Scholar]
- 46.Mattson DH, Roos RP, Arnason BG. Oligoclonal IgG in multiple sclerosis and subacute sclerosing panencephalitis brains. Journal of Neuroimmunology. 1982;2(3-4):261–276. doi: 10.1016/0165-5728(82)90059-5. [DOI] [PubMed] [Google Scholar]
- 47.Whitacre CC, Mattson DH, Paterson PY, Roos RP, Peterson DJ, Arnason BG. Cerebrospinal fluid and serum oligoclonal IgG bands in rabbits with experiment allergic encephalomyelitis. Neurochemical Research. 1981;6:87–96. doi: 10.1007/BF00963909. [DOI] [PubMed] [Google Scholar]
- 48.Walsh MJ, Tourtellotte WW. Temporal invariance and clonal uniformity of brain and cerebrospinal IgG, IgA, and IgM in multiple sclerosis. Journal of Experimental Medicine. 1986;163(1):41–53. doi: 10.1084/jem.163.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goswami R, Marwaha RK, Gupta N, et al. Prevalence of vitamin D deficiency and its relationship with thyroid autoimmunity in Asian Indians: a community-based survey. British Journal of Nutrition. 2009;102(3):382–386. doi: 10.1017/S0007114509220824. [DOI] [PubMed] [Google Scholar]
- 50.Grant WB, Cross HS, Garland CF, et al. Estimated benefit of increased vitamin D status in reducing the economic burden of disease in western Europe. Progress in Biophysics and Molecular Biology. 2009;99(2-3):104–113. doi: 10.1016/j.pbiomolbio.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Kragt J, van Amerongen B, Killestein J, et al. Higher levels of 25-hydroxyvitamin D are associated with a lower incidence of multiple sclerosis only in women. Multiple Sclerosis. 2009;15(1):9–15. doi: 10.1177/1352458508095920. [DOI] [PubMed] [Google Scholar]
- 52.Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. Journal of the American Medical Association. 2006;296(23):2832–2838. doi: 10.1001/jama.296.23.2832. [DOI] [PubMed] [Google Scholar]
- 53.Orton SM, Morris AP, Herrera BM, et al. Evidence for genetic regulation of vitamin D status in twins with multiple sclerosis. American Journal of Clinical Nutrition. 2008;88(2):441–447. doi: 10.1093/ajcn/88.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raghuwanshi A, Joshi SS, Christakos S. Vitamin D and multiple sclerosis. Journal of Cellular Biochemistry. 2008;105(2):338–343. doi: 10.1002/jcb.21858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smolders J, Damoiseaux J, Menheere P, Tervaert JW, Hupperts R. Association study on two vitamin D receptor gene polymorphisms and vitamin D metabolites in multiple sclerosis. Annals of the New York Academy of Sciences. 2009;1173:515–520. doi: 10.1111/j.1749-6632.2009.04656.x. [DOI] [PubMed] [Google Scholar]
- 56.Smolders J, Damoiseaux J, Menheere P, Tervaert JW, Hupperts R. Fok-I vitamin D receptor gene polymorphism (rs10735810) and vitamin D metabolism in multiple sclerosis. Journal of Neuroimmunology. 2009;207(1-2):117–121. doi: 10.1016/j.jneuroim.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 57.Smolders J, Menheere P, Kessels A, Damoiseaux J, Hupperts R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Multiple Sclerosis. 2008;14(9):1220–1224. doi: 10.1177/1352458508094399. [DOI] [PubMed] [Google Scholar]
- 58.Smolders J, Peelen E, Thewissen M, et al. The relevance of vitamin D receptor gene polymorphisms for vitamin D research in multiple sclerosis. Autoimmunity Reviews. 2009;8(7):621–626. doi: 10.1016/j.autrev.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 59.Smolders J, Thewissen M, Peelen E, et al. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS One. 2009;4(8) doi: 10.1371/journal.pone.0006635. Article ID e6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soilu-Hanninen M, Laaksonen M, Laitinen I, Eralinna JP, Lilius EM, Mononen I. A longitudinal study of serum 25-hydroxyvitamin D and intact parathyroid hormone levels indicate the importance of vitamin D and calcium homeostasis regulation in multiple sclerosis. Journal of Neurology, Neurosurgery and Psychiatry. 2008;79(2):152–157. doi: 10.1136/jnnp.2006.105320. [DOI] [PubMed] [Google Scholar]
- 61.van der Mei IA, Ponsonby AL, Dwyer T, et al. Vitamin D levels in people with multiple sclerosis and community controls in Tasmania, Australia. Journal of Neurology. 2007;254(5):581–590. doi: 10.1007/s00415-006-0315-8. [DOI] [PubMed] [Google Scholar]
- 62.Craig TA, Sommer S, Sussman CR, Grande JP, Kumar R. Expression and regulation of the vitamin D receptor in the zebrafish, Danio rerio. Journal of Bone and Mineral Research. 2008;23(9):1486–1496. doi: 10.1359/JBMR.080403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gross M, Kumar R. Physiology and biochemistry of vitamin D-dependent calcium binding proteins. American Journal of Physiology. 1990;259(2):F195–F209. doi: 10.1152/ajprenal.1990.259.2.F195. [DOI] [PubMed] [Google Scholar]
- 64.Warren KG, Catz I. Autoantibodies to myelin basic protein within multiple sclerosis central nervous system tissue. Journal of the Neurological Sciences. 1993;115(2):169–176. doi: 10.1016/0022-510x(93)90221-j. [DOI] [PubMed] [Google Scholar]
- 65.Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nature Medicine. 1999;5(2):170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- 66.O’Connor KC, Appel H, Bregoli L, et al. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. Journal of Immunology. 2005;175(3):1974–1982. doi: 10.4049/jimmunol.175.3.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt S, Haase CG, Bezman L, et al. Serum autoantibody responses to myelin oligodendrocyte glycoprotein and myelin basic protein in X-linked adrenoleukodystrophy and multiple sclerosis. Journal of Neuroimmunology. 2001;119(1):88–94. doi: 10.1016/s0165-5728(01)00345-9. [DOI] [PubMed] [Google Scholar]
- 68.Karni A, Bakimer-Kleiner R, Abramsky O, Ben-Nun A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Archives of Neurology. 1999;56(3):311–315. doi: 10.1001/archneur.56.3.311. [DOI] [PubMed] [Google Scholar]
- 69.Lampasona V, Franciotta D, Furlan R, et al. Similar low frequency of anti-MOG IgG and IgM in MS patients and healthy subjects. Neurology. 2004;62(11):2092–2094. doi: 10.1212/01.wnl.0000127615.15768.ae. [DOI] [PubMed] [Google Scholar]
- 70.Arnon R, Crisp E, Kelley R. Anti-ganglioside antibodies in multiple sclerosis. Journal of the Neurological Sciences. 1980;46(2):179–186. doi: 10.1016/0022-510x(80)90076-3. [DOI] [PubMed] [Google Scholar]
- 71.Endo T, Scott DD, Stewart SS, Kundu SK, Marcus DM. Antibodies to glycosphingolipids in patients with multiple sclerosis and SLE. Journal of Immunology. 1984;132:1793–1797. [PubMed] [Google Scholar]
- 72.Saidi S, Mahjoub T, Almawi WY. Lupus anticoagulants and anti-phospholipid antibodies as risk factors for a first episode of ischemic stroke. Journal of Thrombosis and Haemostasis. 2009;7(7):1075–1080. doi: 10.1111/j.1538-7836.2009.03446.x. [DOI] [PubMed] [Google Scholar]
- 73.Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. Journal of the American Medical Association. 2006;295(9):1050–1057. doi: 10.1001/jama.295.9.1050. [DOI] [PubMed] [Google Scholar]
- 74.Freedman MS, Laks J, Dotan N, Altstock RT, Dukler A, Sindic CJ. Anti-α-glucose-based glycan IgM antibodies predict relapse activity in multiple sclerosis after the first neurological event. Multiple Sclerosis. 2009;15(4):422–430. doi: 10.1177/1352458508101944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwarz M, Spector L, Gortler M, et al. Serum anti-Glc(α1,4)Glc(α) antibodies as a biomarker for relapsing-remitting multiple sclerosis. Journal of the Neurological Sciences. 2006;244(1-2):59–68. doi: 10.1016/j.jns.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 76.Villar LM, Sadaba MC, Roldan E, et al. Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. Journal of Clinical Investigation. 2005;115(1):187–194. doi: 10.1172/JCI22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69(24):2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 78.Hinson SR, Roemer SF, Lucchinetti CF, et al. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down- regulating EAAT2. Journal of Experimental Medicine. 2008;205(11):2473–2481. doi: 10.1084/jem.20081241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. Journal of Experimental Medicine. 2005;202(4):473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Noseworthy JH, O’Brien PC, Petterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology. 2001;56(11):1514–1522. doi: 10.1212/wnl.56.11.1514. [DOI] [PubMed] [Google Scholar]
- 81.Weinstock-Guttman B, Ramanathan M, Lincoff N, et al. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (Devic disease) Archives of Neurology. 2006;63(7):957–963. doi: 10.1001/archneur.63.7.957. [DOI] [PubMed] [Google Scholar]
- 82.Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Archives of Neurology. 2009;66(9):1128–1133. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- 83.Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64(7):1270–1272. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez M. Have we finally identified an autoimmune demyelinating disease? Annals of Neurology. 2009;66(5):572–573. doi: 10.1002/ana.21838. [DOI] [PubMed] [Google Scholar]
- 85.Bailey RE. Diagnosis and treatment of infectious mononucleosis. American Family Physician. 1994;49(4):879–885. [PubMed] [Google Scholar]
- 86.Jenson HB. Virologic diagnosis, viral monitoring, and treatment of Epstein-Barr virus infectious mononucleosis. Current Infectious Disease Reports. 2004;6(3):200–207. doi: 10.1007/s11908-004-0009-2. [DOI] [PubMed] [Google Scholar]
- 87.Cepok S, Zhou D, Srivastava R, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. Journal of Clinical Investigation. 2005;115(5):1352–1360. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One. 2010;5(9):1–5. doi: 10.1371/journal.pone.0012496. Article ID e12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Annals of Neurology. 2006;59(3):499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- 90.Ramagopalan SV, Handel AE, Giovannoni G, Rutherford Siegel S, Ebers GC, Chaplin G. Relationship of UV exposure to prevalence of multiple sclerosis in England. Neurology. 2011;76(16):1410–1414. doi: 10.1212/WNL.0b013e318216715e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goldacre MJ, Wotton CJ, Seagroatt V, Yeates D. Multiple sclerosis after infectious mononucleosis: record linkage study. Journal of Epidemiology and Community Health. 2004;58(12):1032–1035. doi: 10.1136/jech.2003.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nielsen TR, Rostgaard K, Nielsen NM, et al. Multiple sclerosis after infectious mononucleosis. Archives of Neurology. 2007;64(1):72–75. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 93.Ebers GC, Sadovnick AD. The geographic distribution of multiple sclerosis: a review. Neuroepidemiology. 1993;12(1):1–5. doi: 10.1159/000110293. [DOI] [PubMed] [Google Scholar]
- 94.Poser CM, Benedikz J, Hibberd PL. The epidemiology of multiple sclerosis: the Iceland model. Onset-adjusted prevalence rate and other methodological considerations. Journal of the Neurological Sciences. 1992;111(2):143–152. doi: 10.1016/0022-510x(92)90062-p. [DOI] [PubMed] [Google Scholar]
- 95.Rosen LN, Livingstone IR, Rosenthal NE. Multiple sclerosis and latitude: a new perspective on an old association. Medical Hypotheses. 1991;36(4):376–378. doi: 10.1016/0306-9877(91)90014-p. [DOI] [PubMed] [Google Scholar]
- 96.Cantorna MT. Vitamin D and autoimmunity: is vitamin D status an environmental factor affecting autoimmune disease prevalence? Proceedings of the Society for Experimental Biology and Medicine. 2000;223(3):230–233. doi: 10.1046/j.1525-1373.2000.22333.x. [DOI] [PubMed] [Google Scholar]
- 97.Cantorna MT. Vitamin D and multiple sclerosis: an update. Nutrition Reviews. 2008;66(supplement 2):S135–S138. doi: 10.1111/j.1753-4887.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.VanAmerongen BM, Dijkstra CD, Lips P, Polman CH. Multiple sclerosis and vitamin D: an update. European Journal of Clinical Nutrition. 2004;58(8):1095–1109. doi: 10.1038/sj.ejcn.1601952. [DOI] [PubMed] [Google Scholar]
- 99.Haussler MR, Haussler CA, Bartik L, et al. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutrition Reviews. 2008;66(supplement 2):S98–S112. doi: 10.1111/j.1753-4887.2008.00093.x. [DOI] [PubMed] [Google Scholar]
- 100.Johnson JA, Kumar R. Renal and intestinal calcium transport: roles of vitamin D and vitamin D- dependent calcium binding proteins. Seminars in Nephrology. 1994;14(2):119–128. [PubMed] [Google Scholar]
- 101.Jurutka PW, Bartik L, Whitfield GK, et al. Vitamin D receptor: key roles in bone mineral pathophysiology, molecular mechanism of action, and novel nutritional ligands. Journal of Bone and Mineral Research. 2007;22(2):V2–V10. doi: 10.1359/jbmr.07s216. [DOI] [PubMed] [Google Scholar]
- 102.Jurutka PW, Whitfield GK, Hsieh JC, Thompson PD, Haussler CA, Haussler MR. Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Reviews in Endocrine and Metabolic Disorders. 2001;2(2):203–216. doi: 10.1023/a:1010062929140. [DOI] [PubMed] [Google Scholar]
- 103.Kumar R. Metabolism of 1,25-dihydroxyvitamin D3 . Physiological Reviews. 1984;64(2):478–504. doi: 10.1152/physrev.1984.64.2.478. [DOI] [PubMed] [Google Scholar]
- 104.Veenstra TD, Prufer K, Koenigsberger C, Brimijoin SW, Grande JP, Kumar R. 1,25-dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Research. 1998;804(2):193–205. doi: 10.1016/s0006-8993(98)00565-4. [DOI] [PubMed] [Google Scholar]
- 105.Dong X, Lutz W, Schroeder TM, et al. Regulation of relB in dendritic cells by means of modulated association of vitamin D receptor and histone deacetylase 3 with the promoter. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(44):16007–16012. doi: 10.1073/pnas.0506516102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Griffin MD, Dong X, Kumar R. Vitamin D receptor-mediated suppression of RelB in antigen presenting cells: a paradigm for ligand-augmented negative transcriptional regulation. Archives of Biochemistry and Biophysics. 2007;460(2):218–226. doi: 10.1016/j.abb.2007.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Griffin MD, Kumar R. Effects of α,25(OH)2D3 and its analogs on dendritic cell function. Journal of Cellular Biochemistry. 2003;88(2):323–326. doi: 10.1002/jcb.10335. [DOI] [PubMed] [Google Scholar]
- 108.Griffin MD, Kumar R. Multiple potential clinical benefits for 1α,25-dihydroxyvitamin D3 analogs in kidney transplant recipients. Journal of Steroid Biochemistry and Molecular Biology. 2005;97(1-2):213–218. doi: 10.1016/j.jsbmb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 109.Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1α,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(12):6800–6805. doi: 10.1073/pnas.121172198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Griffin MD, Xing N, Kumar R. Vitamin D and its analogs as regulators of immune activation and antigen presentation. Annual Review of Nutrition. 2003;23:117–145. doi: 10.1146/annurev.nutr.23.011702.073114. [DOI] [PubMed] [Google Scholar]
- 111.Xing N, L. Maldonado M, Bachman LA, McKean DJ, Kumar R, Griffin MD. Distinctive dendritic cell modulation by vitamin D3 and glucocorticoid pathways. Biochemical and Biophysical Research Communications. 2002;297(3):645–652. doi: 10.1016/s0006-291x(02)02262-3. [DOI] [PubMed] [Google Scholar]
- 112.Cannell JJ, Zasloff M, Garland CF, Scragg R, Giovannucci E. On the epidemiology of influenza. Virology Journal. 2008;5, article 29 doi: 10.1186/1743-422X-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grant WB. Latitude and multiple sclerosis prevalence: vitamin D reduces risk of Epstein-Barr virus infection. Multiple Sclerosis. 2010;16(3):373–375. doi: 10.1177/1352458509358093. [DOI] [PubMed] [Google Scholar]
- 114.Dal Bianco A, Bradl M, Frischer J, Kutzelnigg A, Jellinger K, Lassmann H. Multiple sclerosis and Alzheimer’s disease. Annals of Neurology. 2008;63(2):174–183. doi: 10.1002/ana.21240. [DOI] [PubMed] [Google Scholar]
- 115.Lindert RB, Haase CG, Brehm U, Linington C, Wekerle H, Hohlfeld R. Multiple sclerosis: B- and T-cell responses to the extracellular domain of the myelin oligodendrocyte glycoprotein. Brain. 1999;122(11):2089–2100. doi: 10.1093/brain/122.11.2089. [DOI] [PubMed] [Google Scholar]
- 116.Wang LY, Fujinami RS. Enhancement of EAE and induction of autoantibodies to T-cell epitopes in mice infected with a recombinant vaccinia virus encoding myelin proteolipid protein. Journal of Neuroimmunology. 1997;75(1-2):75–83. doi: 10.1016/s0165-5728(96)00235-4. [DOI] [PubMed] [Google Scholar]
- 117.Wucherpfennig KW, Catz I, Hausmann S, Strominger JL, Steinman L, Warren KG. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLA-DR2-restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B-cell and T-cell epitopes. Journal of Clinical Investigation. 1997;100(5):1114–1122. doi: 10.1172/JCI119622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Harp CT, Lovett-Racke AE, Racke MK, Frohman EM, Monson NL. Impact of myelin-specific antigen presenting B cells on T cell activation in multiple sclerosis. Clinical Immunology. 2008;128(3):382–391. doi: 10.1016/j.clim.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 119.Bourdette D, Yadav V. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. Current Neurology and Neuroscience Reports. 2008;8:417–418. doi: 10.1007/s11910-008-0064-4. [DOI] [PubMed] [Google Scholar]
- 120.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. New England Journal of Medicine. 2008;358(7):676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 121.Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. Journal of Neuroimmunology. 2006;180(1-2):63–70. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Annals of Neurology. 2010;67(4):452–461. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 123.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nature Immunology. 2002;3(10):944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 124.Duddy M, Niino M, Adatia F, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. Journal of Immunology. 2007;178(10):6092–6099. doi: 10.4049/jimmunol.178.10.6092. [DOI] [PubMed] [Google Scholar]
- 125.Pier GB, Lyczak JB, Wetzler LM. Immunology, Infection, and Immunity. Washington, DC, USA: ASM Press; 2004. [Google Scholar]
- 126.Chippaux JP. Snakebite epidemiology in Benin (West Africa) Toxicon. 1988;27:p. 37. [Google Scholar]
- 127.Uhlig H, Dernick R. Monoclonal autoantibodies derived from multiple sclerosis patients and control persons and their reactivities with antigens of the central nervous system. Autoimmunity. 1989;5(1-2):87–99. doi: 10.3109/08916938909029146. [DOI] [PubMed] [Google Scholar]
- 128.Ravetch JV, Bolland S. IgG Fc receptors. Annual Review of Immunology. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 129.Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement- mediated demyelination. Annals of Neurology. 1998;43(4):465–471. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- 130.Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. American Journal of Pathology. 1993;143(2):555–564. [PMC free article] [PubMed] [Google Scholar]
- 131.Okun E, Mattson MP, Arumugam TV. Involvement of Fc receptors in disorders of the central nervous system. NeuroMolecular Medicine. 2010;12(2):164–178. doi: 10.1007/s12017-009-8099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Neumann J. Ueber das Vorkommen der sogneannten “Mastzellen” bei pathologischen Veraenderungen des Gehirns. Virchows Archiv fur Pathologische Anatomie und Physiologie und fur Klinische Medizin. 1890;122:378–381. [Google Scholar]
- 133.Olsson Y. Mast cells in plaques of multiple sclerosis. Acta Neurologica Scandinavica. 1974;50:611–618. doi: 10.1111/j.1600-0404.1974.tb02806.x. [DOI] [PubMed] [Google Scholar]
- 134.Kruger PG, Bo L, Myhr KM, et al. Mast cells and multiple sclerosis: a light and electron microscopic study of mast cells in multiple sclerosis emphasizing staining procedures. Acta Neurologica Scandinavica. 1990;81(1):31–36. doi: 10.1111/j.1600-0404.1990.tb00927.x. [DOI] [PubMed] [Google Scholar]
- 135.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. Journal of Experimental Medicine. 2000;191(5):813–822. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Frick E, Stickl H. Antibody-dependent lymphocyte cytotoxicity against basic protein of myelin in multiple sclerosis. Journal of the Neurological Sciences. 1980;46(2):187–197. doi: 10.1016/0022-510x(80)90077-5. [DOI] [PubMed] [Google Scholar]
- 137.Frick E, Stickl H. Specificity of antibody-dependent lymphocyte cytotoxicity against cerebral tissue constituents in multiple sclerosis. Studies with basic protein of myelin, encephalitogenic peptide, cerebrosides and gangliosides. Acta Neurologica Scandinavica. 1982;65(1):30–37. doi: 10.1111/j.1600-0404.1982.tb03058.x. [DOI] [PubMed] [Google Scholar]
- 138.Merrill JE, Wahlin B, Siden A, Perlmann P. Elevated direct and IgM enhanced ADCC activity in multiple sclerosis patients. Journal of Immunology. 1982;128(4):1728–1735. [PubMed] [Google Scholar]
- 139.Ponomarenko NA, Durova OM, Vorobiev II, et al. Catalytic antibodies in clinical and experimental pathology: human and mouse models. Journal of Immunological Methods. 2002;269(1-2):197–211. doi: 10.1016/s0022-1759(02)00324-1. [DOI] [PubMed] [Google Scholar]
- 140.Ponomarenko NA, Durova OM, Vorobiev II, et al. Autoantibodies to myelin basic protein catalyze site-specific degradation of their antigen. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(2):281–286. doi: 10.1073/pnas.0509849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Belogurov AA, Kurkova IN, Misikov VK, et al. Substrate specificity of catalytic autoantibodies in neurodegenerative processes. Doklady Biochemistry and Biophysics. 2007;413(1):61–64. doi: 10.1134/s1607672907020068. [DOI] [PubMed] [Google Scholar]
- 142.Lang W, Rodriguez M, Lennon VA, Lampert PW. Demyelination and remyelination in murine viral encephalomyelitis. Annals of the New York Academy of Sciences. 1984;436:98–102. doi: 10.1111/j.1749-6632.1984.tb14779.x. [DOI] [PubMed] [Google Scholar]
- 143.Rodriguez M. Immunoglobulins stimulate central nervous system remyelination: electron microscopic and morphometric analysis of proliferating cells. Laboratory Investigation. 1991;64(3):358–370. [PubMed] [Google Scholar]
- 144.Rodriguez M, Lennon VA. Immunoglobulins promote remyelination in the central nervous system. Annals of Neurology. 1990;27(1):12–17. doi: 10.1002/ana.410270104. [DOI] [PubMed] [Google Scholar]
- 145.Asakura K, Miller DJ, Pease LR, Rodriguez M. Targeting of IgMκ antibodies to oligodendrocytes promotes CNS remyelination. Journal of Neuroscience. 1998;18(19):7700–7708. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Asakura K, Miller DJ, Murray K, Bansal R, Pfeiffer SE, Rodriguez M. Monoclonal autoantibody SCH94.03, which promotes central nervous system remyelination, recognizes an antigen on the surface of oligodendrocytes. Journal of Neuroscience Research. 1996;43(3):273–281. doi: 10.1002/(SICI)1097-4547(19960201)43:3<273::AID-JNR2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 147.Asakura K, Miller DJ, Pogulis RJ, Pease LR, Rodriguez M. Oligodendrocyte-reactive O1, O4, and HNK-1 monoclonal antibodies are encoded by germline immunoglobulin genes. Molecular Brain Research. 1995;34(2):283–293. doi: 10.1016/0169-328x(95)00190-4. [DOI] [PubMed] [Google Scholar]
- 148.Miller DJ, Rodriguez M. A monoclonal autoantibody that promotes central nervous system remyelination in a model of multiple sclerosis is a natural autoantibody encoded by germline immunoglobulin genes. Journal of Immunology. 1995;154(5):2460–2469. [PubMed] [Google Scholar]
- 149.Bieber AJ, Warrington A, Asakura K, et al. Human antibodies accelerate the rate of remyelination following lysolecithin-induced demyelination in mice. Glia. 2002;37(3):241–249. doi: 10.1002/glia.10033. [DOI] [PubMed] [Google Scholar]
- 150.Mitsunaga Y, Ciric B, Van Keulen V, et al. Direct evidence that a human antibody derived from patient serum can promote myelin repair in a mouse model of chronic-progressive demyelinating disease. The FASEB Journal. 2002;16(10):1325–1327. doi: 10.1096/fj.01-0994fje. [DOI] [PubMed] [Google Scholar]
- 151.Van Keulen VP, Ciric B, Radhakrishnan S, et al. Immunomodulation using the recombinant monoclonal human B7-DC cross-linking antibody rHIgM12. Clinical and Experimental Immunology. 2006;143(2):314–321. doi: 10.1111/j.1365-2249.2005.02992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ponomarenko NA, Durova OM, Vorobiev II, et al. Catalytic activity of autoantibodies toward myelin basic protein correlates with the scores on the multiple sclerosis expanded disability status scale. Immunology Letters. 2006;103(1):45–50. doi: 10.1016/j.imlet.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 153.Warrington AE, Bieber AJ, Ciric B, Pease LR, Van Keulen V, Rodriguez M. A recombinant human IgM promotes myelin repair after a single, very low dose. Journal of Neuroscience Research. 2007;85(5):967–976. doi: 10.1002/jnr.21217. [DOI] [PubMed] [Google Scholar]
- 154.Pirko I, Ciric B, Gamez J, et al. A human antibody that promotes remyelination enters the CNS and decreases lesion load as detected by T2-weighted spinal cord MRI in a virus-induced murine model of MS. The FASEB Journal. 2004;18(13):1577–1579. doi: 10.1096/fj.04-2026fje. [DOI] [PubMed] [Google Scholar]
- 155.Paz Soldan MM, Warrington AE, Bieber AJ, et al. Remyelination-promoting antibodies activate distinct Ca2+ influx pathways in astrocytes and oligodendrocytes: relationship to the mechanism of myelin repair. Molecular and Cellular Neuroscience. 2003;22(1):14–24. doi: 10.1016/s1044-7431(02)00018-0. [DOI] [PubMed] [Google Scholar]
- 156.Howe CL, Bieber AJ, Warrington AE, Pease LR, Rodriguez M. Antiapoptotic signaling by a remyelination-promoting human antimyelin antibody. Neurobiology of Disease. 2004;15(1):120–131. doi: 10.1016/j.nbd.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 157.Watzlawik J, Holicky E, Edberg DD, et al. Human remyelination promoting antibody inhibits apoptotic signaling and differentiation through Lyn kinase in primary rat oligodendrocytes. Glia. 2010;58(15):1782–1793. doi: 10.1002/glia.21048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vrethem M, Skogh T, Berlin G, Holmgren H, Ernerudh J. Antibodies to peripheral nerve myelin may occur without clinical neuropathy in healthy persons. Journal of Neuroimmunology. 1991;32(3):219–222. doi: 10.1016/0165-5728(91)90191-9. [DOI] [PubMed] [Google Scholar]
- 159.Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. Journal of Neuroscience. 2006;26(1):328–332. doi: 10.1523/JNEUROSCI.2615-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Howe CL, Mayoral S, Rodriguez M. Activated microglia stimulate transcriptional changes in primary oligodendrocytes via IL-1β . Neurobiology of Disease. 2006;23(3):731–739. doi: 10.1016/j.nbd.2006.06.012. [DOI] [PubMed] [Google Scholar]