

Abstract
Current diagnostic tools limit a clinician’s ability to discriminate between many possible causes of sensorineural hearing loss. This constraint leads to the frequent diagnosis of the idiopathic condition, leaving patients without a clear prognosis and only general treatment options. As a first step toward developing new diagnostic tools and improving patient care, we report the first use of liquid chromatography-tandem mass-spectrometry (LC-MS/MS) to map the proteome of human perilymph. Using LC-MS/MS, we analyzed four samples, two collected from patients with vestibular schwannoma (VS) and two from patients undergoing cochlear implantation (CI). For each cohort, one sample contained pooled specimens collected from five patients and the second contained a specimen obtained from a single patient. Of the 271 proteins identified with high confidence among the samples, 71 proteins were common in every sample, and used to conservatively define the proteome of human perilymph. Comparison to human cerebrospinal fluid and blood plasma, as well as murine perilymph, showed significant similarity in protein content across fluids; however, a quantitative comparison was not possible. Fifteen candidate biomarkers of VS were identified by comparing VS and CI samples. This list will be used in future investigations targeted at discriminating between VS tumors associated with good versus poor hearing.
Keywords: perilymph, proteomics, mass spectrometry, human, vestibular schwannoma
Introduction
Hearing is initiated when sound-induced vibrations of the eardrum and middle-ear ossicles are transmitted to the fluids of the inner ear, leading to stimulation of sensory hair cells and excitation of the auditory nerve. Hearing loss is broadly classified into two categories: (1) conductive hearing loss (CHL), in which mechanical energy transfer from the air to the inner ear is impeded, and (2) sensorineural hearing loss (SNHL), in which tissue pathology in the inner ear or central auditory pathways hamper signal transduction or neural conduction. SNHL can be the result of damage to almost any of the inner ear’s approximately 30 different cell types, as well as the nerves connecting the cochlea to the brain. Although SNHL can be routinely differentiated from CHL, diagnosing the specific pathology invoking an individual patient’s SNHL is a major challenge. No method currently exists to directly evaluate the inner ear, leaving the clinician blind to the underlying pathologic condition in SNHL. Indirect measures, such as audiograms, word recognition scores1, auditory brainstem responses and otoacoustic emissions2 can provide some insight, but are unable to make a specific determination.
Clinically, the limitation described above results in a frequent diagnosis of idiopathic SNHL leaving the patient and physician with no clear prognosis and general, and often ineffective, treatment options. A diagnostic tool capable of providing insight into inner ear pathology would enable the formulation of individualized treatment strategies (“personalized medicine”) tailored to each patient’s specific pathology. The need for such a tool is immediately apparent from the large number of patients who have unsatisfactory results with general treatment options. Furthermore, a diagnostic platform is critical for successful implementation of preservative and restorative therapies that may emerge from ongoing research in inner ear development and regeneration3. Positive patient outcomes will only result if specific disease states are known and targeted, making a diagnostic tool absolutely critical to treatment success.
To lay the groundwork for development of a diagnostic platform capable of filling this clinical need, we present the first analysis of the human perilymph proteome using mass spectrometric (MS) techniques. Perilymph, a proximal fluid of the inner ear, bathes spiral ganglion cell bodies of the auditory nerve and nearly all of the tissues vital to sound transduction. Due to its localization, any protein secreted by a damaged cell or released during an apoptotic or necrotic event will be found in perilymph at higher concentrations than in more peripheral fluids such as blood or cerebrospinal fluid (CSF).
Previous work has demonstrated the utility of perilymph as a diagnostic fluid. Prior to modern imaging technologies, vestibular schwannoma (VS), an important cause of SNHL in clinical practice, was diagnosed via a significant, i.e. >2.5-fold increase in the total protein content of perilymph4,5. This example demonstrates the potential for significant change in perilymphatic protein levels as a result of pathology, encouraging our search for relevant diagnostic information. Identification and validation of a specific set of biomarkers coupled with the development of a refined collection technique, minimizing the risk to hearing, may facilitate the collection and analysis of perilymph for diagnostic purposes in a clinical setting.
The present investigation is, to the best of our knowledge, the first attempt to define the proteome of human perilymph using mass spectrometry. Previous knowledge of the perilymphatic proteome is derived mainly from 2-D gel electrophoresis work aimed at diagnosis of perilymphatic fistula6–9. These early investigations established the presence of over 100 proteins in the fluid, nearly 30 of which were subsequently identified. Our work extends the existing proteomic characterization and compares the protein profile of perilymph to other bodily fluids. We have compared protein content in different pathologic states as a first step towards the discovery of disease biomarkers. The similarity of human and mouse perilymph has also been analyzed to explore the potential applicability of mouse models for discovery of biomarkers relevant for human SNHL.
Beyond the previously mentioned diagnostic possibilities, the knowledge generated in this investigation will be of significant utility to basic auditory scientists and inner ear pharmacologists. Characterization of the protein content of perilymph may provide insight into the molecular mechanisms that function to maintain the inner ear’s unique environment. Knowledge of the fluid content will also allow better understanding and prediction of protein-drug interactions, aiding in assessment of pharmacological efficacy and drug delivery within the highly specialized organ.
Methods
Collection of Samples
Perilymph specimens were obtained from 12 patients undergoing clinically indicated surgeries. All patients had profound sensorineural hearing loss with speech discrimination score < 40%, where 100% is normal. Although detailed medical histories are not available, all patients were healthy enough to undergo general anesthesia without complications. All procedures, six translabyrinthine craniotomies for resection of vestibular schwannomas (VS) and six cochlear implantations (CI), involved surgical opening of the cochlea and collection of approximately 1 μl of perilymph prior to it being displaced. Perilymph samples obtained from CI patients generally have protein concentrations near 2 μg/μl and those from VS patients 15–30 μg/μl4,5. The specimens were placed in 0.2 ml phosphate buffered saline (PBS) and immediately stored at −80°C. The study was approved by the Institutional Review Board of the Massachusetts Eye and Ear Infirmary. Five VS specimens were a generous gift from Dr. Jose Fayad (House Ear Clinic, Los Angeles).
Utilizing these 12 specimens, four samples were prepared for MS/MS analysis. Sample CI_1 contained the perilymph specimen from one CI patient and CI_2 contained the pooled specimens from the remaining five CI patients (Fig. 1). Similarly, sample VS_1 contained the perilymph specimen from one VS patient and VS_2 contained the pooled specimens of the five remaining VS patients. Samples were grouped in this manner to determine if pooling samples would result in more protein identifications per analysis. The CI samples, representing a heterogeneous disease group, served as controls for the VS biomarker search (a homogeneous disease group), as hearing loss in VS may be associated with elevated protein content of perilymph, presumably due to an unknown toxic substance produced by the VS10,11.
Figure 1.
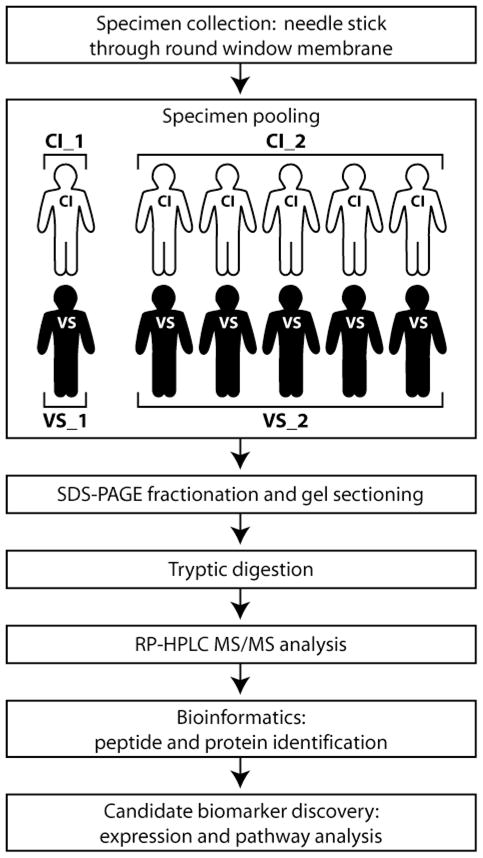
Workflow diagram of perilymph analysis outlining specimen collection, grouping, preparation, and analysis. RP-HPLC MS/MS: reversed-phase high performance liquid chromatography tandem mass spectrometry.
Mass Spectrometric Analysis
Samples were fractionated using polyacrylamide gel electrophoresis and subjected to in-gel tryptic digestion followed by reversed-phase liquid chromatography in-line with a tandem mass spectrometer (GeLC-MS/MS) (Fig. 1). In brief, entire gel lanes were divided into 7–10 sections (VS_1: 8, VS_2: 10, CI_1: 7, CI_2: 10) and proteins in each gel section were digested with trypsin12,13. Peptides extracted from each gel section were analyzed by nanoflow reversed-phase high-performance liquid chromatography (HPLC) system (Eksigent) hyphenated with an LTQ-Orbitrap mass spectrometer (Thermo Scientific) (samples CI_1 and VS_1) or microscale capillary HPLC (Surveyor, Thermo Scientific) hyphenated with an LTQ mass spectrometer (Thermo Scientific) (samples CI_2 and VS_2). The LC columns (15 cm × 100 μm ID, New Objective) were packed in-house (Magic C18, 5 μm, 100 Å, Michrom BioResources). Samples were analyzed with a 60-minute linear gradient (0–35% acetonitrile with 0.2% formic acid) and data were acquired in a data-dependent fashion, with six MS/MS scans for every full scan spectrum.
All data generated from the gel sections were searched against the IPI-human database (v3.61)14 using the Paragon Algorithm15 integrated into the ProteinPilot search engine (v.3; AB/Sciex). Search parameters were set as follows: sample type, identification; Cys alkylation, iodoacetamide; Instrument, Orbitrap/FT (1–3 ppm) (CI_1 and VS_1) or LTQ (CI_2 and VS_2); special factors, gel-based ID; ID focus, none; database, international protein index (IPI) human (v.3.61); detection protein threshold, 99.0%; and search effort, thorough ID. To be considered a valid identification, proteins were required to score above a threshold established by a 1% global false detection rate (corresponding to “Unused” scores in sample VS_1 ≥ 2.01, VS_2 ≥ 7.23, CI_1 ≥ 2.47, and CI_2 ≥ 5.58). This criterion enforced confidence intervals exceeding 99% for each identification. Additionally, each valid protein identification was required to have a minimum of two high confidence (≥95%) peptide identifications attributed to it.
Quantitative comparison of protein expression was accomplished via analysis of spectral count data using the statistical framework QSPEC16. QSPEC employs hierarchical Bayes estimation of a generalized linear mixed effects model to identify proteins with differential expression across data sets. A protein was considered differentially expressed across disease state if QSPEC returned a Bayes factor larger than 10 and the fold change exceeded 1.5.
QSPEC analysis of a 2 disease state data set, each with 2 replicates, would typically couple the spectral count data of all 4 samples into one analysis to maximize the amount of information utilized and increase the statistical power. However, the differences in sample preparation and instrumentation (described previously) make the optimal analytic approach ambiguous. To gain maximal insight into the data, three different implementations of the QSPEC package were tested and the results compared. The three analysis paradigms studied were: (1) spectral counts from all 4 samples were coupled into a single 2-disease-state/2-replicate analysis, (2) spectral counts were summed within each disease state, prior to analysis, to generate a single 2-disease-state/1-replicate data set, and (3) two 2-disease-state/1-replicate QSPEC analyses were conducted and the results were combined using a rudimentary scoring metric to determine expression characteristics. Samples prepared/analyzed similarly were directly compared such that the first QSPEC analysis compared samples CI_1 and VS_1 and the second compared CI_2 and VS_2.
These three paradigms have different benefits and drawbacks. Method 1 is generally the most favorable as it utilizes all of the spectral count data generated in the 4 MS/MS analyses along with the spectra distribution characteristics of each sample. However, this method can fail when replicates have significantly different statistical properties.
Method 2 includes all of the available spectral data into one analysis but fails to utilize information contained within the individual statistical properties of each sample. The creators of QSPEC analyzed algorithm performance with summed vs. distinct replicates and demonstrated, with replicates having similar statistical distributions, that this type of combination yielded satisfactorily equivalent results to method 116.
Method 3 has the benefit of utilizing all of the available spectral and sample distribution information but suffers because the data is divided between two separate analyses. To merge the results of method (3), a simple scoring metric was employed. Proteins found to be over-expressed in the VS sample of either analysis were given a score of +1, those up-regulated in the CI samples a −1, and those without differential expression were given a score of 0. Scores were summed across the two analyses resulting in individual protein scores ranging from −2 to +2.
Results
A total of 271 proteins were identified at high confidence within the four perilymph samples (Fig. 2). The number of proteins identified (MS/MS spectra generated) per sample was: CI_1: 225 (35,027), VS_1: 167 (21,272), CI_2: 106 (112,465), VS_2: 90 (111,127). As none of the samples can be considered clinically normal, we began our analysis with the conservative requirement that a protein must be present in all four samples to be considered part of the core proteome. This analysis generated a list of 71 proteins identified with high confidence in all four samples (Supplementary Table 1). Comparison of this list to previous work9, using 2-D gel electrophoresis of perilymph obtained during stapedectomy procedures, shows strong similarity. Our list of 71 proteins contains 92% of those previously identified, and adds another 46 to the knowledgebase.
Figure 2.
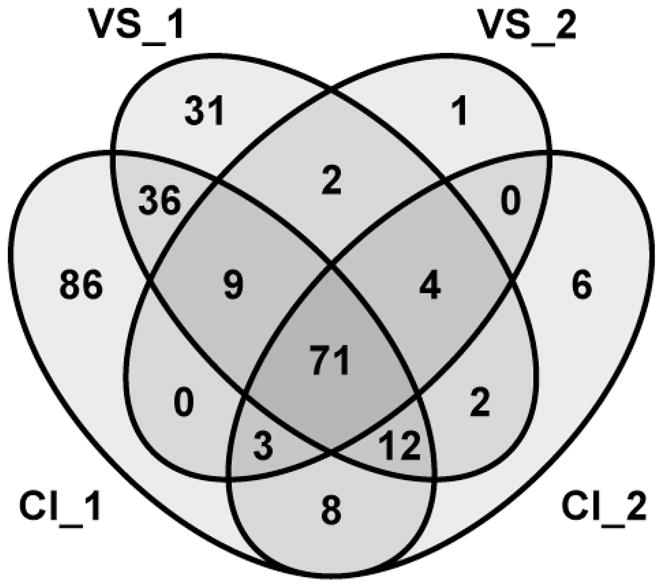
Distribution of 271 proteins identified at high confidence within four samples of human perilymph.
If the ‘normal’ proteome inclusion requirement is loosened to include proteins identified in only three of the four samples the number of identifications increases to 99 (lower panel of Supplementary Table 1). This number is similar to the number of protein spots separated by Thalmann et al.9 and includes 100% of the proteins previously identified. Such a reduction of inclusion criteria may be warranted based on the low levels of reproducibility observed in MS analyses (<60% similarity across repeated trials17), suggesting the requirement that a protein be present in all four samples is overly stringent.
Similarity to CSF, blood plasma, and mouse perilymph
Comparing the 71 common protein identifications with published proteomes of human cerebrospinal fluid (CSF) and blood plasma, we note a strong similarity across the fluids (Supplementary Table 1). The CSF database downloaded from the Max-Planck Unified Proteome Database (MAPU)18,19 contained 56 of 71 of the proteins we identified in perilymph while the plasma database obtained from Plasma Proteome Institute (PPI)20,21 contained 56 of 71. In each case, the majority of the non-common proteins are suspected contaminants (keratins, carbonic anhydrases). This similarity among human fluids was expected as animal models of the fluids contain similar protein complements. However, the quantity of individual proteins and the total protein content varies widely8.
Comparison of our perilymph dataset with the proteome of mouse perilymph published by Swan et al.22 revealed moderate similarity across the two fluids (Supplementary Table 1). Orthologues of 31 of the 52 proteins identified in that investigation were found in our list of human perilymphatic proteins (orthology determined using OrthoDB23).
Quantitative comparison of CI and VS samples
The first iteration of QSPEC analysis, employing method 1, resulted in the identification of 104 proteins with differential expression between CI and VS samples. As mentioned above, this method was favorable due to its utilization of all available spectral count data in one analysis. However, the high degree of dissimilarity in spectral distribution properties, owing to the large difference in the number of spectra acquired by each MS/MS platform, resulted in poor model behavior and unreliable Bayes factors. The normalizing constant (Nj) employed in the generalized linear mixed effects model within QSPEC can adequately account for small differences in total spectral counts between replicates, but in this comparison the difference was large (>4 fold). At the protein level this causes heterogeneous counts within and across disease states. This results in poor model fit and inflation of the Bayes factor due to increased model flexibility when the differential expression term is included. Such false identifications can be partly filtered by only considering fold changes in excess of 50%16 as valid, as this reduces the chances of over-fitting, but the extreme deviation in these data sets may render such methods insufficient.
QSPEC analysis using method 2 resulted in identification of 324 differentially-expressed proteins. Summing counts within disease state had the benefit of generating two data sets with similar statistical properties (resulting in better stability of the model), but did so at the cost of information about each sample’s spectral distribution statistics. This informational loss induced the large discrepancy between the results of methods 1 and 2 and caused very different model behavior than QSPEC’s authors experienced with their statistically more similar replicates16. To illustrate a common conflict, consider the protein hornerin (HRNR). HRNR was identified based on 22 spectra in sample CI_1 and was not found in any other sample. Using method 1, HRNR was not determined to be differentially expressed because the zero spectra from CI_2 suppressed the Bayes factor below significance. However in method 2, QSPEC compared 22 spectra from the CI samples vs. 0 spectra from the VS samples and determined it to be differentially expressed. Furthermore, because the number of spectra associated with samples CI_2 and VS_2 was significantly greater than CI_1 and VS_1, this analysis paradigm gave heavy weighting to the second set of samples.
The final QSPEC analysis, method 3, identified 25 proteins scoring ±2 (indicating up-regulation in both VS or CI samples respectively) and 271 proteins scoring ±1 (up-regulated in one VS or CI sample while the other analysis did not reach significance). In addition to the hundreds of proteins which scored ‘0’ because neither analysis reached significance, twenty proteins scored ‘0’ due to conflicting expression patterns across the two sample groups. Despite the drawback of splitting the data, this approach worked well as each analysis compared data sets with similar statistical parameters and took full advantage of information imbedded within sample spectral distributions. This resulted in improved model stability and more confident determinations of differential expression. As such, the results of method 3 were selected for further analysis.
It is important to note that all of the spectra generated in the MS/MS analysis of the 4 samples, with no limitations on spectra confidence, were analyzed by QSPEC. In cases where a protein was evidenced by many low-confidence (but no high-confidence) spectra this resulted in a QSPEC report of differential expression, even though the protein in question did not meet requirements for a confident identification.
When the results of method 3 are filtered to remove these ‘non-significant’ returns, isoform conflicts and immunological proteins, 3 proteins remained that scored ±2 (CRYM, FN1, and KRT10) and 65 that scored ±1. Of these 68 proteins, 14 were selected as particularly interesting due to their differential expression and biological function, Table 1.
Table 1.
Candidate biomarkers of human vestibular schwannomas associated with poor hearing
| Candidate Biomarkers Identified via Uniqueness Criteria and Spectral Count analysis
| ||
|---|---|---|
| Symbol | Name | Entrez Gene ID |
| CRYM | μ-Crystallin | 1428 |
| FN1 | Fibronectin 1 | 2335 |
| KRT10 | Keratin 10 | 3858 |
| APOC3 | Apolipoprotein C-III | 345 |
| VCAN | Versican | 1462 |
| DCD | Dermcidin | 117159 |
| SERPINB12 | Serpin peptidase inhibitor, clade B, member 12 | 89777 |
| CTSD | Cathepsin D | 1509 |
| SERPINB3 | Serpin peptidase inhibitor, clade B, member 3 | 6317 |
| SERPINA5 | Serpin peptidase inhibitor, clade A, member 5 | 5104 |
| SOD3 | Superoxide dismutase 3, extracellular | 6649 |
| PARK7 | Parkinson disease protein 7 | 11315 |
| SERPINF1 | Serpin peptidase inhibitor, clade F, member 1 | 5176 |
| CHI3L1 | Cartilage glycoprotein 39 | 1116 |
| LRP2 | Low density lipoprotein receptor-related protein 2 | 4036 |
italics: found only in VS samples; bold: scored ±2 in QSPEC analysis
Discussion
Interestingly, while the total protein content of the VS samples, as determined by SDS-PAGE analysis, was greater than the CI samples (due to higher protein concentration in perilymph from VS patients, data not shown), more identifications were made within the latter group based on our search criteria (combined VS samples: 171; combined CI samples: 237). This difference may be real, indicating decreased complexity of the VS perilymph proteome due to reduced protein secretion (possibly because of tissue loss), or may be an artifactual result from limitations of the dynamic range of detection. Tumor induced up-regulation of a small number of proteins may have suppressed the signal of lower abundance proteins, thus impeding their detection. Separate analysis of each section of the electrophoresis gel likely reduced, but did not eliminate, the potential signal suppression due to high abundance proteins.
Comparison across samples demonstrates that more proteins were identified in each of the samples derived from the specimen of one patient (CI_1 and VS_1) than in the samples derived from the specimens of five patients (CI_2 and VS_2). This disparity may have a similar cause as above in that pooling of samples led to suppression of the signal from low abundance proteins. However, the higher resolution and mass accuracy of the LTQ/Orbitrap MS used to analyze samples CI_1 and VS_1 is the most likely reason for the observed increase in identifications. In the context of the experiment, this discontinuity would likely serve to reduce the number of identifications common across all samples and individual disease groups.
Beyond the obvious limitations induced by the use of pathologic specimens, additional evidence for the need to perform a deeper characterization of perilymph comes from the comparison with published proteomes of human CSF and blood plasma. Both databases are significantly larger than the currently observed perilymph proteome, as each is composed of several hundred proteins. However, these body fluids are plentiful and large volumes in the ml-range are available in contrast to perilymph, of which only single digit μl-amounts can be obtained. Thus, multidimensional separation strategies that are optimized for minimized sample losses would be highly advantageous for the comprehensive analysis of the perilymph proteome.
Direct and quantitative comparison of perilymph with CSF and/or plasma, similar to previous work with mice22, would improve our understanding of human perilymph as a functional fluid. Such work would also be useful to assess the possibility that the similarity between perilymph and plasma is due to blood contamination of the perilymph sample.
It is worth noting that the list of 71 common proteins contains several that may not be of perilymphatic origin. The robust presence of carbonic anhydrases (common in red blood cells), multiple keratins and hemoglobins suggest that samples have suffered contamination from blood and cellular debris. However, carbonic anhydrase is known to be abundant in the cochlea, comprising about 1% of the protein of the membranous lateral wall24. In this analysis, we tried to minimize blood contamination by using visibly transparent samples of perilymph and excluding obviously pink specimens. Standardized methodologies must be carefully implemented in the future to limit sources of contamination and establish whether these proteins are in fact typical components of perilymph.
Candidate Biomarkers of VS
Exact mechanisms of hearing loss associated with VS are not known as some VS induce hearing loss while others do not, a phenomenon that shows poor correlation with tumor size or growth10,25,26. Evidence suggests that these tumors cause hearing loss not only by compressing the auditory nerve but also by secreting a substance toxic to the cochlear nerve or inner ear11,27. In line with this hypothesis, we compared the VS and CI samples to generate an initial list of candidate biomarkers for VS associated with poor hearing (all samples were collected from patients with severe to profound deafness in the affected ear). Candidates were identified using uniqueness criterion, i.e. identified within both VS but neither CI sample, and differential expression analysis using the QSPEC algorithm for spectral counting.
Two proteins where identified in both VS samples but neither CI sample, Fig. 2. The first, μ-crystallin (CRYM, encoded by CRYM) is also known as NADPH-regulated thyroid hormone binding protein. CRYM is found in the cytoplasm where it binds and promotes transcriptional activity of the thyroid hormone triiodothyronine (T3)28. Expression occurs within many cells of the spiral ligament29 and mutation results in autosomal dominant deafness through changes in intracellular localization and the loss of its T3 binding ability, potentially leading to impaired K+ recycling30,31.
The second protein found within both VS samples and neither CI sample was low density lipoprotein-related protein 2 (LRP2, encoded by LRP2, also known as megalin). LRP2 is a trans-membrane receptor protein found primarily in absorptive epithelial cells, including those of the ear and kidney32–35. LRP2 can bind a wide variety of structurally dissimilar ligands and is a key player in mediating endocytosis of many substances including lipoproteins, sterols, vitamin binding proteins, and hormones. Mutations in LRP2 result in Donnai-Barrow and facio-oculo-acoustico-renal (FOAR) syndromes36, both of which present with sensorineural hearing loss. Surprisingly, LRP2 was not found to be differentially expressed by QSPEC because its spectral count in each VS sample was small. Despite this, LRP2 has been included in the list of potential candidates because of its detection pattern; LRP2 will be carefully validated in future work.
Analysis of the remaining proteins identified in Table 1 shows enrichment for many serine protease inhibitors. This class of molecules plays a critical role in regulating the inflammation response. Similarly, PARK7 and SOD3 both play active roles in regulating oxidative stress while CTSD and VCAN have been implicated in both cancer and neurological disorders. These molecules therefore appear particularly relevant to our interest in illuminating mechanisms of inner ear damage and will be investigated in future work.
Conclusion
Analysis of human perilymph specimens has provided novel insight into the protein content of the inner ear fluid microenvironment. However, the specimens originate from clinically diseased ears, and thus may not represent the complete proteome of normal perilymph. Using mass spectrometry, we have more than doubled the known proteome to include over 70 proteins. This number is less than the approximately 100 proteins previously separated using 2D gel electrophoresis9, although the gap may be narrowed by loosening the stringency of our inclusion criteria, as supported by known limitations of MS analysis. The proteins in perilymph are found to be similar to CSF and plasma, but quantitative comparison is not possible at this time. Human perilymph proteins are also similar to those in mouse perilymph, lending potential support to the use of a mouse model as a vehicle for biomarker discovery. Finally, a list of 15 candidate biomarkers of VS is generated, with some candidates having known roles in hearing and deafness, and other candidates showing significant up-regulation as determined via spectral count analysis with QSPEC.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Jose Fayad for the kind donation of five perilymph specimens. This project was supported by the American Otologic Society (AOS), Massachusetts Life Sciences Center (MLSC), NIDCD grants T32 DC00038 and K08 DC010419, NIH grant U24 DC008559 and Shore Fellowship at Harvard Medical School. This work is solely the responsibility of the authors and does not necessarily represent the official views of the AOS, MLSC, NIDCD or NIH.
References
- 1.Halpin C, Rauch SD. Clinical implications of a damaged cochlea: pure tone thresholds vs information-carrying capacity. Otolaryngol Head Neck Surg. 2009;140:473–476. doi: 10.1016/j.otohns.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 2.Ahmmed A, Brockbank C, Adshead J. Cochlear microphonics in sensorineural hearing loss: lesson from newborn hearing screening. Int J Pediatr Otorhinolaryngol. 2008;72:1281–1285. doi: 10.1016/j.ijporl.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Wei D, Yamoah EN. Regeneration of the mammalian inner ear sensory epithelium. Curr Opin Otolaryngol Head Neck Surg. 2009;17:373–380. doi: 10.1097/MOO.0b013e328330345b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverstein H. A rapid protein test for acoustic neurinoma. Arch Otolaryngol. 1972;95:202–204. doi: 10.1001/archotol.1972.00770080344003. [DOI] [PubMed] [Google Scholar]
- 5.Silverstein H. Labyrinthine tap as a diagnostic test for acoustic neurinoma. Otolaryngol Clin North Am. 1973;6:229–244. [PubMed] [Google Scholar]
- 6.Arrer E, Oberascher G, Gibitz HJ. A map of cochlear perilymph protein based on high-resolution two-dimensional electrophoresis. Eur Arch Otorhinolaryngol. 1990;247:271–273. doi: 10.1007/BF00176534. [DOI] [PubMed] [Google Scholar]
- 7.Paugh DR, Telian SA, Disher MJ. Identification of perilymph proteins by two-dimensional gel electrophoresis. Otolaryngol Head Neck Surg. 1991;104:517–525. doi: 10.1177/019459989110400416. [DOI] [PubMed] [Google Scholar]
- 8.Thalmann I, Comegys TH, Liu SZ, Ito Z, Thalmann R. Protein profiles of perilymph and endolymph of the guinea pig. Hear Res. 1992;63:37–42. doi: 10.1016/0378-5955(92)90071-t. [DOI] [PubMed] [Google Scholar]
- 9.Thalmann I, et al. Protein profile of human perilymph: in search of markers for the diagnosis of perilymph fistula and other inner ear disease. Otolaryngol Head Neck Surg. 1994;111:273–280. doi: 10.1177/01945998941113P117. [DOI] [PubMed] [Google Scholar]
- 10.Nadol JB, Jr, Diamond PF, Thornton AR. Correlation of hearing loss and radiologic dimensions of vestibular schwannomas (acoustic Neuromas) Am J Otol. 1996;17:312–316. [PubMed] [Google Scholar]
- 11.Stankovic KM, et al. Genetic determinants of hearing loss associated with vestibular schwannomas. Otol Neurotol. 2009;30:661–667. doi: 10.1097/MAO.0b013e3181a66ece. [DOI] [PubMed] [Google Scholar]
- 12.Neubauer G, Mann M. Mapping of phosphorylation sites of gel-isolated proteins by nanoelectrospray tandem mass spectrometry: potentials and limitations. Anal Chem. 1999;71:235–242. doi: 10.1021/ac9804902. [DOI] [PubMed] [Google Scholar]
- 13.Steen H, Kuster B, Fernandez M, Pandey A, Mann M. Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode. Anal Chem. 2001;73:1440–1448. doi: 10.1021/ac001318c. [DOI] [PubMed] [Google Scholar]
- 14.Kersey PJ, et al. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- 15.Shilov IV, et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6:1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics. 2008;7:2373–2385. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J. Proteomics of human cerebrospinal fluid - the good, the bad, and the ugly. PROTEOMICS - CLINICAL APPLICATIONS. 2007;1:805–819. doi: 10.1002/prca.200700081. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, et al. MAPU: Max-Planck Unified database of organellar, cellular, tissue and body fluid proteomes. Nucleic Acids Res. 2007;35:D771–779. doi: 10.1093/nar/gkl784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zougman A, et al. Integrated analysis of the cerebrospinal fluid peptidome and proteome. J Proteome Res. 2008;7:386–399. doi: 10.1021/pr070501k. [DOI] [PubMed] [Google Scholar]
- 20.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 21.Anderson NL, et al. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol Cell Proteomics. 2004;3:311–326. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Swan EE, et al. Proteomics analysis of perilymph and cerebrospinal fluid in mouse. Laryngoscope. 2009;119:953–958. doi: 10.1002/lary.20209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2011;39:D283–288. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drescher DG. Purification of a carbonic anhydrase from the inner ear of the guinea pig. Proc Natl Acad Sci U S A. 1977;74:892–896. doi: 10.1073/pnas.74.3.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckermeier L, Pirsig W, Mueller D. Histopathology of 30 non-operated acoustic schwannomas. Arch Otorhinolaryngol. 1979;222:1–9. doi: 10.1007/BF00456332. [DOI] [PubMed] [Google Scholar]
- 26.Fisher LM, Doherty JK, Lev MH, Slattery WH. Concordance of bilateral vestibular schwannoma growth and hearing changes in neurofibromatosis 2: neurofibromatosis 2 natural history consortium. Otol Neurotol. 2009;30:835–841. doi: 10.1097/MAO.0b013e3181b2364c. [DOI] [PubMed] [Google Scholar]
- 27.Suga F, Lindsay JR. Inner ear degeneration in acoustic neurinoma. Ann Otol Rhinol Laryngol. 1976;85:343–358. doi: 10.1177/000348947608500305. [DOI] [PubMed] [Google Scholar]
- 28.Mori J, et al. Nicotinamide adenine dinucleotide phosphate-dependent cytosolic T(3) binding protein as a regulator for T(3)-mediated transactivation. Endocrinology. 2002;143:1538–1544. doi: 10.1210/endo.143.4.8736. [DOI] [PubMed] [Google Scholar]
- 29.Usami S, et al. The localization of proteins encoded by CRYM, KIAA1199, UBA52, COL9A3, and COL9A1, genes highly expressed in the cochlea. Neuroscience. 2008;154:22–28. doi: 10.1016/j.neuroscience.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 30.Abe S, et al. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet. 2003;72:73–82. doi: 10.1086/345398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshima A, et al. CRYM mutations cause deafness through thyroid hormone binding properties in the fibrocytes of the cochlea. J Med Genet. 2006;43:e25. doi: 10.1136/jmg.2005.034397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol. 1995;6:35–47. doi: 10.1681/ASN.V6135. [DOI] [PubMed] [Google Scholar]
- 33.Kerjaschki D, Farquhar MG. Immunocytochemical localization of the Heymann nephritis antigen (GP330) in glomerular epithelial cells of normal Lewis rats. J Exp Med. 1983;157:667–686. doi: 10.1084/jem.157.2.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatelet F, Brianti E, Ronco P, Roland J, Verroust P. Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. II. Extrarenal distribution. Am J Pathol. 1986;122:512–519. [PMC free article] [PubMed] [Google Scholar]
- 35.Raychowdhury R, Niles JL, McCluskey RT, Smith JA. Autoimmune target in Heymann nephritis is a glycoprotein with homology to the LDL receptor. Science. 1989;244:1163–1165. doi: 10.1126/science.2786251. [DOI] [PubMed] [Google Scholar]
- 36.Kantarci S, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957–959. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.