

Abstract
Characterizing protein–protein interactions in a biologically-relevant context is important for understanding the mechanisms of signal transduction. Most signal transduction systems are membrane-associated and consist of large, multi-protein complexes that undergo rapid reorganization, circumstances that present challenges to traditional structure determination methods. To study protein–protein interactions in biologically relevant, complex milieu, we employed a protein footprinting strategy based on isotope-coded affinity tag (ICAT) reagents. ICAT reagents are valuable tools for proteomics. Here, we show their utility in an alternative application—they are ideal for protein footprinting in complex backgrounds because the affinity tag moiety allows for enrichment of alkylated species prior to analysis. We employed a water-soluble ICAT reagent to monitor cysteine accessibility and thereby identify residues involved in two different protein–protein interactions in the Escherichia coli chemotaxis signaling system. The chemotaxis system is an archetypal transmembrane signaling pathway in which complex protein superstructure underlies sophisticated sensory performance. The formation of this superstructure depends upon the adaptor protein CheW, which mediates a functionally important bridging interaction between the transmembrane receptors and the histidine kinase. ICAT footprinting was used to map the surfaces of CheW that interact with the large, multi-domain histidine kinase CheA as well as the transmembrane chemoreceptor Tsr in native E. coli membranes. By leveraging the affinity tag, we successfully identified CheW surfaces responsible for CheA and Tsr interaction. The proximity of the CheA and Tsr binding sites on CheW suggests the formation of a composite Tsr-CheW surface for recruitment of the signaling kinase to the chemoreceptor complex.
Introduction
High resolution protein structures are appearing at unprecedented rates. As structures of the subunits of complex biological systems accumulate, interest in the higher-order organization of proteins in native environments is growing. The organization of proteins in supramolecular complexes, also called protein suprastructure, has emerged as a new frontier. Advances in traditional approaches for high resolution structure determination (e.g., multidimensional NMR and X-ray crystallography) are providing insight into suprastructure. Still, some systems--especially those involving membrane proteins--remain challenging. We sought to address this challenge by expanding the repertoire of tools for analyzing protein suprastructure. New methods are a prerequisite to understanding the organization, regulation, and function of higher-order protein assemblies.
In principle, protein footprinting strategies offer intriguing possibilities for analyzing suprastructure. Footprinting involves measuring the susceptibility of protein residues to chemical modification, a parameter that reports on residue chemical environment and solvent accessibility. Changes in the propensity of a residue to undergo modification under specific conditions can be mapped onto protein structures to define changes in protein conformation, folding, dynamics, and interactions. Several chemical modification strategies have been exploited. For example, hydrogen-deuterium exchange of peptide bond amide protons can reveal differences in the chemical environment of sections of the protein backbone.1,2 By the same principle, modifications to amino acid side chains (e.g., lysine or methionine) can be monitored to reveal residues protected by protein interactions.3–6 Similarly, the accessibility of protein regions can be evaluated by using limited proteolysis or radiolytic cleavage.7–11 These footprinting methods are powerful when applied to simple single-protein questions (e.g., folding, conformational changes). Still, methods for exploring higher-order protein structure in complex milieu are needed.
Our interest in methods to extend the utility of protein footprinting grew out of a desire to illuminate protein–protein interactions within a multi-component signaling cascade. To this end, we focused on bacterial chemotaxis. The bacterial chemotaxis signaling system has emerged as a model of transmembrane signaling, information processing, and motility behavior. The functions and structures of most of the individual signaling proteins are well defined. The transmembrane chemoreceptors form the core of a two-component phosphorelay signaling system.12,13 The cytoplasmic signaling domains of the chemoreceptors associate with the adaptor protein CheW and the histidine kinase CheA.14,15 This ternary complex is organized further into a dense, interconnected signaling lattice that localizes to the cell poles.16–18 Interestingly, recent investigations into the signaling performance of the chemotaxis system have revealed unexpected complexity in the higher-order organization of its components.19–24 These studies highlight the importance of supramolecular assemblies for coupling temporal changes in the extracellular chemical environment to swimming behavior. Despite a wealth of knowledge regarding signal transmission and propagation, the architecture and higher-order organization of this lattice remains a subject of active interest.
The chemotaxis system presents challenges to structural characterization common to all transmembrane signaling systems. The core signaling unit is a higher-order complex of large, multi-domain proteins associated with transmembrane receptors. To probe this complicated suprastructure requires a sensitive method that functions in a high background of protein and membranes alike. We postulated that a footprinting strategy could satisfy these requirements.
Cysteine residues are particularly effective targets for selective, residue-specific footprinting because they occur infrequently yet possess unique reactivity.25–30 Thiolates can undergo oxidation to disulfides, engage in disulfide exchange reactions, and readily attack electrophilic carbon centers. Cysteines engineered at sites of interest can be footprinted by measuring the rates of disulfide exchange or alkylation. Diverse strategies have been developed for effecting and monitoring Cys modification.28,29,31–34 To date most applications of Cys footprinting have focused on protein folding and protein–ligand interactions.26,28,29,32 We were intrigued by the possibility of probing Cys reactivity in complex environments with ICAT reagents.
ICAT reagents are stable isotope electrophilic mass tags.35,36 Cysteine alkylation rates can be measured by initiating a footprinting reaction with a “heavy” (13C) ICAT reagent and counter-labeling with “light” (12C) ICAT at a range of time points (Fig. 1). The chemically-identical “light” and “heavy” ICAT-conjugates ionize with equal efficiency during mass spectrometry analysis. As a result, the signal intensities of the ICAT-conjugate pairs can be directly compared to yield quantitative measurements of the fractional labeling at each time point. A key feature of ICAT reagents is their affinity tag moiety, which can facilitate post-labeling enrichment of ICAT-conjugates. Sample enrichment removes non-labeled peptides that complicate mass spectrometry analysis. Carbohydrate-based affinity tags are ideally suited for ICAT footprinting probes because they are small, water-soluble, and can serve dual roles as both the mass label and affinity tag.28,33 The vicinal diols of the carbohydrate moiety can form reversible covalent bonds with a boronate affinity resin (Fig 1A). Previously, we reported the design and synthesis of a toolkit of ICAT reagents suitable for diverse footprinting applications.33 We reasoned that probing interface residues of protein–protein interactions in a complex milieu would be accessible by leveraging the ICAT affinity tag functionality and the sensitivity of mass spectrometry. Thus, we envisioned that mapping the interactions of the chemotactic proteins would serve as a challenging test of the ICAT footprinting method.
Fig. 1.

Overview of Cys footprinting using ICAT reagents and mass spectrometry. A, Structure of bromoacetamide ICAT reagents. Dots represent 12C for light or 13C for heavy reagents. B, Cysteine alkylation rates can be determined by pulse-chase labeling with ICAT isotope pairs in the complex milieu of a transmembrane protein preparation. Multiple pooled Cys variants of the protein of interest (green, orange ovals) and labeled with 13C-ICAT for time tAlk, and then excess ICAT is quenched. Proteins are denatured and remaining free thiol is counter-labeled with a light 12C ICAT. After labeling, samples are digested with trypsin, modified peptides are enriched on boronate affinity resin, and the resulting mixtures analyzed by mass spectrometry. Each reporter Cys is identified by their predicted tryptic peptide mass (green, orange). Relative abundance of the heavy and light labels at various tAlk define the alkylation rate profiles.
We chose the adaptor protein CheW as our footprinting target because it mediates bridging interactions between the chemoreceptors and the histidine kinase CheA. CheW is essential to the structure and function of the chemotaxis signaling lattice, yet its role in signaling remains poorly understood.14,37 It is known that CheW interacts simultaneously with the C-terminal domain of CheA and the membrane-distal signaling domain of the chemoreceptors.38,39 In addition, CheW is necessary for the ligand-dependent chemoreceptor regulation of CheA kinase activity.40–42 Still, the arrangement of the ternary complex remains unclear,16,21,43,44 as does the mechanism by which CheW mediates signal coupling. Delineating the two interaction surfaces of CheW may provide a rationale for why CheW is required for communication between CheA and the chemoreceptors.
The size, complexity, and membrane localization of the CheW interaction partners preclude the use of traditional footprinting approaches. Investigating the protein—protein interactions of a transmembrane protein, such as a chemoreceptor, presents special challenges.45–49 The presence of the membranes themselves and the abundant protein background within them are major hurdles to analysis. As a result, characterizing the interactions of membrane proteins often requires problematic solubilization steps or reconstitution into artificial surfactant systems. Examining membrane proteins in their native membranes, however, preserves biologically-relevant membrane composition, protein topology, and structure. No structure mapping approaches amenable to these challenging conditions have been reported. We predicted that leveraging the ICAT affinity tag to enrich for our protein of interest would enable direct footprinting measurements in the high protein background of large interaction partners and protein-rich membranes.
Results
To map the two different interaction surfaces of CheW, we carried out footprinting experiments with CheW in the presence of either the kinase CheA or the transmembrane chemoreceptors. Experiments focused on the CheA–CheW interactions were designed to afford guidelines for mapping the interface of two soluble yet low affinity signaling complex components. The resulting data also provide the means to compare the regions of CheW that interact with CheA to those that interact with the chemoreceptors. Experiments focused on the chemoreceptor interactions with Tsr demonstrate the utility of ICAT footprinting for probing transmembrane protein interactions.
CheW Cys variants for footprinting
ICAT footprinting relies on Cys variants of the footprinting target. Methods have been developed for rapid generation of protein Cys variants for high-coverage footprinting screens.28 Still, we employed a targeted footprinting strategy because existing genetic and structural information collected on CheW suggested residues that could provide insight.21,39,50,51 Nine CheW variants were engineered using site-directed mutagenesis to afford proteins with individual Cys substitutions at surface residues. The abilities of the CheW Cys variants to function in reconstituted signaling complexes were characterized prior to their evaluation by footprinting (see Supplemental Fig. 1). Seven variants were capable of forming functional signaling complexes. Two variants, I65C and N60C, resulted in assemblies that exhibit diminished signaling yet retain significantly higher activity than complexes lacking CheW.
Footprinting the CheW-CheA interaction
We predicted that ICAT footprinting would offer advantages for high resolution mapping of protein–protein interactions. ICAT pulse–chase labeling can be used to selectively measure Cys reactivity in the protein of interest, notwithstanding the presence of binding partners or other species in the sample. This feature is especially important when characterizing binding surfaces in signaling complexes, wherein interactions are often transient and affinities relatively weak. In theory, ICAT footprinting is also amenable to the characterization of large proteins without necessitating high sample purity. Indeed, ICAT footprinting can measure alkylation rates of multiple Cys residues concurrently, as LC-MS can be used to monitor simultaneously the ionization of multiple ICAT-conjugate ion pairs.28,33 We previously developed a toolkit of ICAT reagents that can be used to determine alkylation rate differences over a wide range of Cys residue solvent accessibilities.33 By employing an ICAT reagent with a relatively slow intrinsic reaction rate, we could detect even a subtle decrease in the reactivity of a highly-solvent exposed Cys residue at the CheW—CheA binding interface. As a prelude to conducting ICAT footprinting on a complex mixture, we evaluated its capacity to map a full protein—protein interface.
We tested whether ICAT protein footprinting could overcome the difficulties of mapping the dynamic, reversible interaction between CheW and the full-length, multi-domain interaction partner CheA and therefore a high CheA concentration. By surveying a large number of CheW surface residues targeted at the putative interaction region, we sought to define the extent of the interaction surface between the two proteins. The weak interaction between CheW and CheA (KD ~ 17 μM)52 presents challenges. Driving the equilibrium toward >50% complex formation necessitates a large (5–10 fold) molar excess of binding-partner. In addition, full-length CheA is a large protein (146 kDa dimer) with a complex, five-domain organization. These factors result in an extremely high protein background, with the targeted CheW Cys variants representing less than 1% (w/w) of the total protein in the footprinting sample. We predicted that affinity tag enrichment of the ICAT label would allow us to distinguish the CheW residues affected by CheA binding despite the high protein background. This enrichment step also provides the means toexamine several protein variants in a single reaction, thereby streamlining data collection.
To explore the CheA-binding surface of CheW, we footprinted our battery of CheW Cys variants in the presence of a functional CheA variant lacking its native cysteine (hereafter referred to as CheA*). Although our footprinting strategy is compatible with the presence of background Cys residues, the extra reactive thiol contributed by native CheA necessitated the use of a higher concentration of ICAT reagent. The use of CheA* avoids this complication. Functional assays in reconstituted signaling complexes confirmed that the Cys substitutions did not affect CheA* activity significantly (Supplemental Fig. 2). Combinations of two to four Cys variants of CheW were pooled and incubated in equal volumes of either buffer or a molar excess (5-fold) of CheA*. Footprinting was initiated with 13C-ICAT reagent. At various time points, reaction aliquots were withdrawn and quenched with dithiothreitol, which scavenges excess electrophile. Unmodified cysteine residues were exposed upon CheW denaturation with urea and counter-labeled with 12C-ICAT reagent (Fig. 1). Labeled samples were digested with trypsin; the modified peptide products were enriched on boronate resin, and the resulting products were analyzed by ESI-LC-MS. The fraction of Cys labeled with 13C-ICAT at each time point was calculated from the relative signal intensities of the “light” and “heavy” peak envelopes derived from each ICAT-conjugated tryptic fragment.
The extent of alkylation by the 13C-labled bromacetamide (Fig. 1A) is influenced by the environment of the thiolate. If the formation of a protein–protein complex buries a cysteine side chain, it will be protected from modification. This protection will be manifested at short reaction times; longer reaction times should allow for higher modification levels of buried residues because the alkylating agent can access the thiolate upon complex dissociation. Thus, the ratio of alkylated products (heavy versus light) changes over time and can be fit to a pseudo-first order decay model to determine the rate of alkylation of each residue in the presence and absence of CheA* (Supplemental Fig. 3). Cys residues for which alkylation rates did not significantly differ were interpreted to be either disruptive of binding or uninvolved in the CheA interaction. For some residues, CheA* causes a decrease in the rate of Cys alkylation, and this kind of protection was expected for residues buried by protein interaction. Interestingly, in one case, we detected an increase in the modification rate. We attributed all differences in the modification of specific CheW residues to their involvement in the interaction with CheA.
The rates of alkylation of four residues were unaffected by CheA* addition (Fig. 2A). Three of these Cys variants (Q37C, N50C, and R62C) also retain function in signaling complexes. These results indicate that the aforementioned residues do not participate in the interaction with CheA (Supplemental Fig. 1). The remaining residue, I65C, is insensitive to the presence of CheA*; however, Cys substitution at Ile-65 disrupts CheW activity in reconstituted signaling complexes (Supplemental Fig. 1). These data imply that Cys substitution at Ile-65 disrupts functional CheW—CheA interaction. The combination of footprinting and functional data provides the means to identify specific residues involved in complexation.
Fig. 2.
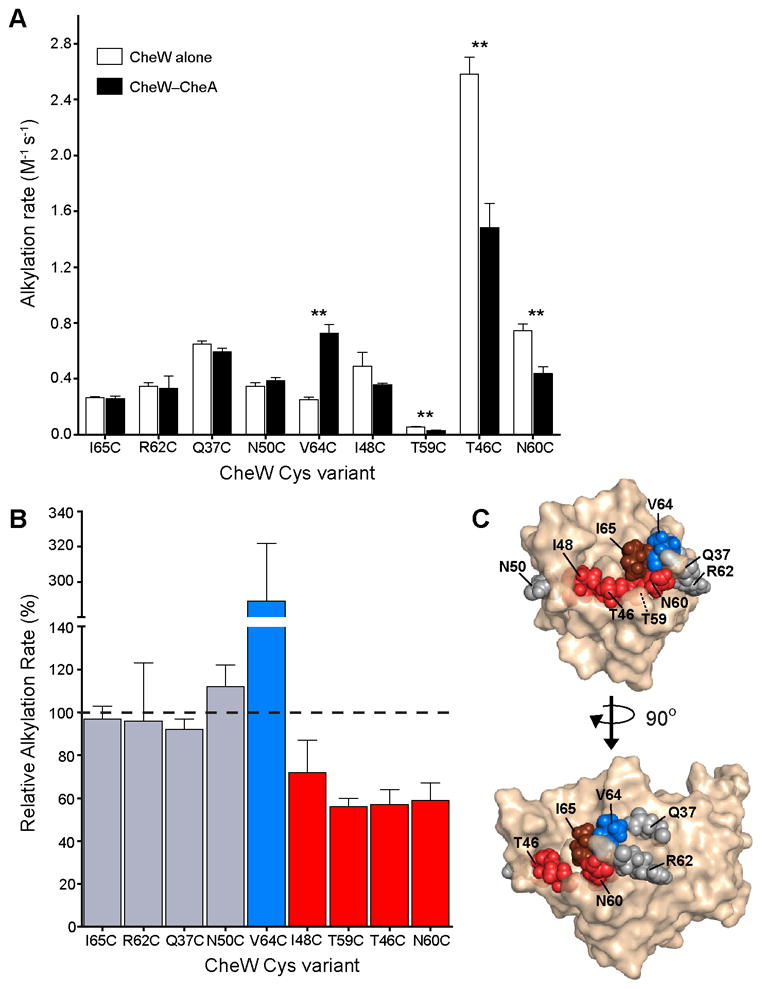
Footprinting the CheW-CheA interaction surface. A, CheW Cys variants were footprinted in the presence (black bars) and absence (white bars) of CheA*. Alkylation rates were determined from footprinting curves fit to a first order exponential decay. Error bars represent the standard deviation of the rates determined from three independent footprinting timecourses. Double asterisks denote p<0.002. B, Changes in Cys alkylation rates induced by CheA* are reported as the percentage of the ratio of the CheA-bound and free CheW rates. C, Footprinting results were mapped to the structure of E. coli CheW (PDB: 2HO9). Changes in alkylation rate induced by the presence of CheA* were classified as either unchanged (less than 25% difference, gray), decreased (red), or increased (blue). I65C, which disrupts active complex formation, is colored brown.
Of the residues that exhibit CheA*-induced changes in alkylation rate, four appeared less reactive, and one, more reactive (Fig. 2A). The presence of CheA* significantly reduces the rates of alkylation of residues T46C, T59C, and N60C (Fig. 2B). CheA* also slows the rate of alkylation of I48C by over 25%, but the difference was only weakly significant. Interestingly, the rate of alkylation of the buried CheW residue T59C is significantly changed upon CheA* binding. This result suggests that the binding of a protein influences the accessibility and reactivity of both surface and buried residues. Moreover, this finding demonstrates that while Cys substitution at residues involved in the core of the protein—protein interface (e.g., I65C) may be disruptive, the residues beneath the interface surface (e.g., T59C) can be used to diagnose interactions. In principle, changes in Cys reactivity can be caused by either protein interactions or conformational changes. Accordingly, integrating footprinting results from multiple reporter residues facilitates interpretation of changes in reactivity. In our experiments, the clustered pattern of CheA*-sensitive residues points to an interaction surface rather than conformational change. Interestingly, the functionally important residue Ile-65 occupies the center of this cluster.
We anticipated that protein–protein interactions would bury surface area and thereby elicit a decrease in the rate of reaction of Cys residues at the interface. The residue V64C is notable, however, as its alkylation rate increased in the presence of CheA*. It perhaps is not immediately apparent that protein binding might result in such an increase, but there are two scenarios in which such a change should occur. First, the rate increase may correspond to an increase in solvent accessibility due to a conformational change in the CheW-CheA complex. Second, CheA*-binding may change the local chemical environment around V64C, increasing the nucleophilicity of the thiol.53 In either case, a change in alkylation rate indicates that the residue V64C contributes to the interaction of CheW with CheA.
Footprinting the CheW-Tsr interaction in native E. coli membranes
With footprinting data on the CheW—CheA complex, we sought to localize the region of CheW that binds to the chemoreceptors. The chemoreceptor is the core of the transmembrane signaling complex, responsible for binding extracellular ligand and communicating ligand-occupancy to its associated histidine kinase. E. coli produce a complement of five chemoreceptors that form mixed signaling teams in vivo54. We chose the serine receptor Tsr as the object of our study because it engages in highly cooperative signaling in reconstituted in vitro systems, paralleling the strong inter-receptor communication measured in vivo.24,55 To preserve biologically-relevant receptor topology and membrane orientation, we isolated E. coli membranes from a strain over-producing Tsr. There are two important considerations in generating functional signaling complexes. First, the presence extremely high levels of chemoreceptor can be problematic, as chemoreceptors can cluster to form non-functional complexes.57 The relative ratio of signaling components is another important factor.14,56 We therefore optimized our reconstitutions to ensure they afforded biologically-relevant function. When CheA and CheW were added to the membrane preparations generate ternary complexes, the resulting assemblies exhibited biologically-relevant activation of CheA kinase activity (Supplemental Fig. 1A). Moreover, dose-dependent kinase responses to the Tsr ligand serine (Supplemental Fig. 1B) were observed. Relative ratios of the three signaling components were optimized to maximize kinase activity and the cooperativity of serine response (data not shown). In this way, reconstituted systems were obtained in which a productive Tsr—CheW interaction is preserved in E. coli membranes.
To date, no high resolution structural data are available to direct our choice of residues to footprint, but mutagenesis studies have implicated some CheW residues in chemoreceptor coupling.39,50,58 Interestingly, several of the residues identified by genetic screening cluster near the CheW surface involved in the CheA interaction, as defined by our footprinting and other studies.21,50,58 Nevertheless, the extent of this putative CheW—Tsr interaction surface is ambiguous, and the boundaries between the site of CheA interaction and Tsr-interaction have not been elucidated. We generated several CheW variants with Cys engineered in the vicinity of the surfaces implicated in CheW signaling function and subjected these proteins to footprinting in the presence of the chemoreceptor Tsr. We also included a variant (I65C) that reports on a core residue within the CheW-CheA interaction surface. Because Ile-65 is at the center of the CheA—CheW interaction, it is unlikely to be involved in the Tsr—CheW interface. Importantly, the selected CheW variants afford complexes that retain signaling activity as assessed by the kinase activation assay (Supplemental Fig. 1A).
The level of modified CheW peptide is low in the CheA–CheW footprinting experiment; consequently, the extremely high protein background contributed by the reconstituted signaling complex within the E. coli membranes renders the Cys modification difficult to detect (Fig. 3A). We therefore wanted to ensure that we were monitoring CheW fully incorporated into complexes. To this end, we tested for specificity of the CheW—Tsr interaction by assessing whether CheW interactions with E. coli membranes depend on the presence of the chemoreceptor. In the absence of Tsr, the association of CheW with the membranes was undetectable, but when Tsr is present CheW is incorporated into complexes (Supplemental Fig. 4). These results indicate that the interaction of CheW with Tsr in E. coli membranes is specific.
Fig 3.
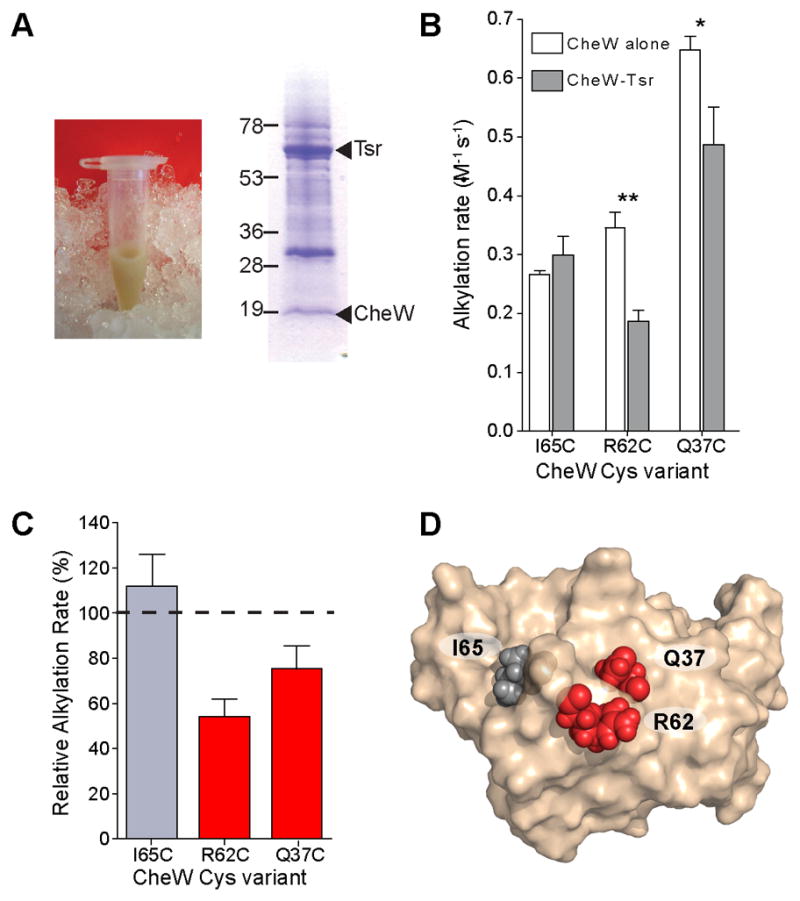
Footprinting the CheW-Tsr interaction surface in native membranes. A, Native membranes contribute considerable background, as evidenced by the photograph of a Tsr-containing membrane preparation. SDS-PAGE analysis of a typical footprinting reaction reveals that the footprinting target is a minor component in the reaction mixture. Expected molecular weights of CheW and Tsr are 18.1 kDa and 59.4 kDa, respectively. B, CheW Cys variants were subjected to footprinting in the presence (dark gray bars) and absence (white bars) of Tsr-containing native membranes. Alkylation rates were determined from footprinting curves fit to a first order exponential decay. Error bars represent the standard deviation of the rates determined from three independent footprinting time courses. Double asterisks denote p<0.002, single asterisk, p<0.02 C, Changes in Cys alkylation rates induced by Tsr membranes are reported as the percent difference of the Tsr-bound rate normalized to the rate of CheW alone. D, Footprinting results were mapped to the structure of E. coli CheW. Changes in alkylation rate induced by the presence of Tsr were classified as either unchanged (less than 25% difference, gray) or decreased (red).
The CheW Cys variants were subjected to footprinting in the presence and absence of Tsr-containing native membranes. Even in this complex system, protocols similar to those described for the CheW-CheA experiments were successful. Of the three CheW residues selected, two (Q37C and R62C) had significantly slower alkylation rates (Fig. 3B,C). As predicted, the residue (I65C) that maps to the center of the CheW—CheA interface did not show significant changes in its rate of alkylation in the presence and absence of Tsr. Two residues that were affected, Q37C and R62C, cluster closely together when mapped to the high-resolution structure of CheW. These results identify a contiguous patch of CheW as a Tsr-binding interface (Fig. 3D).
Discussion
ICAT reagents are proteomics tools that can surmount the challenges of identifying and quantifying low-abundance proteins in complex biological samples.36,59,60 The ICAT affinity tag moiety allows for selective enrichment of labeled peptides from the vast proteome background. The stable isotope tags enable high precision, sensitive quantitation of relative abundances with mass spectrometry. We have leveraged these two ICAT features to footprint protein—protein interactions in chemotaxis signaling complexes. The enrichment of ICAT-labeled peptides on boronate resin enabled the identification of ICAT alkylation products in the presence of complex footprinting samples, including those composed of protein-rich E. coli inner membranes (native membranes). Moreover, encoding alkylation states with isotope labels enables the reproducible detection of subtle differences in residue environments that arise from transient protein—protein interactions.
In principal, the application of ICAT reagents to protein footprinting opens up investigations of protein function in extremely complex biological systems. With affinity tag enrichment, sample complexity not a limiting factor in designing a footprinting study. The primary factor in the design of an ICAT footprinting study is the incorporation and distribution of Cys reporter residues. We introduced Cys residues individually to probe targeted regions of the CheW surface. Engineering each Cys variant is practical for focused studies and enables secondary assays to assess the functional effects of each cysteine variant. Cysteine translational misincorporation can be employed for higher coverage footprinting screens.28,32 This approach trades control over reporter residue placement for rapid generation of mixed populations of cysteine variants. The use of alkylating reagents for footprinting introduces a second point of control. Changes in the accessibility of deeply buried residues can be probed with rapidly reacting (highly electrophilic) alkylating reagents. Conversely, the accessibility of solvent-exposed residues can be assessed with reagents with low reactivity, as those reagents more readily distinguish between subtle differences in alkylation rate. The reactivity of ICAT reagents can be tuned to suit the application.33 An advantage of changing the electrophilicity of the ICAT reagent provides the means to probe cysteine residues in different environments without dramatically changing conditions (i.e., pH).
Mapping footprinting results to structures
ICAT protein footprinting proved effective for mapping the two interaction surfaces of the chemotaxis adaptor protein CheW. The results of the footprinting studies reveal seven residues that exhibit substantial changes (>25%) in alkylation rate in the presence of either CheA* or Tsr (Fig. 4A). When mapped to the high-resolution structure of E. coli CheW, the residues implicated in Tsr and CheA interaction localize to two distinct patches (Fig. 4B).61 Interestingly, these patches are proximal. The nearly contiguous binding surfaces imply that CheW can mediate or reinforce direct contacts between CheA and Tsr.
Fig. 4.

Summary of footprinting results. A, Changes in Cys alkylation rates induced by CheA* or Tsr-containing E. coli membranes are reported as the percent difference of the binding-partner-induced rate normalized to the rate of CheW alone. B, Footprinting results were mapped to the structure of E. coli CheW (PDB: 2HO9). Residues that undergo alkylation rate changes greater than 25% in the presence of CheA* (purple) or Tsr membranes (green) are highlighted. The white residue did not afford significant changes in the presence of binding partners. Ile-65 (light purple) did not exhibit changes in reactivity but was implicated in the CheA interaction by a combination of footprinting and functional screens. Comparison of footprinting and genetic approaches to mapping interaction surfaces C, Results from genetic screens for functional perturbations. Residues identified as functionally important for CheA-binding (cyan) or Tsr-binding (yellow) are highlighted.50,58 One residue (Arg-62, grey) was not identified by a functional perturbation screen yet its substitution disrupts E. coli chemotaxis. Asterisks, daggers, and double-daggers denote residues Ile-65, Arg-62, and Gln-37, respectively. D, Footprinting results were mapped to the co-structure of truncated CheA (dark gray) and CheW (beige) from T. maritima (PDB: 1B3Q). T. maritima residues corresponding to the footprinted E. coli CheW residues were identified by ClustalW amino acid sequence alignment.67
Contrasting protein footprinting with loss-of-function screening
Genetic screens for loss-of-function mutations are now classic approaches for identifying functionally important protein residues. Typically, the protein of interest is subjected to random mutagenesis, and the pool of protein variants is screened to identify residue changes that disrupt function. Several strategies for screening functional perturbations have been developed specifically for mapping protein–protein interactions. Alanine scanning mutagenesis allows for analysis of the binding energy contributions of individual sidechains by systematically substituting interface residues with alanine.62,63 The related PICM (protein-interactions-by-cysteine-modification) and SCAM (substituted-cysteine accessibility method) strategies involve engineering Cys substitutions throughout a protein of interest and assessing the functional consequences of alkylation with bulky, perturbing substituents.34,42,64–66 Functional perturbation approaches hinge on the interpretation of the disruptive residue changes to identify protein regions involved in interactions. Still, interpreting the functional changes can be difficult. Remote residues can have long-distance consequences on folding and conformation that affect protein docking interfaces. Moreover, some residues buried by protein—protein interaction can be insensitive to perturbation.
Functional perturbation mapping is a valuable complement to protein footprinting, but protein footprinting can identify residues (Fig. 4B) that the former method does not (Fig. 4C).50,58 The CheA-interaction surface revealed by protein footprinting extends further than that deduced from functional perturbation screens. Although the functional perturbation screen successfully identified the core CheW resides responsible for interaction with CheA, residues at the periphery of the interaction interface were not detected, apparently because they tolerate variation. The success of the protein footprinting approach in uncovering a role for these peripheral residues in the CheW-CheA interface can be attributed to its ability to probe for subtle changes in the environment of a residue. The method is effective even when residue substitution preserves functional interactions. The residues identified by functional perturbation screening also may include false-positives, as amino acid substitutions that destabilize the protein can affect remote interactions. Protein footprinting, however, is not prone to misidentification of interaction surface residues. Only cysteine substitutions that preserve function will exhibit changes in accessibility in the presence of binding partner.
The sensitivity of ICAT footprinting to peripheral interaction residues proved useful for probing the proximity of the CheW docking sites for CheA and Tsr. Functional perturbation screens identified spatially distinct clusters of CheW residue variants that disrupt interactions with either CheA or chemoreceptors (Fig. 4C). We examined CheW residues between these regions previously implicated in CheA or Tsr binding. ICAT footprinting implicates CheW residues Gln-37 and Arg-62 in the interaction with Tsr (Fig. 4B,C).50,58 Interestingly, one of the two residues (Arg-62) was also identified by functional perturbation screens for disruptive mutations. The CheW variant R62H exhibits minor decreases in affinity for both CheA and Tsr yet entirely disrupts signaling in vivo.50 Taken together, our footprinting results and the R62H perturbation suggest a key role for this residue in ternary complex activation. These results are reinforced by those of a genetic suppression screen39. CheW mutations that rescue signaling defects observed with Tsr mutants map to a broad, discontinuous surface, yet many of these residue changes cluster around those identified by our footprinting studies and the functional perturbation studies.
Comparisons with a homologous co-structure
Although the structure of the complex of CheA with CheW from E. coli has not been determined, that of the corresponding hyperthermophile Thermotoga maritima proteins has been solved.21 We compared our footprinting study of the E. coli CheW–CheA interaction with the T. maritima co-structure to ascertain whether the interaction surfaces are conserved (Fig. 6)67. CheW residues that manifested alkylation rate changes in the presence of CheA or Tsr were mapped to the corresponding residues of the T. maritima CheW in the structure of the complex. The results matched extremely well. The E. coli CheW residues that were found to undergo CheA-induced alkylation rate changes mapped directly to the CheW–CheA interface. The parallels between the T. maritima co-structure and our E. coli footprinting results demonstrate highly conserved interaction sites in the signaling components of these distantly related organisms. Electron cryotomography experiments have revealed a conservation of higher order signaling complex assembly, 19 and our experiments indicate that the conservation extends to protein–protein interactions within the signaling complexes.
The residues implicated in CheW interaction with Tsr group to an exposed CheW surface abutting the CheA interface. The proximity of the Tsr interaction surface of CheW with CheA suggests the formation of a contiguous surface for chemoreceptor docking that spans both CheA and CheW. A cooperative interaction between the three core signaling components is consistent with the role of CheW as a bridging component that augments the intrinsically weak interactions between CheA and Tsr (Supplemental Fig. 2A,B).14 In addition to its role as an adaptor molecule, CheW may serve a direct role in signaling propagation. The mechanism by which the chemoreceptor signaling state is communicated to CheA is largely unknown. The contiguous CheW interaction surfaces revealed by our footprinting suggest that the conformational changes controlling kinase activity may involve coordinated rearrangements around this point of ternary contact.
Our explorations of the CheW bridging interactions in the chemotaxis system highlight the utility of ICAT reagents as robust tools for footprinting proteins in complex native environments. We therefore anticipate that ICAT footprinting will be a valuable method for determining protein–protein interaction surfaces in transmembrane signaling systems. Dynamic, transient complexes of protein components are hallmarks of signaling and regulation, and protein footprinting provides the means to probe the architecture of these complexes, even in a complex, biologically-relevant environment.
Materials and Methods
Footprinting Methods
E. coli chemotaxis proteins CheW, CheA variants were produced and purified to >85% homogeneity. Tsr (E. coli) was over-produced and prepared in bacterial membranes. All proteins were dialyzed in a buffer composed of 50 mM Tris pH 8.0, 50 mM KCl, 5 mM MgCl2, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP). Master reaction mixes were prepared by combining Cys-variant target proteins (0.5 nmol each) with or without binding partner proteins (3.5 to 10-fold molar excess over total CheW) and equilibrating at room temperature (26 °C) for 45 min. Multiple Cys-variants were included in one master reaction whenever expected product masses were predicted to be non-overlapping. Footprinting time courses (8 time points, through 300 min) were initiated by adding the 13C-ICAT reagent to a 400 – 500-fold molar excess over protein thiols. At each time point, aliquots of the master footprinting reaction were withdrawn, and remaining 13C-ICAT was quenched by addition of excess of DTT. To remove DTT and quenched 13C-ICAT, protein was precipitated by adding 400 μL of 0.05% sodium deoxycholate followed by TCA to a final concentration of 10% (w/v). Precipitated protein was pelleted by centrifugation and washed twice with acetone. Protein pellets were dried briefly by vacuum centrifugation and resuspended in 5 M urea (30 μL). 12C-ICAT was added to a final concentration of 10 mM and incubated for 10 min. Note, to improve the resuspension of protein precipitate, footprinting reactions with extremely high protein content (e.g., bacterial membranes) were divided into two or more tubes before TCA precipitation, to be recombined after 12C-ICAT labeling. Prior to protease digestion, the urea concentration in the samples was diluted to <0.5 M by adding 50 mM Tris pH 7.5, 1 mM CaCl2 to each footprinting sample. The samples were treated with 1 μg sequencing-grade modified trypsin (Promega) for 14 h. at 26 °C to generate proteolytic fragments for analysis.
ICAT-labeled peptides were purified on Affi-Gel boronate resin (Bio-Rad Laboratories). Each footprinting time point sample was incubated with 50 mg boronate resin, pre-swelled in 5 mL boronate buffer (50 mM HEPES pH 9, 500 mM NaF, 10% acetonitrile). The resin was washed 6–7 times with 5 mL boronate buffer by connecting chromatography columns to a vacuum manifold. ICAT-labeled peptides were eluted by incubating the resin samples with 2× 500 μL elution buffer (20 mM Tris pH 8, 200 mM sorbitol, 10% acetonitrile), followed by 500 μL MilliQ water. Eluates were concentrated by vacuum centrifugation. Samples were desalted for mass spectrometry using C18-Carbon TopTips (Glygen Corp.) according to manufacturer’s instructions.
Footprinting time point samples were injected onto a Shimadzu LC-MS containing a C18 column (Supelco Discovery, 2.1 × 150 mm) equilibrated with 0.2% formic acid. ICAT-labeled peptides were separated over a 15 min. linear gradient of acetonitrile and 0.2% (v/v) formic acid (5–95%), while positive-ion mass spectra were collected for masses between 400 and 1,000 amu. Cys residues were identified from their predicted tryptic digest mass plus the mass of the ICAT label (ExPASy PeptideMass, http://www.expasy.ch/tools/peptide-mass.html).68
The fraction of each Cys residues of each timepoint labeled with 12C-ICAT was calculated as the ratio of the integrated peaks of the 12C-labeled mass envelope over the total 12C/13C mass envelope [12C/(12C+13C)]. These data were plotted versus time and fit to a pseudo-first order decay equation to derive rate constants. Statistical significance was calculated using a two-tailed, unpaired Student’s t-test.
Additional Methods
Detailed protocols for site-directed mutagenesis, protein production, and functional assays reconstituted chemotaxis signaling complexes containing CheW variants are available in Supplemental Information.
Supplementary Material
Acknowledgments
This research was supported by the NIH (GM55984). E.S.U thanks the Molecular Biosciences Training Grant for support (GM07215). We thank J.S. Parkinson for helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Krishna MMG, Hoang L, Lin Y, Englander SW. Hydrogen exchange methods to study protein folding. Methods. 2004;34:51–64. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Smith DL, Deng YZ, Zhang ZQ. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SFJ. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA: tRNA complex by mass spectrometry and a primary amine selective reagent. Proc Natl Acad Sci U S A. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanai R, Wang JC. Protein footprinting by the combined use of reversible and irreversible lysine modifications. Proc Natl Acad Sci U S A. 1994;91:11904–11908. doi: 10.1073/pnas.91.25.11904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Arruda MV, Bazari H, Wallek M, Matsudaira P. An actin footprint on villin - single site substitutions in a cluster of basic residues inhibit the actin severing but not capping activity of villin. J Biol Chem. 1992;267:13079–13085. [PubMed] [Google Scholar]
- 6.West GM, Thompson JW, Soderblom EJ, Dubois LG, DeArmond PD, Moseley MA, Fitzgerald MC. Mass Spectrometry-Based Thermal Shift Assay for Protein-Ligand Binding Analysis. Anal Chem. 2010;82:5573–5581. doi: 10.1021/ac100465a. [DOI] [PubMed] [Google Scholar]
- 7.Liu RT, Guan JQ, Zak O, Aisen P, Chance MR. Structural reorganization of the transferrin C-lobe and transferrin receptor upon complex formation: The C-lobe binds to the receptor helical domain. Biochemistry. 2003;42:12447–12454. doi: 10.1021/bi0352973. [DOI] [PubMed] [Google Scholar]
- 8.Dokudovskaya S, Williams R, Devos D, Sali A, Chait BT, Rout MP. Protease accessibility laddering: A proteomic tool for probing protein structure. Structure. 2006;14:653–660. doi: 10.1016/j.str.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Xu GZ, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 10.Zhong M, Lin L, Kallenbach NR. A method for probing the topography and interactions of proteins - footprinting of myoglobin. Proc Natl Acad Sci U S A. 1995;92:2111–2115. doi: 10.1073/pnas.92.6.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angel TE, Gupta S, Jastrzebska B, Palczewski K, Chance MR. Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc Natl Acad Sci U S A. 2009;106:14367–14372. doi: 10.1073/pnas.0901074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hazelbauer GL, Falke JJ, Parkinson JS. Bacterial chemoreceptors: high-performance signaling in networked arrays. Trends Biochem Sci. 2008;33:9–19. doi: 10.1016/j.tibs.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sourjik V. Receptor clustering and signal processing in E coli chemotaxis. Trends Microbiol. 2004;12:569–576. doi: 10.1016/j.tim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Gegner JA, Graham DR, Roth AF, Dahlquist FW. Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell. 1992;70:975–982. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- 15.Erbse AH, Falke JJ. The Core Signaling Proteins of Bacterial Chemotaxis Assemble To Form an Ultrastable Complex. Biochemistry. 2009;48:6975–6987. doi: 10.1021/bi900641c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu TS, Le Novere N, Levin MD, Beavil AJ, Sutton BJ, Bray D. Molecular model of a lattice of signalling proteins involved in bacterial chemotaxis. Nat Cell Biol. 2000;2:792–796. doi: 10.1038/35041030. [DOI] [PubMed] [Google Scholar]
- 17.Maddock JR, Shapiro L. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science. 1993;259:1717–1723. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- 18.Massazza DA, Parkinson JS, Studdert CA. Crosslinking Evidence for Motional Constraints within Chemoreceptor Trimers of Dimers. Biochemistry. 2010 doi: 10.1021/bi101483r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briegel A, Ortega DR, Tocheva EI, Wuichet K, Li Z, Chen SY, Muller A, Iancu CV, Murphy GE, Dobro MJ, Zhulin IB, Jensen GJ. Universal architecture of bacterial chemoreceptor arrays. Proc Natl Acad Sci U S A. 2009;106:17181–17186. doi: 10.1073/pnas.0905181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khursigara CM, Wu XW, Zhang PJ, Lefman J, Subramaniam S. Role of HAMP domains in chemotaxis signaling by bacterial chemoreceptors. Proc Natl Acad Sci U S A. 2008;105:16555–16560. doi: 10.1073/pnas.0806401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park SY, Borbat PP, Gonzalez-Bonet G, Bhatnagar J, Pollard AM, Freed JH, Bilwes AM, Crane BR. Reconstruction of the chemotaxis receptor-kinase assembly. Nat Struct Mol Biol. 2006;13:400–407. doi: 10.1038/nsmb1085. [DOI] [PubMed] [Google Scholar]
- 22.Gestwicki JE, Kiessling LL. Inter-receptor communication through arrays of bacterial chemoreceptors. Nature. 2002;415:81–84. doi: 10.1038/415081a. [DOI] [PubMed] [Google Scholar]
- 23.Lamanna AC, Ordal GW, Kiessling LL. Large increases in attractant concentration disrupt the polar localization of bacterial chemoreceptors. Mol Microbiol. 2005;57:774–785. doi: 10.1111/j.1365-2958.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- 24.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 25.Abramczyk O, Rainey MA, Barnes R, Martin L, Dalby KN. Expanding the repertoire of an ERK2 recruitment site: Cysteine Footprinting identifies the D-recruitment site as a mediator of Ets-1 binding. Biochemistry. 2007;46:9174–9186. doi: 10.1021/bi7002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha JH, Loh SN. Changes in side chain packing during apomyoglobin folding characterized by pulsed thiol-disulfide exchange. Nat Struct Biol. 1998;5:730–737. doi: 10.1038/1436. [DOI] [PubMed] [Google Scholar]
- 27.Johnson CP, Tang HY, Carag C, Speicher DW, Discher DE. Forced unfolding of proteins within cells. Science. 2007;317:663–666. doi: 10.1126/science.1139857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silverman JA, Harbury PB. Rapid mapping of protein structure, interactions, and ligand binding by misincorporation proton-alkyl exchange. J Biol Chem. 2002;277:30968–30975. doi: 10.1074/jbc.M203172200. [DOI] [PubMed] [Google Scholar]
- 29.Tu BP, Wang JC. Protein footprinting at cysteines: Probing ATP-modulated contacts in cysteine-substitution mutants of yeast DNA topoisomerase II. Proc Natl Acad Sci U S A. 1999;96:4862–4867. doi: 10.1073/pnas.96.9.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cliff MJ, Alizadeh T, Jelinska C, Craven CJ, Staniforth RA, Waltho JP. A thiol labelling competition experiment as a probe for sidechain packing in the kinetic folding intermediate of N-PGK. J Mol Biol. 2006;364:810–823. doi: 10.1016/j.jmb.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 31.Burguete AS, Harbury PB, Pfeffer SR. In vitro selection and prediction of TIP47 protein-interaction interfaces. Nat Methods. 2004;1:55–60. doi: 10.1038/nmeth702. [DOI] [PubMed] [Google Scholar]
- 32.Silverman JA, Harbury PB. The equilibrium unfolding pathway of a (beta/alpha)(8) barrel. J Mol Biol. 2002;324:1031–1040. doi: 10.1016/s0022-2836(02)01100-2. [DOI] [PubMed] [Google Scholar]
- 33.Underbakke ES, Zhu Y, Kiessling LL. Isotope-Coded Affinity Tags with Tunable Reactivities for Protein Footprinting. Angew Chem Int Ed. 2008;47:9677–9680. doi: 10.1002/anie.200803378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 35.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 36.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 37.Wolfe AJ, Conley MP, Kramer TJ, Berg HC. Reconstitution of signaling in bacterial chemotaxis. J Bacteriol. 1987;169:1878–1885. doi: 10.1128/jb.169.5.1878-1885.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourret RB, Davagnino J, Simon MI. The carboxy-terminal portion of the CheA kinase mediates regulation of autophosphorylatoin by transducer and CheW. J Bacteriol. 1993;175:2097–2101. doi: 10.1128/jb.175.7.2097-2101.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu JD, Parkinson JS. Genetic-evidence for interaction between the CheW and Tsr proteins during chemoreceptor signaling by Escherichia coli. J Bacteriol. 1991;173:4941–4951. doi: 10.1128/jb.173.16.4941-4951.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borkovich KA, Kaplan N, Hess JF, Simon MI. Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc Natl Acad Sci U S A. 1989;86:1208–1212. doi: 10.1073/pnas.86.4.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ninfa EG, Stock A, Mowbray S, Stock J. Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J Biol Chem. 1991;266:9764–9770. [PubMed] [Google Scholar]
- 42.Zhao JS, Parkinson JS. Cysteine-scanning analysis of the chemoreceptor-coupling domain of the Escherichia coli chemotaxis signaling kinase CheA. J Bacteriol. 2006;188:4321–4330. doi: 10.1128/JB.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briegel A, Ding HJ, Li Z, Werner J, Gitai Z, Dias DP, Jensen RB, Jensen GJ. Location and architecture of the Caulobacter crescentus chemoreceptor array. Mol Microbiol. 2008;69:30–41. doi: 10.1111/j.1365-2958.2008.06219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhatnagar J, Borbat PP, Pollard AM, Bilwes AM, Freed JH, Crane BR. Structure of the Ternary Complex Formed by a Chemotaxis Receptor Signaling Domain, the CheA Histidine Kinase, and the Coupling Protein CheW As Determined by Pulsed Dipolar ESR Spectroscopy. Biochemistry. 2010;49:3824–3841. doi: 10.1021/bi100055m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Werten PJL, Remigy HW, de Groot BL, Fotiadis D, Philippsen A, Stahlberg H, Grubmuller H, Engel A. Progress in the analysis of membrane protein structure and function. FEBS Lett. 2002;529:65–72. doi: 10.1016/s0014-5793(02)03290-8. [DOI] [PubMed] [Google Scholar]
- 46.Loll PJ. Membrane protein structural biology: the high throughput challenge. J Struct Biol. 2003;142:144–153. doi: 10.1016/s1047-8477(03)00045-5. [DOI] [PubMed] [Google Scholar]
- 47.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carpenter EP, Beis K, Cameron AD, Iwata S. Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol. 2008;18:581–586. doi: 10.1016/j.sbi.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leis A, Rockel B, Andrees L, Baumeister W. Visualizing cells at the nanoscale. Trends Biochem Sci. 2009;34:60–70. doi: 10.1016/j.tibs.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 50.Boukhvalova MS, Dahlquist FW, Stewart RC. CheW binding interactions with CheA and Tar - Importance for chemotaxis signaling in Escherichia coli. J Biol Chem. 2002;277:22251–22259. doi: 10.1074/jbc.M110908200. [DOI] [PubMed] [Google Scholar]
- 51.Hamel DJ, Dahlquist FW. The contact interface of a 120 kD CheA-CheW complex by methyl TROSY interaction spectroscopy. J Am Chem Soc. 2005;127:9676–9677. doi: 10.1021/ja052517m. [DOI] [PubMed] [Google Scholar]
- 52.Gegner JA, Dahlquist FW. Signal Transduction in Bacteria - Chew Forms a Reversible Complex with the Protein-Kinase Chea. Proc Natl Acad Sci U S A. 1991;88:750–754. doi: 10.1073/pnas.88.3.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weerapana E, Simon GM, Cravatt BF. Disparate proteome reactivity profiles of carbon electrophiles. Nat Chem Biol. 2008;4:405–407. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:7060–7065. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li GY, Weis RM. Covalent modification regulates ligand binding to receptor complexes in the chemosensory system of Escherichia coli. Cell. 2000;100:357–365. doi: 10.1016/s0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]
- 56.Li MS, Hazelbauer GL. Cellular stoichiometry of the components of the chemotaxis signaling complex. J Bacteriol. 2004;186:3687–3694. doi: 10.1128/JB.186.12.3687-3694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang PJ, Khursigara CM, Hartnell LM, Subramaniam S. Direct visualization of Escherichia coli chemotaxis receptor arrays using cryo-electron microscopy. Proc Natl Acad Sci U S A. 2007;104:3777–3781. doi: 10.1073/pnas.0610106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boukhvalova M, VanBruggen R, Stewart RC. CheA kinase and chemoreceptor interaction surfaces on CheW. J Biol Chem. 2002;277:23596–23603. doi: 10.1074/jbc.M202288200. [DOI] [PubMed] [Google Scholar]
- 59.Han DK, Eng J, Zhou HL, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ranish JA, Yi EC, Leslie DM, Purvine SO, Goodlett DR, Eng J, Aebersold R. The study of macromolecular complexes by quantitative proteomics. Nat Genet. 2003;33:349–355. doi: 10.1038/ng1101. [DOI] [PubMed] [Google Scholar]
- 61.Li Y, Hu YF, Fu WY, Xia B, Jin CW. Solution structure of the bacterial chemotaxis adaptor protein CheW from Escherichia coli. Biochem Biophys Res Commun. 2007;360:863–867. doi: 10.1016/j.bbrc.2007.06.146. [DOI] [PubMed] [Google Scholar]
- 62.Clackson T, Wells JA. A hot-spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 63.Wells JA. Binding in the growth hormone receptor complex. Proc Natl Acad Sci U S A. 1996;93:1–6. doi: 10.1073/pnas.93.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller AS, Kohout SC, Gilman KA, Falke JJ. CheA kinase of bacterial chemotaxis: Chemical mapping of four essential docking sites. Biochemistry. 2006;45:8699–8711. doi: 10.1021/bi060580y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bass RB, Miller AS, Gloor S, Falke JJ. The PICM chemical scanning method for identifying domain-domain and protein-protein interfaces: Applications to the core signaling complex of E-coli chemotaxis. Two-Component Signaling Systems, Pt B. 2007;423:3–24. doi: 10.1016/S0076-6879(07)23001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehan RS, White NC, Falke JJ. Mapping out regions on the surface of the aspartate receptor that are essential for kinase activation. Biochemistry. 2003;42:2952–2959. doi: 10.1021/bi027127g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson JD, Higgins DG, Gibson TJ. Clustal-W - Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilkins MR, Lindskog I, Gasteiger E, Bairoch A, Sanchez JC, Hochstrasser DF, Appel RD. Detailed peptide characterization using PEPTIDEMASS - A World-Wide-Web-accessible tool. Electrophoresis. 1997;18:403–408. doi: 10.1002/elps.1150180314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.