

Abstract
Carbohydrate polymerases are abundant in nature. Although they play vital physiological roles, the molecular mechanisms that they use for the controlled assembly of polymers are largely unknown. One fundamental issue is whether an enzyme utilizes a processive or distributive mechanism for chain elongation. The shortage of mechanistic information on polysaccharide-generating glycosyltransferases became apparent when we sought to carry out investigations of GlfT2, a glycosyltransferase essential for cell wall biosynthesis in Mycobacterium tuberculosis. GlfT2 catalyzes the formation of the cell wall galactan, which is a linear polysaccharide consisting of 20-40 repeating D-galactofuranose (Galf) residues. Recombinant GlfT2 can act on synthetic acceptors to produce polymers with lengths similar to those of endogenous galactan, indicating that GlfT2 has an intrinsic ability to control polymer length. To address whether GlfT2 utilizes a processive or distributive mechanism, we developed a mass spectrometry assay. Our approach, which relies on acceptors labeled with stable isotopes, provides direct evidence that GlfT2 is a processive polymerase that maintains contact with the glycan substrate through successive monomer additions. Given this finding, we probed further the catalytic mechanism of GlfT2 to address the basis of an observed kinetic lag phase. These studies suggest that GlfT2 possesses subsites for Galf residue binding and that substrates that can fill these subsites undergo efficient processive polymerization. The presence of these subsites and the kinetic lag phase are common features of processive enzymes. We anticipate that the strategies described herein can be applied to mechanistic studies of other carbohydrate polymerization reactions.
Introduction
The sequence, length, and dispersity of biological polymers are critical determinants of their physiological roles. These properties are controlled by the mechanisms that biosynthetic enzymes employ during the initiation, elongation, and termination stages of the polymerization reaction. For template-dependent polymerases, such as those that generate nucleic acids or proteins, mechanistic investigations have provided a clear picture of how length is controlled. Most of these enzymes utilize processive chain elongation, in which the polymerase remains bound to the template and growing polymer through multiple rounds of monomer addition.1,2 In fewer cases, polymer elongation occurs in a distributive manner, such that each catalytic event is separated by dissociation and re-association of the enzyme and product/substrate.3 The degree of processivity of DNA polymerases, in particular, can be quantified, and some of these enzymes perform thousands of bond-forming events without dissociation from the template.2-4 Monodisperse polymers are formed because the encoding template dictates chain termination.
Although carbohydrate polymers are the most abundant organic compounds in nature,5 little is known about the mechanisms that govern length control in polysaccharide biosynthesis. Polysaccharides are synthesized by glycosyltransferases that catalyze the transfer of tens to thousands of saccharide residues from an activated sugar donor onto a glycoside acceptor. These enzymes coordinate biosynthesis in the absence of a template. Typically, the polysaccharides generated have some level of polydispersity, and many biologically active polysaccharides are able to function over a span of defined lengths.6-10 Still, length control is a feature of carbohydrate polymerases. Certain polysaccharides, like cellulose, are very long (>10,000 residues) while others, such as the mycobacterial galactan, are shorter (<100 residues). Because there is not a specific signal for chain termination, the mechanism of elongation plays a significant role in determining how product length is controlled. For example, in a distributive template-independent polymerization, product length is governed by the concentration of activated donor and exposure of the enzyme to the growing chain. Long polymers are generated only after many cycles of substrate association, bond formation, and polymer release. Conversely, length in a processive polymerization is dictated largely by the strength of interaction of the enzyme with the growing strand, provided that the saccharide donor is not limiting.
At present, very few glycosyltransferases have been evaluated for processivity. Existing methods to explore glycosyltransferase mechanisms can be insensitive, rely on end-point product analysis, and typically do not provide single product resolution. Additional confusion arises because current classification schemes have generally denoted glycosyltransferases as either ‘nonprocessive’ or ‘processive,’ using a distinction that often is based not on mechanism but rather on whether single or multiple glycosidic bonds are generated.11,12 As a result, many issues remain unexplored: whether polymerizing glycosyltransferases generate polysaccharide products through processive or distributive modes of catalysis, what molecular or cellular factors govern the mode of catalysis, and how specific mechanisms contribute to the ability of individual glycosyltransferases to modulate product length.
The need for mechanistic studies is underscored by data indicating that carbohydrate polymer length can be a critical determinant of biological function. The length of polysaccharides and their conjugates influences their physiological roles, including their functions in energy storage, cellular structure and protection, cell differentiation, cell proliferation, and immune responses.13,14 Long-chain polysaccharides containing thousands of monomeric units serve as structural scaffolds (chitin, cellulose),15,16 and are vital for energy storage (starch, glycogen).17,18 Alternatively, shorter polysaccharides and oligosaccharides containing tens or hundreds of residues (polysialic acids, certain hyaluronan chains, and glycosaminoglycans) mediate a variety of signaling processes.7,19-22 Chains of short and intermediate lengths also commonly serve as linker units between biomacromolecular components, as in the case of bacterial peptidoglycan23 and mycobacterial arabinogalactan.24,25
To illuminate how polymerizing glycosyltransferases control and modulate polysaccharide length, we investigated the enzyme GlfT2.26 GlfT2, encoded by the glfT2 (or Rv3808c) gene in Mycobacterium tuberculosis, is a galactosyltransferase involved in cell wall biosynthesis.27 The enzyme catalyzes the synthesis of a linear polymer consisting of approximately 20-40 galactofuranose (Galf) residues linked via alternating β-(1→5) and β-(1→6) glycosidic bonds.28 This polymer, termed galactan, is an essential portion of the mycobacterial cell wall that connects the peptidoglycan to the mycolic acid–arabinan layer.29 Understanding the enzymes that mediate mycobacterial cell wall biosynthesis is important because the process is a validated target for the clinical treatment of diseases such as tuberculosis.30 In addition, compounds that inhibit the enzyme UDP-galactopyranose mutase, which generates UDP-Galf, the glycosyl donor for GlfT2, prevent mycobacterial growth.31 Thus, mechanistic investigations into GlfT2 catalysis can yield new antimycobacterial strategies.
GlfT2 catalyzes successive transfer of Galf residues from the activated donor UDP-Galf to the non-reducing end of a polyprenol-linked oligosaccharide acceptor (1, Figure 1).32-36 We previously demonstrated that recombinant His6-GlfT2 can extend synthetic acceptor mimics, such as 2, to afford polymers with lengths similar to those isolated from mycobacteria.26 Interestingly, the length of polymeric product can be altered by the acceptor lipid substituent. Substrates containing long lipid moieties yield products composed up to 50 Galf residues, whereas substrates containing shorter lipid groups were elongated by only a few Galf units.26 These results led to a proposal for length control by GlfT2, in which the lipid substituent is critical for maintaining binding of the growing polysaccharide chain to the enzyme through multiple rounds of catalysis. Dissociation of polysaccharide products occurs when this dual point binding, or tethering, is no longer favorable.37 This model is consistent with the observation that GlfT2 does not form polymers of a single length, but rather generates a distribution of products centered on a particular length. Our observations in this in vitro system are consistent with the length and polydispersity of galactan polysaccharides isolated from mycobacteria.28,32
Figure 1.

The reaction catalyzed by GlfT2 in mycobacterial galactan biosynthesis. GlfT2 transfers 20 to 40 D-galactofuranose (Galf) residues (n = 10–20) from UDP-Galf to the nonreducing end of a lipid-linked acceptor (1). Synthetic acceptor mimics such as 2 are elongated by GlfT2 to provide degrees of polymerization similar to those of endogenous galactan. Rha, rhamnose; GlcNAc, N-acetylglucosamine.
An underlying assumption of the tethering mechanism is that GlfT2 is a processive enzyme. The catalytic mechanism of GlfT2 was examined using traditional methods, which afforded results consistent with a processive mechanism.26 This conclusion, however, was tentative, because the data generated were neither definitive nor quantitative. Thus, we devised a general strategy to directly distinguish between distributive and processive enzyme-catalyzed reactions. Application of this approach reveals that GlfT2 does indeed catalyze processive chain elongation. Moreover, the assay provides quantitative data on carbohydrate polymerase processivity of the kind that has been used to describe DNA polymerases. Mechanistic insights afforded by this quantitative assay illuminated critical aspects of galactan polymerization by GlfT2, including an observed kinetic lag phase. We propose that this lag phase arises when Galf-binding subsites are not fully occupied by the acceptor. While occupation of these subsites is important for processivity, it does not have an effect on the length of polymers synthesized. Combined, these experiments provide a blueprint to for elucidating the molecular mechanisms of other carbohydrate polymerases, and even other processive enzymes.
Results and Discussion
Single-hit processivity assay
The most common method used to evaluate enzymatic processivity is to analyze the enzyme under conditions that minimize multiple binding events between enzyme and the initiating substrate, that is, with a large substrate to enzyme ratio.3,4,38 When reactions are conducted under “single-hit” conditions, oligomeric products should be generated only if the enzyme retains the elongated substrate through multiple catalytic events. This type of analysis is most commonly used to evaluate DNA polymerases, but it has been used to assess processivity for other enzymes, including protein kinases39 and polymerizing glycosyltransferases.7,40-44 In our previous investigations, the processivity of GlfT2 was interrogated under single-hit conditions, with acceptor substrate 2 in 1000-fold molar excess over enzyme. When reactions were analyzed by matrix-assisted laser-desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry, products elongated by as many as 11 Galf residues were observed at early time points (2 minutes).26 These results suggest GlfT2 is processive.
For the few cases wherein processivity in carbohydrate polymerizations has been investigated, the presence of polymeric products under single-hit conditions has been interpreted as processive catalysis. For example, radiolabeled products have been subjected to chromatographic or electrophoretic separation, and this analysis has revealed a number of plant and bacterial glycosyltransferases are processive, including enzymes that catalyze the production of peptidoglycan,42 dextran,43 N-linked glycan,40 and the Streptococcus pneumoniae type 3 capsular polysaccharide.45 Similarly, polysialyltransferases from Escherichia coli and Neisseria meningitidis and pectin polymerases46 have been examined by assessing the reaction products of fluorescently labeled acceptors using chromatography.41,44 In these assays, a polymerase is determined to be processive when no polymers of intermediate length are observed. Although such an observation supports processive catalysis, it does not provide direct evidence for processivity.
A small number of studies have provided evidence that certain polymerizing glycosyltransferases catalyze distributive polymerization. An example is the hyaluronan (HA) synthase from Pasteurella multocida (PmHAS). PmHAS is a dual-action polymerase that utilizes two separate active sites to add alternating N-acetylglucosamine (GlcNAc) and glucuronic acid (GlcUA) residues.47 PmHAS has been converted into two single-action glycosyltransferases (GlcNAc transferase and GlcUA transferase) that can act in concert to synthesize HA polymers.19 The ability to separate the catalytic domains is consistent with distributive polymerization. Binding data on another enzyme, the chondroitin polymerase K4CP from E. coli, suggests it too employs a distributive mechanism. K4CP catalyzes the synthesis of a polymer consisting of a disaccharide repeat of alternating GlcUA and N-acetylgalactosamine (GalNAc) residues. K4CP, which contains two distinct N- and C-terminal active sites, is capable of binding only a single donor substrate (UDP-GlcUA, UDP-GalNAc, or UDP) at a time.48 This binding mode suggests a means by which the enzyme controls the alternating transfer of GlcUA and GalNAc residues. Chondroitin production likely requires that the acceptor glycan dissociate after each elongation step, so that it can bind to the appropriate active site, but the evidence supporting this conclusion is indirect. Clarifying the catalytic mechanisms of of PmHAS and K4CP depends upon the development of assays that can distinguish between processive and distributive catalytic processes.
A mass spectrometry assay for glycosyltransferase processivity
Our objectives were to develop an assay that would report on whether a polymerizing enzyme is processive or distributive. We also wanted an assay that would afford quantitative data, so that different enzymes, enzyme variants, and substrates could be compared directly. We drew inspiration from the landmark biochemical assays utilized by Meselson and Stahl to probe the mechanisms of DNA replication. They showed that isotopic labeling could be used to differentiate between biochemical mechanisms. By first replicating E. coli to afford uniformly 15N-labeled DNA strands and then switching to an 14N-labeled growth medium, Meselson and Stahl provided compelling evidence that the mechanism of DNA replication is semi-conservative.49 A key feature of their isotopic labeling strategy is that it distinguishes between different mechanistic possibilities. We postulated that substrates differentiated only by mass could be used to distinguish between processive versus distributive carbohydrate polymer biosynthesis. A method that could exploit mass spectrometry would be especially valuable because of its high sensitivity, ease of use, and ability to resolve individual polymeric products. Stable isotope labeling of small molecules and proteins has been utilized extensively for numerous quantitative and comparative mass spectrometry applications,50,51 but there is little precedent for their use in mass spectrometry to decipher enzyme mechanisms.
We envisioned using light and heavy labeled substrates in an order-of-addition experiment. By controlling when a given substrate was present, we could probe for the formation of a processive enzyme-substrate complex (Figure 2). Specifically, GlfT2 can be exposed initially to a “light,” or unlabeled, glycosyl acceptor substrate. Subsequent addition of a “heavy,” or labeled, substrate to the same reaction is then used to assess whether the substrate-bound GlfT2 can be distracted from the initial substrate.52 The ratio of light and heavy products detected with mass spectrometry can discriminate between distributive and processive mechanisms. For the heavy substrate, we synthesized a version of acceptor 2 bearing a d5-phenolic ether at the terminus of its lipid chain (3). We reasoned that this substrate would be chemically equivalent to 2 in enzymatic assays because the deuterium labels would be incorporated at positions distal to the site of bond formation. Most significantly, this substrate should provide a sufficient mass difference to use mass spectrometry for quantitation.
Figure 2.

A distraction assay developed to investigate GlfT2 processivity. GlfT2 and UDP-Galf are exposed to “light” (unlabeled) acceptor 2 for time t1 to initiate polymerization. “Heavy” (isotopically labeled) acceptor 3 is then added and the reaction is allowed to proceed for an additional period t2 (t2 > t1). The reaction products are analyzed by MALDI-TOF mass spectrometry. A bias between heavy and light products is expected if the heavy acceptor cannot compete for GlfT2 elongation with the light acceptor, an outcome consistent with a processive mechanism. A more equal product distribution is expected if the heavy acceptor has equal access to GlfT2, a situation that would result in a distributive mechanism.
To test the feasibility of this approach, the polymerization experiments were conducted by incubating GlfT2 with UDP-Galf and light acceptor 2 for a preincubation time, t1, and then adding heavy acceptor 3. The reaction was allowed to proceed for an additional period, t2 (t2 > t1), prior to analysis by mass spectrometry. Anticipated outcomes for processive versus distributive mechanisms are depicted in Figure 2. If GlfT2 forms a processive catalytic complex, it will maintain contact with the light acceptor added initially, even after the heavy acceptor is added. If the heavy acceptor cannot distract substrate-bound GlfT2, we expect that products from processive polymerization reactions should contain fewer heavy polymers. Conversely, if the polymerization reaction is distributive, the light acceptors will dissociate from the enzyme after each bond-forming event; therefore, both acceptors have equal access to the enzyme. In this case, both acceptors will give rise to products of similar lengths. A requirement for this analysis is that the light and heavy acceptors behave identically with GlfT2. The validity of this assumption was tested using a coupled kinetic assay, which confirmed that GlfT2 catalyzes the elongation of each acceptor with the same rate (Supporting Information). As expected, GlfT2 exposure provided identical degrees of polymerization with either acceptor (Figure S1).
A competitive distraction assay indicates GlfT2 is processive
Execution of the competitive distraction assay required determining appropriate incubation times, t1 and t2. The initiation time t1 needed to be long enough to allow GlfT2 to form a catalytic complex with the light acceptor; extended t1 times, however, would provide a head-start with the light acceptor that would not be indicative of a single binding event. Meanwhile, the incubation time following heavy acceptor addition (t2) must be sufficient for detectable levels of products to accumulate, but short enough that a lag in heavy product formation would be observable. We therefore scanned a range of t1 and t2 times to find conditions that report accurately on the mechanism of GlfT2.
We sought to find as short a t1 time as possible. First, we tested 15 sec and the assay products were evaluated after t2 incubation times of 15 sec and 30 sec. At these early time points, the only products observable by MALDI-TOF mass spectrometry contained an additional two Galf residues. There was little difference in the ratios of light or heavy reaction products following additional incubation of 15 sec (Figure S2). There are two possible explanations for this result: either GlfT2 utilizes a distributive mechanism, or a pre-incubation time of 15 sec is not sufficient for the detection of products from a processive GlfT2–substrate complex. To test the latter possibility, we carried out a distraction assay with a pre-incubation time of 30 sec and a series of t2 times ranging from 30 sec to 90 sec. Following incubation for a total time of 120 sec (t2 = 90 sec), the longest products observed by MALDI mass spectrometry contained an additional 13 Galf residues (Figure S3). Products elongated by 7, 9, and 11 Galf residues were the most abundant and served as the best diagnostic peaks for evaluating processivity (Figure 3A). Under these conditions, a clear distinction in the abundance of light and heavy products was observed. Very little, if any, heavy products were detected at the longest polymer lengths (+12 or +13 Galf). These results are consistent with a processive mechanism. In conjunction with the previous data, they indicate that the processive complex forms in 15-30 sec. The expected isotopic bias in products also was observed when polymerization was initiated with the heavy acceptor (Figure S4), demonstrating that the increase in the relative production of light and heavy products does not arise from the isotopic composition of the initial substrate added.
Figure 3.

GlfT2 catalyzes processive polymer formation. Data shown are derived from MALDI-TOF mass spectral analyses of assays containing isotopically labeled acceptor substrates 2 and 3 and UDP-Galf. Peaks that correspond to m/z values of [M + Na]+, where M equals the mass of compounds 2 or 3 possessing an additional n Galf residues are shown. (A) The competitive distraction assay, wherein polymerization was initiated with light acceptor 2 for 30 sec and chased with heavy acceptor 3 for 90 sec, shows a significant lag in the formation of heavy products (red). Long products (e.g. +13 Galf) resulting from elongation of the heavy acceptor were not detected. (B) A non-competitive assay where GlfT2 was reacted with either 2 for 120 sec or 3 for 90 sec in separate reactions. The two reactions were combined, and then analyzed. Little bias is observed in the polymeric products derived from heavy and light acceptors, including products containing as many as 17 additional Galf residues.
To test whether the product distribution was a result of the time of substrate exposure to GlfT2, we treated the two substrates with GlfT2 in parallel reactions. In the first reaction, the light acceptor 2 was exposed to GlfT2 for 120 sec (t1 + t2) while in a separate reaction heavy acceptor 3 was incubated with GlfT2 for 90 sec (t2). The reactions were quenched, then combined and analyzed by MALDI mass spectrometry. These reactions showed the abundance of light and heavy polymers was virtually identical, an outcome that starkly contrasts with the competitive distraction experiment (Figure 3B, full spectra Figure S5). The greatest differences in light versus heavy products were apparent in the longest polymers produced (those elongated by 17 Galf units). Together, the data reveal that the preponderance of elongated light products observed in the competitive experiment results from the inability of the heavy acceptor to distract GlfT2 from the light acceptor once polymerization has initiated.
The mass spectrometry-based method described herein should be generally applicable for dissecting the molecular mechanisms of other carbohydrate polymerases. To implement such an assay, acceptor substrates bearing isotopic labels are needed. A number of other polymerases catalyze the formation of lipid-linked polysaccharides, including those involved in bacterial peptidoglycan and capsular polysaccharide biosynthesis.8,53,54 For these systems, isotopic labels can also be incorporated in the lipid aglycone, at positions distal from where bond formation occurs. Other strategies can be employed to install labels for polymerases that act on acceptor substrates that do not contain an aglycone. For example, the reducing end of an acceptor glycan can be readily modified with alkyl hydrazides or aminooxy nucleophiles to furnish isotopically differentiated substrates.
Single-hit polymer extension by GlfT2 and evaluation of microscopic processivity parameters
The data indicating that GlfT2 catalyzes processive galactan extension led us to seek a quantitative measure of processivity. A general quantitative description could be used to compare the mechanism of polymerizing glycosyltransferases in different contexts. Unfortunately, in the few cases that processivity in carbohydrate polymerization reactions have been investigated, a quantitative description was lacking. In contrast, gel electrophoresis has provided the means of conducting quantitative analyses of several DNA polymerases.3,4,55 In this way, a processivity factor, Pn, can be calculated for each reaction product observed (possessing an additional n monomer units) under single-hit conditions. The value Pn describes the probability that the polymerase will continue catalysis to form product n + 1 rather than dissociate. To determine quantitative processivity parameters using mass spectrometry, GlfT2 was incubated with 500-fold molar excess of acceptor 3 and UDP-Galf for 3 min, and the reaction products were analyzed by MALDI-TOF mass spectrometry (Figure 4). Products extended by as many as 13 Galf residues were observed under these conditions. The percent of active polymerases and Pn value for each + n Galf addition product was calculated from the mass spectral peak intensity (Table 1). Because no products extended by a single Galf residue were observed, the percentage of active polymerases adding at least 2 Galf residues was set to 100%. By definition, the percentage of active polymerases decreased after each subsequent Galf addition due to dissociation of substrate from some proportion of enzymes (Equation 1, Experimental Procedures). With these conditions, a percentage of GlfT2 enzymes fail to incorporate more than 2 Galf monomers prior to dissociation of the product, but approximately 40% of the GlfT2 enzymes incorporate at least 6 Galf residues before dissociating from the acceptor substrate.
Figure 4.

Results from a single-hit assay of GlfT2 and acceptor 3 that were used to calculate processivity parameters. Acceptor 3 was incubated in 500-fold molar excess to GlfT2 along with UDP-Galf and the products were analyzed by MALDI-TOF mass spectrometry after 3 min. Peaks that correspond to m/z values of [M + Na]+, where M equals the mass of compounds 3 possessing an additional n Galf residues are shown. Products are observed from n = 2–13.
Table 1. Processivity parameters derived from 3 min. single-hit reaction of GlfT2 with UDP-Galf and acceptor 3.
| # of Galf residues Added | Active Polymerases (%) | Pn* |
|---|---|---|
| 2 | 100.0 | 0.57 |
| 3 | 57.4 | 1.00 |
| 4 | 57.4 | 0.78 |
| 5 | 44.9 | 0.90 |
| 6 | 40.6 | 0.90 |
| 7 | 36.5 | 0.79 |
| 8 | 28.7 | 0.86 |
| 9 | 24.7 | 0.55 |
| 10 | 13.5 | 0.75 |
| 11 | 10.1 | 0.33 |
| 12 | 3.3 | 0.58 |
| 13 | 1.9 | 0.00 |
An average Pn value of 0.73 ± 0.20 was calculated for 2-12 Galf additions
The processivity factor, Pn, was calculated for each Galf addition. Equation 2 provided the fraction of active polymerases, at any position n, that add at least one additional Galf residue (Table 1). Of the GlfT2 enzymes that incorporated 4 Galf monomers, 78% added at least one more Galf, meaning that P4 = 0.78. The Pn values ranged from 1.00 for n = 3 to 0.31 for n = 11, with an average value of 0.73 ± 0.20. For context, the average Pn value for yeast DNA polymerase η, which possesses moderate processivity, was calculated to be 0.76 ± 0.20. 55 On the other hand, many DNA polymerases, such as the E. coli DNA polymerases I and II, exhibit extremely high processivity (Pn > 0.99).56,57 We anticipate that a collection of processivity values can provide insight into the mechanistic attributes of different glycosyltransferases. Specifically, access to these Pn values could reveal the connections between polysaccharide length and polymerase mechanism. Moreover, such data can be used to compare variants of a glycosyltransferase to ascertain whether specific mutations affect processivity or to evaluate the processivity of a single enzyme with multiple substrates.58
Access to a quantitative description of processivity raises interesting questions about GlfT2-catalyzed chain elongation. The data suggest that the early stages of polymerization are relatively inefficient. For example, many active GlfT2 enzymes dissociate from the acceptor substrate following only 2 Galf additions (43 %, P2 = 0.57). This observation is consistent with previous findings that GlfT2 exhibits a kinetic lag phase with synthetic disaccharide acceptors.26 Experiments using a continuous assay indicate that the lag phase lasts for approximately 1-2 min with acceptor 2. The rate of conversion of UDP-Galf to UDP in this period is approximately 10-fold less than that observed under steady-state conditions. We hypothesize that the lag phase likely arises from the lower affinity of substrates containing only a few Galf residues. With acceptors that occupy these subsites, we postulate that GlfT2 reaches a steady-state rate of Galf addition and achieves high processivity.
Abrogation of kinetic lag phase by extended GlfT2 acceptor substrates
To test whether the lag phase exhibited by GlfT2 results from unoccupied Galf-binding subsites, we compared disaccharide 4 with a tetrasaccharide acceptor in a coupled kinetic assay. If our hypothesis is correct, a faster initial rate should be measured with a tetrasaccharide acceptor because it can fill more subsites. The tetrasaccharide GlfT2 acceptor needed for these studies was prepared by a chemoenzymatic approach. Reaction mixtures containing GlfT2, disaccharide 4, and UDP-Galf were optimized to favor short oligomeric products in which only 2 to 4 Galf residues had been added. Specifically, equimolar concentrations of UDP-Galf and 4 were employed. After the reaction was allowed to proceed for one hour, the products were purified by analytical C18 reverse-phase HPLC, and the fractions obtained were analyzed by MALDI-TOF mass spectrometry (Figures S6-S8). We generated samples with tetrasaccharide or hexasaccharide acceptors (5 and 6, Figure 6A), and a sample containing a mixture of acceptors with 4 -19 Galf residues (7).
Figure 6.
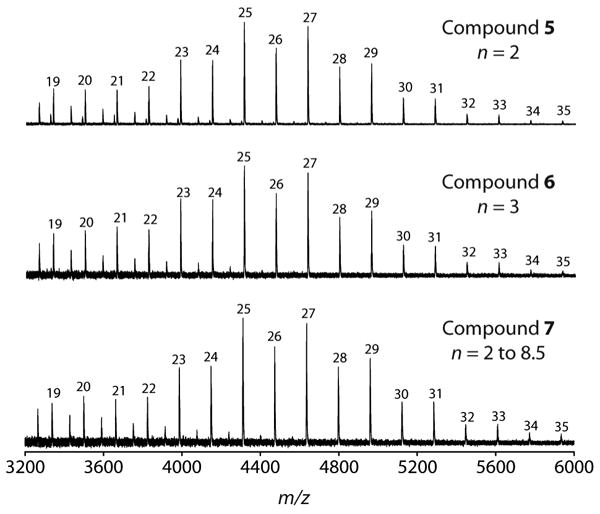
GlfT2 catalyzes the synthesis of polymers with the same degrees of polymerization irrespective of the number of Galf residues on the initiating substrate. GlfT2 was exposed to acceptor substrates 5, 6, or 7 and UDP-Galf for 20 h. Nearly identical degrees of polymerization were observed by MALDI-TOF mass spectrometry for each reaction. Peaks that correspond to m/z values of [M + Na]+, where M equals the mass of products possessing a total of m Galf residues are shown.
In agreement with our prediction, there was no kinetic lag phase when tetrasaccharide 5 was exposed to GlfT2 and UDP-Galf. The initial rate of UDP formation with 5 was similar to the steady-state rate achieved with the disaccharide acceptor 4 (1.4 μM/min, Figure 5B). We next conducted single-hit assays with acceptors 4 and 5 to investigate whether there was a difference in the length of polymers produced in this initial phase of catalysis. We anticipated that these conditions would result in longer polymeric products from tetrasaccharide 5. Approximately 100-fold molar excess of 4 or 5 was exposed to GlfT2 for 10 minutes, and the resulting products were analyzed by MALDI-TOF mass spectrometry. After this relatively short incubation time, both acceptors gave rise to only a small proportion of elongated products, yet differences were observed. Longer products were detected with the tetrasaccharide acceptor. Specifically, polymers possessing as many as 15 Galf residues were obtained from the tetrasaccharide acceptor (Figure 5C), but the longest polysaccharides detected with the disaccharide acceptor were extended by only 9 Galf units (Figure 5D). These results are consistent with the processivity parameters and suggest that GlfT2 is more processive when more subsites are occupied.
Figure 5.
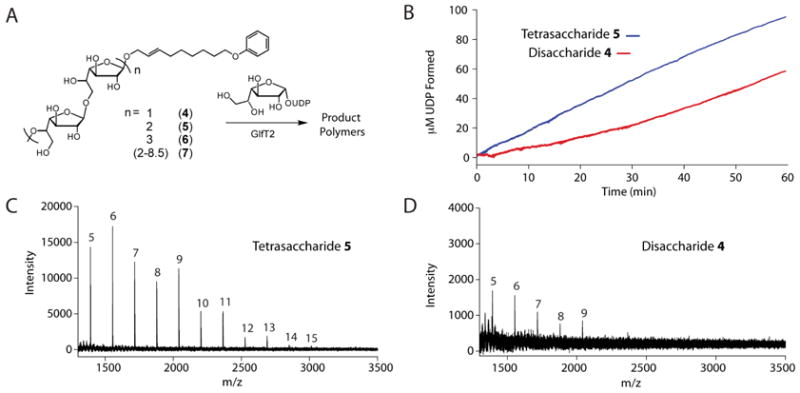
GlfT2 prefers tetrasaccharide acceptor 5 to disaccharide acceptor 4 in kinetic and single-hit assays. (A) Acceptor substrates were prepared that possess varying numbers of Galf residues to probe GlfT2 mechanism. Acceptor substrates composed of 4, 6, or a mixture of 4-17 Galf residues were isolated. (B) Treatment of UDP-Galf and disaccharide acceptor 4 (red) with GlfT2 resulted in a pronounced lag in the formation of UDP in a coupled enzyme assay. This lag phase is not present with tetrasaccharide acceptor 5 (blue). (C) When GlfT2 was exposed to 5 and UDP-Galf under single-hit conditions, products elongated by as many as 15 Galf residues were observed. (D) When GlfT2 was mixed with 4 and UDP-Galf under single-hit conditions, products elongated by as many as 9 Galf residues were observed. For both (C) and (D), peaks that correspond to m/z values of [M + Na]+, where M equals the mass of compounds 4 or 5 possessing a total of m Galf residues are shown.
GlfT2 shares common features with processive enzymes
The observation of a kinetic lag phase is not unique to GlfT2.1,5,41,59 We postulate that this kinetic lag phase is an intrinsic feature of processive enzymes. Indeed, enzymes exhibiting increased processivity with substrates that occupy all monomer subsites is observed for processive enzymes, ranging from DNA polymerases,1 to polyhydroxyalkanoate synthases,5 to ubiquitin ligases.59 A test of the subsite model was conducted by delCardayre and Raines who transformed a distributive ribonuclease into one that was processive.52 In this case, the processive enzyme possessed nucleotide-binding subsites, which allow the enzyme to maintain contact with its RNA substrate through multiple cleavage steps. For processive polymerizing enzymes, substrates that fill all of the subsites will be polymerized rapidly—those that cannot will display a lag phase. Many polymerizing glycosyltransferases exhibit a lag phase. For example, the transfer of sialic acid residues by E. coli K92 polysialyltransferase occurs in two stages distinguished by the kinetics; initial accumulation of products containing 1-8 additional sialic acid residues is followed by the rapid production of high molecular weight polymers.41 Similar initiation events have also been reported for the hyaluronan synthase from P. multocida,60 peptidoglycan glycosyltransferases from E. coli and Aquifex aeolicus,42,61,62 and the synthase responsible for synthesizing the type 3 capsular polysaccharide in S. pneumoniae.45 Given our observations and those in the literature,52 we anticipate that, as with our synthetic acceptors, the physiological GlfT2 acceptors will exhibit a kinetic lag phase.
GlfT2 exerts length control
Our previous studies suggested that GlfT2 produces polymers of controlled length and that product length depends on the presence and features of the lipid substituent of the initiating substrate.26 Our findings that GlfT2 is a processive polymerase are consistent with this model. With the development of our chemoenzymatic route to oligomers, we were poised to probe whether the degrees of polymerization on the initiating acceptor substrate influence the length of products GlfT2 generates. If the ability of GlfT2 to control polymer length is intrinsic and mediated by dual-point substrate binding, we expect that the number of Galf residues on the initiating substrate should not influence final product length. To test this idea, GlfT2 was exposed to UDP-Galf and equimolar concentrations of acceptor substrates 5, 6 or 7 (25 μM). Characterization of products by MALDI-TOF mass spectrometry from 20 h reactions revealed that each of the initiating acceptor substrates was processed to afford long galactan chains. Most importantly, the degrees of polymerization were indistinguishable for each of these reactions (Figure 6). The most abundant products observed by mass spectrometry for each assay possessed a total of 25 and 27 Galf residues, while the longest products observed contained a total of 35 Galf sugars. These results provide evidence that length control is an intrinsic feature of GlfT2 catalysis and the number of Galf residues on the initiating acceptor substrate does not influence final product length.
Conclusions
We have developed a general assay for processivity. This approach was applied to illuminate the molecular mechanism of GlfT2, an essential cell wall biosynthetic enzyme from M. tuberculosis. The data indicate that GlfT2 synthesizes polymers of controlled length that are commensurate with those isolated from mycobacteria. Mechanistic investigations of the GlfT2 polymerization reaction were conducted using a distraction assay with isotopically labeled acceptor substrates. The data unambiguously indicate that GlfT2 is a processive polymerase. To provide a quantitative benchmark of this processivity for comparison to other enzymes, we adapted a description of processivity that has been traditionally applied to nucleic acid polymerases. We found that galactan polymerization by GlfT2 exhibits a kinetic lag phase. This lag phase is eliminated when GlfT2 is reacted with acceptor substrates containing additional Galf residues, suggesting that GlfT2 possesses Galf-binding subsites that contribute to efficient processive chain elongation. This attribute of GlfT2 appears to be a general feature of processive enzymes. We anticipate that the tools implemented herein can be used to correlate and compare various aspects of carbohydrate polymerization reactions, including the structure and mechanism of polymerizing enzymes and how these factors impact control of polymer length.
Experimental Procedures
Synthesis of substrates
Acceptor substrates 2, 3, and 4 used in this work were synthesized as reported previously.26,63 UDP-Galf was prepared according to a published procedure.64 Elongated His6-GlfT2 reaction products were generated as described below.
Production of His6-GlfT2
The gene encoding His6-GlfT2 was overexpressed, and the protein product was purified according to published procedures.26 Briefly, a pET-24a plasmid encoding His6-GlfT2 was transformed into Tuner (DE3) E. coli cells (Novagen) by electroporation and plated onto Luria-Bertani agar with 50 μg/mL kanamycin. A 50 mL starter culture (LB with 50 μg/mL kanamycin) was grown for 12 h at 37 °C. A 4 mL portion of the starter culture was then used to inoculate larger-volume cultures (1 L of LB with 50 μg/mL kanamycin), which were incubated at 37 °C until an optical density at 600 nm exceeded 0.8. At this time, the cultures were placed in an ice bath for 1 h, at which time overexpression was induced with 0.3 mM β-thiogalactopyranoside (IPTG) and incubated at 15 °C for 18-20 h. Cultures were harvested by centrifugation at 5,000 rpm and the cell pellets were frozen at -80 °C until further use. Cell pellets were thawed on ice in a lysis buffer that contained 50 mM Hepes, pH 7.4, 25 mM imidazole, and 500 mM sodium chloride (NaCl). Protease inhibitor cocktail III (Calbiochem) and lysozyme were also added to the lysis mixture. Cells were lysed by sonication (4 × 10s cycles at 90% duty cycle, Branson Sonifier 450, tip setting 7) and the lysate clarified by centrifugation (22,000 × g for 1 h). The soluble lysate was filtered through a 0.45 μm nylon filter (Millipore) and loaded onto a pre-equilibrated (in lysis buffer) 5 mL HisTrap column (GE Healthcare) at 1.0 mL/min using an AKTA FPLC system. The column was washed until the UV Absorbance at 260 nm reached a baseline level. At this point, a linear gradient from 0% to 100% elution buffer (50 mM Hepes, pH 7.4, 500 mM imidazole, 500 mM NaCl) was applied to the column over 20 min to elute bound protein. Fractions containing His6-GlfT2 were identified by SDS-PAGE. Glycerol (10% v/v) was added to H6-GlfT2 fractions, which were flash-frozen and stored at -80 °C until use. For enzyme assays, a sample of His6-GlfT2 was dialyzed twice into 2 L of 50 mM Hepes, pH 7.4, 100 mM NaCl and 5 mM ethylenediaminetetraacetic acid (EDTA) using 10,000 molecular weight cut-off dialysis cassettes (Pierce Biotechnology). Protein concentration was determined using the BCA assay (Pierce Biotechnology) with bovine serum albumin as standard.
General procedure for MALDI mass spectrometric analysis of GlfT2 reaction products
Typically, reactions of 120 μL total volume contained final concentrations of 0.2 μM His6-GlfT2, 50-200 μM acceptor, 150-1250 μM UDP-Galf in 50 mM Hepes, pH 7.0, 25 mM magnesium chloride (MgCl2), and 100 mM NaCl. Reactions were incubated at room temperature for a specified time, then quenched with 120 μL of a 1:1 mixture of methanol:chloroform. Quenched reaction mixtures were evaporated to dryness under vacuum in a SpeedVac SC100 (Varian) and the products were resuspended in 50 μL 50% acetonitrile:water for MALDI mass spectrometry analysis. Samples for MALDI mass spectrometry analysis were spotted as a 1:3 mixture with α-cyano-4-hydroxycinnamic acid matrix and spectra were recorded in positive linear mode using a Bruker Ultraflex III mass spectrometer. Substrate concentrations and special considerations for specific assays are described below.
Isolation of extended GlfT2 reaction products
In order to generate short reaction products, several reactions with a total volume of 120 μL, containing 1.0 mM UDP-Galf, 1.0 mM acceptor 4, and 0.2 μM His6-GlfT2 were incubated at room temperature. Following one hour, the reactions were quenched with an equal volume of a mixture of 1:1 methanol:chloroform and evaporated to dryness. The samples were resuspended in 50 μL of 50 % acetonitrile:water and reaction progress was assessed by MALDI-TOF mass spectrometry. The products obtained were a mixture consisting mainly of short polysaccharide products (ca. 2 to 9 Galf additions), which were then separated by analytical C18 HPLC (Thermo Scientific Hypersil Gold column, 5 μm, 250 × 4.6 mm ID). Gradient elution from 20 – 80 % acetonitrile (v/v), 0.1 % acetic acid (v/v) was employed for product separation at a flow rate of 1.0 mL/min. Due to low abundance of reaction products, the chromatograph was monitored at 200 nm. The identity of each collected fraction was analyzed by MALDI-TOF mass spectrometry (Figures S6-S8).
GlfT2 elongation of oligomeric acceptor substrates
Reactions contained acceptors 5, 6, or 7 with approximately 25 μM acceptor and 1.25 mM UDP-Galf. The samples were exposed to 0.2 μM His6-GlfT2 for 20 h at 25 °C, then quenched with an equal volume of a 1:1 mixture of chloroform:methanol. The samples were resuspended in 50 μL of 50 % acetonitrile:water and analyzed by MALDI-TOF mass spectrometry.
Distraction assays
Assays were conducted in a total volume of 240 μL that contained 0.2 μM His6-GlfT2, 100 μM of acceptors 2 and 3, and 150 μM UDP-Galf. Acceptor 2 and UDP-Galf were exposed to His6-GlfT2 for time t1 (15 or 30 sec) at room temperature. Acceptor 3 was added at this time. Following an additional incubation period, t2, a 120 μL portion of the reaction was quenched with an equal volume of a 1:1 mixture of methanol:chloroform. In the assay where t1 was 15 sec, the t2 timepoints were 15 sec and 30 sec. For the assay where t1 was 30 sec, the t2 timepoints were 75 sec or 90 sec. For the distraction assay control, two separate 240 μL assays were prepared, containing either 100 μM of either acceptor 2 or acceptor 3 and 150 μM UDP-Galf. A 120 μL portion the assay containing acceptor 2 was quenched after 105 sec and combined with a 120 μL portion of the assay combining acceptor 3 that had been incubated for 75 sec. Meanwhile, the 120 sec assay containing acceptor 2 and 90 sec assay containing acceptor 3 were combined in a similar manner. The samples were resuspended in 50 μL of 50 % acetonitrile:water and analyzed by MALDI-TOF mass spectrometry.
Single-hit GlfT2 assays
The assay with acceptor 3 contained 0.2 μM His6-GlfT2, 100 μM acceptor (500-fold acceptor to enzyme ratio) and 150 μM UDP-Galf and was incubated for 3 min prior to organic quench. Single-hit assays with 4 or elongated tetrasaccharide acceptor 5 contained 0.2 μM His6-GlfT2, 200 μM acceptor (500-fold acceptor to enzyme ratio) and 150 μM UDP-Galf. Due to slower rate of product formation with these substrates, the assays were exposed to His6-GlfT2 for 10 min prior to an organic quench with an equal volume of a mixture of 1:1 chloroform:methanol. Intensities of each elongation product were obtained using Bruker FlexAnalysis software. For each Galf addition n, the percentage of active polymerase molecules incorporating at least n Galf residues is given by Equation 1,55
| (Equation 1) |
where I1 is the intensity of the product extended by 1 Galf residue, In is the intensity of the product extended by n Galf residues, etc. For each product extended by n Galf residues, the probability that GlfT2 will incorporate an additional Galf rather than dissociating was given by the processivity factor Pn, which is described by Equation 2,
| (Equation 2) |
Combining Equations 1 and 2 provides an expression for Pn in terms of mass spectral intensity,
| (Equation 3) |
Coupled assay to measure UDP production by H6-GlfT2
The following were mixed together in a 1 cm quartz cuvette to give a total volume of 120 μL: 50 mM Hepes, pH 7.0, 25 mM MgCl2, 100 mM NaCl, 300 units of pyruvate kinase (Sigma), 20 units of lactate dehydrogenase (Sigma), 250 μM nicotinamide adenine dinucleotide (NADH), 500 μM phosphoenolpyruvate, and 0.2 μM His6-GlfT2. The formation of NAD+ was monitored at an absorbance of 340 nm over time in a Cary 50 Bio UV-Visible Spectrophotometer (Varian), until a steady baseline was observed. UDP-Galf was then added to a final concentration of 1.25 mM. Again, after a steady baseline was observed, acceptor substrate was added to the desired concentration. Absorbance at 340 nm was monitored over time and the steady-state rate was calculated from the slope of the linear portion of the decrease in absorbance over time using ε = 6300 M-1 cm-1 for NADH.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health (R01 AI063596). M.R.L. was supported by an NIH Postdoctoral Fellowship (F32GM089219). R.A.S. was supported by an American Chemical Society Division of Medicinal Chemistry Graduate Fellowship. MALDI-TOF mass spectrometry data were obtained at the University of Wisconsin–Madison Chemistry Instrument Center Mass Spectrometry Facility on a Bruker Ultraflex III Instrument that was supported by the NIH (NIH NCRR 1S10RR024601). We thank Dr. M.M. Vestling for technical assistance, and Dr. J.F. May for conducting preliminary experiments regarding the kinetic lag phase of GlfT2 and helpful discussions.
Footnotes
Supporting Information Available. Supplementary Figures S1-S8, kinetic characterization of acceptor substrates 2 and 3, and processivity parameters for single-hit GlfT2 elongation of substrates 4 and 5 are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Breyer WA, Matthews BW. Protein Sci. 2001;10:1699–1711. doi: 10.1110/ps.10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornberg A, Baker TA. DNA Replication. W.H. Freeman; New York: 1992. [Google Scholar]
- 3.McClure WR, Chow Y. Methods Enzymol. 1980;64:277–297. doi: 10.1016/s0076-6879(80)64013-0. [DOI] [PubMed] [Google Scholar]
- 4.von Hippel PH, Fairfield FR, Dolejsi MK. Ann NY Acad Sci. 1994;726:118–131. doi: 10.1111/j.1749-6632.1994.tb52803.x. [DOI] [PubMed] [Google Scholar]
- 5.Stubbe J, Tian J, He A, Sinskey AJ, Lawrence AG, Liu P. Annu Rev Biochem. 2005;74:433–480. doi: 10.1146/annurev.biochem.74.082803.133013. [DOI] [PubMed] [Google Scholar]
- 6.Somerville C. Annu Rev Cell Dev Biol. 2006;22:53–78. doi: 10.1146/annurev.cellbio.22.022206.160206. [DOI] [PubMed] [Google Scholar]
- 7.Weigel PH, DeAngelis PL. J Biol Chem. 2007;282:36777–36781. doi: 10.1074/jbc.R700036200. [DOI] [PubMed] [Google Scholar]
- 8.Whitfield C. Annu Rev Biochem. 2006;75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 9.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang TS, Manning SA, Walker S, Kahne D. J Am Chem Soc. 2008;130:14068–14069. doi: 10.1021/ja806016y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coutinho PM, Deleury E, Davies GJ, Henrissat B. J Mol Biol. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- 12.Saxena IM, Brown RM, Jr, Fevre M, Geremia RA, Henrissat B. J Bacteriol. 1995;177:1419–1424. doi: 10.1128/jb.177.6.1419-1424.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiessling LL, Splain RA. Annu Rev Biochem. 2010;79:619–653. doi: 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]
- 15.Ross P, Mayer R, Benziman M. Microbiol Rev. 1991;55:35–58. doi: 10.1128/mr.55.1.35-58.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minke R, Blackwell J. J Mol Biol. 1978;120:167–181. doi: 10.1016/0022-2836(78)90063-3. [DOI] [PubMed] [Google Scholar]
- 17.Ball SG, Morell MK. Annu Rev Plant Biol. 2003;54:207–233. doi: 10.1146/annurev.arplant.54.031902.134927. [DOI] [PubMed] [Google Scholar]
- 18.Smith AM. Biomacromolecules. 2001;2:335–341. doi: 10.1021/bm000133c. [DOI] [PubMed] [Google Scholar]
- 19.DeAngelis PL, Oatman LC, Gay DF. J Biol Chem. 2003;278:35199–35203. doi: 10.1074/jbc.M306431200. [DOI] [PubMed] [Google Scholar]
- 20.Jann K, Jann B. Can J Microbiol. 1992;38:705–710. doi: 10.1139/m92-116. [DOI] [PubMed] [Google Scholar]
- 21.DeAngelis PL. Glycobiology. 2002;12:9R–16R. doi: 10.1093/glycob/12.1.9r. [DOI] [PubMed] [Google Scholar]
- 22.Stern R. Glycobiology. 2003;13:105R–115R. doi: 10.1093/glycob/cwg112. [DOI] [PubMed] [Google Scholar]
- 23.van Heijenoort J. Glycobiology. 2001;11:25R–36R. doi: 10.1093/glycob/11.3.25r. [DOI] [PubMed] [Google Scholar]
- 24.Berg S, Kaur D, Jackson M, Brennan PJ. Glycobiology. 2007;17:35–56R. doi: 10.1093/glycob/cwm010. [DOI] [PubMed] [Google Scholar]
- 25.Brennan PJ, Nikaido H. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 26.May JF, Splain RA, Brotschi C, Kiessling LL. Proc Natl Acad Sci USA. 2009;106:11851–11856. doi: 10.1073/pnas.0901407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sassetti CM, Boyd DH, Rubin EJ. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 28.Besra GS, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. Biochemistry. 1995;34:4257–4266. doi: 10.1021/bi00013a015. [DOI] [PubMed] [Google Scholar]
- 29.Pan F, Jackson M, Ma Y, McNeil M. J Bacteriol. 2001;183:3991–3998. doi: 10.1128/JB.183.13.3991-3998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belanger AE, Besra GS, Ford ME, Mikusova K, Belisle JT, Brennan PJ, Inamine JM. Proc Natl Acad Sci USA. 1996;93:11919–11924. doi: 10.1073/pnas.93.21.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dykhuizen EC, May JF, Tongpenyai A, Kiessling LL. J Am Chem Soc. 2008;130:6706–6707. doi: 10.1021/ja8018687. [DOI] [PubMed] [Google Scholar]
- 32.Belanova M, Dianiskova P, Brennan PJ, Completo GC, Rose NL, Lowary TL, Mikusova K. J Bacteriol. 2008;190:1141–1145. doi: 10.1128/JB.01326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kremer L, Dover LG, Morehouse C, Hitchin P, Everett M, Morris HR, Dell A, Brennan PJ, McNeil MR, Flaherty C, Duncan K, Besra GS. J Biol Chem. 2001;276:26430–26440. doi: 10.1074/jbc.M102022200. [DOI] [PubMed] [Google Scholar]
- 34.Mikusova K, Belanova M, Kordulakova J, Honda K, McNeil MR, Mahapatra S, Crick DC, Brennan PJ. J Bacteriol. 2006;188:6592–6598. doi: 10.1128/JB.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mikusova K, Yagi T, Stern R, McNeil MR, Besra GS, Crick DC, Brennan PJ. J Biol Chem. 2000;275:33890–33897. doi: 10.1074/jbc.M006875200. [DOI] [PubMed] [Google Scholar]
- 36.Rose NL, Completo GC, Lin SJ, McNeil M, Palcic MM, Lowary TL. J Am Chem Soc. 2006;128:6721–6729. doi: 10.1021/ja058254d. [DOI] [PubMed] [Google Scholar]
- 37.Kramer RH, Karpen JW. Nature. 1998;395:710–713. doi: 10.1038/27227. [DOI] [PubMed] [Google Scholar]
- 38.Bambara RA, Fay PJ, Mallaber LM. Methods Enzymol. 1995;262:270–280. doi: 10.1016/0076-6879(95)62023-0. [DOI] [PubMed] [Google Scholar]
- 39.Aubol BE, Chakrabarti S, Ngo J, Shaffer J, Nolen B, Fu XD, Ghosh G, Adams JA. Proc Natl Acad Sci USA. 2003;100:12601–12606. doi: 10.1073/pnas.1635129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Troutman JM, Imperiali B. Biochemistry. 2009;48:2807–2816. doi: 10.1021/bi802284d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vionnet J, Vann WF. Glycobiology. 2007;17:735–743. doi: 10.1093/glycob/cwm032. [DOI] [PubMed] [Google Scholar]
- 42.Barrett D, Wang TS, Yuan Y, Zhang Y, Kahne D, Walker S. J Biol Chem. 2007;282:31964–31971. doi: 10.1074/jbc.M705440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robyt JF, Yoon SH, Mukerjea R. Carbohydr Res. 2008;343:3039–3048. doi: 10.1016/j.carres.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 44.Freiberger F, Claus H, Gunzel A, Oltmann-Norden I, Vionnet J, Muhlenhoff M, Vogel U, Vann WF, Gerardy-Schahn R, Stummeyer K. Mol Microbiol. 2007;65:1258–1275. doi: 10.1111/j.1365-2958.2007.05862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forsee WT, Cartee RT, Yother J. J Biol Chem. 2006;281:6283–6289. doi: 10.1074/jbc.M511124200. [DOI] [PubMed] [Google Scholar]
- 46.Akita K, Ishimizu T, Tsukamoto T, Ando T, Hase S. Plant Physiol. 2002;130:374–379. doi: 10.1104/pp.005587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jing W, DeAngelis PL. Glycobiology. 2000;10:883–889. doi: 10.1093/glycob/10.9.883. [DOI] [PubMed] [Google Scholar]
- 48.Sobhany M, Kakuta Y, Sugiura N, Kimata K, Negishi M. J Biol Chem. 2008;283:32328–32333. doi: 10.1074/jbc.M804332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meselson M, Stahl FW. Proc Natl Acad Sci USA. 1958;44:671–682. doi: 10.1073/pnas.44.7.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ong SE, Mann M. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 51.Tao WA, Aebersold R. Curr Opin Biotechnol. 2003;14:110–118. doi: 10.1016/s0958-1669(02)00018-6. [DOI] [PubMed] [Google Scholar]
- 52.delCardayre SB, Raines RT. Biochemistry. 1994;33:6031–6037. doi: 10.1021/bi00186a001. [DOI] [PubMed] [Google Scholar]
- 53.Vollmer W, Blanot D, de Pedro MA. FEMS Microbiol Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 54.Perlstein DL, Wang TS, Doud EH, Kahne D, Walker S. J Am Chem Soc. 2010;132:48–49. doi: 10.1021/ja909325m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Washington MT, Johnson RE, Prakash S, Prakash L. J Biol Chem. 1999;274:36835–36838. doi: 10.1074/jbc.274.52.36835. [DOI] [PubMed] [Google Scholar]
- 56.Bambara RA, Uyemura D, Choi T. J Biol Chem. 1978;253:413–423. [PubMed] [Google Scholar]
- 57.Neff NF, Chamberlin MJ. J Biol Chem. 1978;253:2455–2460. [PubMed] [Google Scholar]
- 58.We note that the ionization efficiency of short and long polymers may be different, which would lead to an underestimation in the calculated processivity values. Preliminary data from HPLC analysis of the polymeric products, suggest that the ionization efficiency does not vary greatly for shorter and longer polymers, but we cannot rule out subtle differences. Nevertheless, this analysis provides a quantitative description of glycosyltransferase processivity that is generally applicable.
- 59.Deshaies RJ, Joazeiro CA. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 60.Jing W, DeAngelis PL. J Biol Chem. 2004;279:42345–42349. doi: 10.1074/jbc.M402744200. [DOI] [PubMed] [Google Scholar]
- 61.Schwartz B, Markwalder JA, Seitz SP, Wang Y, Stein RL. Biochemistry. 2002;41:12552–12561. doi: 10.1021/bi026205x. [DOI] [PubMed] [Google Scholar]
- 62.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Proc Natl Acad Sci USA. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Splain RA, Kiessling LL. Bioorg Med Chem. 2010;18:3753–3759. doi: 10.1016/j.bmc.2010.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marlow AL, Kiessling LL. Org Let. 2001;3:2517–2519. doi: 10.1021/ol016170d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.