

Abstract
Background
Coxiella burnetii is an obligate intracellular bacterium and the etiologic agent of Q fever; both coxiella outer membrane protein 1 (Com1) and heat shock protein B (HspB) are its major immunodominant antigens. It is not clear whether Com1 and HspB have the ability to mount immune responses against C. burnetii infection.
Results
The recombinant proteins Com1 and HspB were applied to pulse human monocyte-derived dendritic cells (HMDCs), and the pulsed HMDCs were used to stimulate isogenic T cells. Com1-pulsed HMDCs expressed substantially higher levels of surface molecules (CD83, CD40, CD80, CD86, CD54, and CD58) and a higher level of interleukin-12 than HspB-pulsed HMDCs. Moreover, Com1-pulsed HMDCs induced high-level proliferation and activation of CD4+ and CD8+ cells, which expressed high levels of T-cell activation marker CD69 and inflammatory cytokines IFN-γ and TNF-α. In contrast, HspB-pulsed HMDCs were unable to induce efficient T-cell proliferation and activation.
Conclusions
Our results demonstrate that Com1-pulsed HMDCs are able to induce efficient T-cell proliferation and drive T cells toward Th1 and Tc1 polarization; however, HspB-pulsed HMDCs are unable to do so. Unlike HspB, Com1 is a protective antigen, which was demonstrated by the adoptive transfer of Com1-pulsed bone marrow dendritic cells into naive BALB/c mice.
Background
Dendritic cells (DCs) are potent antigen-presenting cells (APCs) that bridge the innate and adaptive immune responses through direct pathogen neutralization, cytokine production, and T-cell activation [1]. Activated DCs express major histocompatibility complex (MHC) I and MHC II molecules and T-cell costimulatory molecules, which possess the unique ability to activate naive T cells [2,3]. Immature DCs (iDCs) reside in the peripheral epithelial tissues, where they serve as sentinels against invading microorganisms [4]. Contact with a pathogen typically elicits stimulation of iDCs via pattern-recognition receptors, such as the toll-like receptor (TLR), and subsequent conversion of iDCs to mature DCs (mDCs) [5,6]. mDCs exhibit a reduction in phagocytic ability and an increase in surface expression of MHC II and costimulatory molecules, and they switch in chemokine receptor expression, which results in mDC migration to the local lymph nodes to induce adaptive immunity [2,7,8].
Coxiella burnetii is a Gram-negative, obligate intracellular bacterium, which survives inside large replication vacuoles that display phagolysosomal characteristics [9,10]. C. burnetii is the etiologic agent of Q fever, a disease with a worldwide distribution [11,12]. Acute Q fever is usually self-limited in immunocompetent hosts, whereas the chronic form of the disease develops in individuals defective in cell-mediated immunity [11,13]. Owing to its very low infectious dose, known environment stability, and aerosol transmission route, C. burnetii is recognized as a potential biological weapon agent, and it has been classified as a category B bioterrorism agent [13,14]. An inactivated C. burnetii vaccine against acute Q fever is effective, but the vaccination remains problematic owing to its significant side effects in individuals who have already had Q fever [15]. Second-generation Q fever vaccine, offering significant relief from adverse reactions, has been developed by means of chloroform-methanol extraction of phase I organisms by Williams and colleagues [16,17]; however, the complex procedure and biosecurity requirements in dealing with the propagation and purification of C. burnetii organisms are a hindrance to large-scale production.
A safe, effective subunit vaccine against Q fever would clearly be desirable, and a number of potential protective antigens have been identified toward developing such a vaccine [18-22]. Among them, coxiella outer-membrane protein 1 (Com1) and heat-shock protein B (HspB) were strongly recognized by sera from Q fever patients or C. burnetii-infected animals [20-22]. HspB is a member of the Hsp60 family, and Hsp60 in Legionella pneumophila (a facultative intracellular pathogen largely similar to C. burnetii in phylogenesis) is located in the periplasm and outer membrane, and the surface-associated Hsp60 is involved in bacterial virulence [23,24]. Both Com1 and HspB of C. burnetii are viewed as major immunodominant antigens and important surface-associated molecules that can interact with surface molecules of innate/adaptive immune cells [18-20,25]. However, it is not clear whether Com1 and HspB possess the ability to mount immune responses against C. burnetii infection.
In the present study, the recombinant proteins Com1 and HspB were used to pulse human monocyte-derived DCs (HMDCs) in vitro, and the maturation and activation status of the pulsed HMDCs were measured by flow cytometry. Since the protective response to intracellular bacteria is mainly dependent on a cell-mediated immune reaction, the pulsed HMDCs were used to stimulate isogenic T cells in vitro; the maturation and activation status of CD4+ and CD8+ cells in the T-cell population were analyzed after interaction of the HMDCs with the T cells. We found that Com1 induced efficient maturation and activation of HMDCs and that Com1-pulsed cells were able to initiate the adaptive T-cell response by skewing CD4+ and CD8+ cells toward, respectively, the Th1 and Tc1 pathway. In contrast, HspB elicited incomplete maturation of HMDCs, and HspB-pulsed cells were unable to induce efficient T-cell proliferation and activation.
Results
Immunoblot analysis of Com1 and HspB
The purified recombinant proteins Com1 and HspB were analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE; Figure 1A) and incubated with sera from mice experimentally infected with C. burnetii (Figure 1B). The recombinant Com1 and HspB were strongly recognized by infected mouse sera obtained at 21 and 28 days postinfection.
Figure 1.
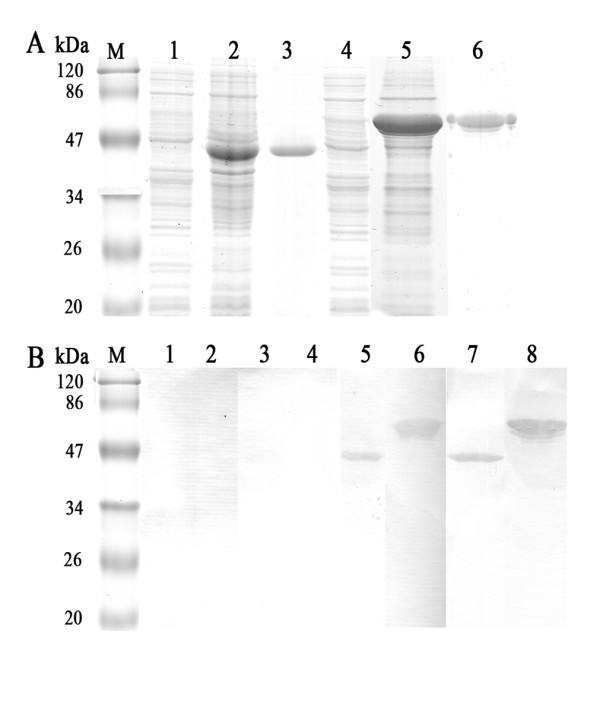
SDS-PAGE and immunoblot analysis of recombinant proteins Com1 and HspB. A SDS-PAGE analysis of Com1 and HspB: lane M, protein molecular mass markers; lane 1, lysate of E. coli cells harboring plasmid; lane 2, lysate of E. coli cells harboring com1-recombined plasmid; lane 3, purified 41 kDa Com1 fused with 17 kDa tagged-peptide from pET32a expression plasmid; lane 4, lysate of E. coli cells harboring plasmid; lane 5, lysate of E. coli cells harboring hspB-recombined plasmid; lane 6, purified 54 kDa HspB. B Immunoblot analysis of proteins Com1 and HspB with sera from mice experimentally infected with C. burnetii. Lanes 1, 3, 5, and 7, Com1 immunoblot assay with mouse sera obtained at days 7, 14, 21, and 28 postinfection, respectively, with C. burnetii. Lanes 2, 4, 6, and 8, HspB immunoblot assay with mouse sera obtained at days 7, 14, 21, and 28, respectively, postinfection with C. burnetii.
Maturation and activation of HMDCs induced by Com1 and HspB
To determine the capability of Com1 and HspB to induce maturation and activation of HMDCs, immature HMDCs (iHMDCs) were treated with Com1, HspB, or Escherichia coli lipopolysaccharide (LPS) for 24 h. The resulting populations of HMDCs were analyzed by flow cytometry to determine their expression of surface molecules. As shown in Figure 2, Com1-treated HMDCs exhibited substantially increased expression of surface molecules, and the expression levels of surface molecules (CD40, CD54, CD58, CD80, CD83, and CD86) were equal to or higher than those of LPS-treated HMDCs. However, with HspB-treated HMDCs, CD83 expression remained at baseline levels, while expression of the other surface molecules was much lower than with Com1-pulsed HMDCs (Figure 2).
Figure 2.
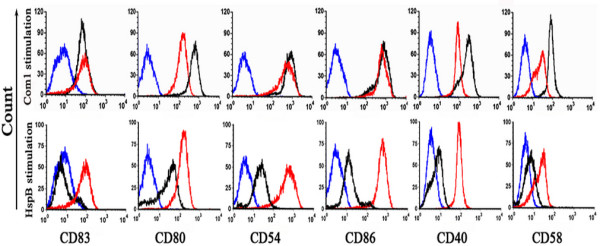
Expression of surface molecules on antigen-treated HMDCs. After 24 h of Com1 or HspB stimulation (black histograms), HMDCs were stained with monoclonal antibody to CD83, CD80, CD54, CD86, CD40, or CD58 and the expression of the surface molecules was measured by flow cytometry. LPS-treated HMDCs (red histograms) and mock-stimulated HMDCs (blue histograms) were applied as positive and negative controls, respectively. The results are representative of three independent experiments, each using cells derived from a different donor.
Cytokine expression of HMDC elicited by Com1 and HspB
Antigen-activated DCs play an important role in controlling differentiation orientation of naive T cells by differential expression of IL-12 and IL-10. To investigate the possible effector function of Com1- or HspB-pulsed HMDCs on T cells, the production of intracellular IL-12p70 and IL-10 was determined in the antigen-treated HMDCs. As seen in Figure 3, the percentages of IL-12 p70-positive cells in the Com1-pulsed HMDCs were approximately 23-, 30-, and 35-fold higher, respectively, than in the mock-pulsed HMDCs, LPS-pulsed HMDCs, and HspB-pulsed HMDCs. The percentage of IL-12p70-positive cells in the HspB-pulsed HMDCs was 36-fold lower than in the Com1-pulsed HMDCs, whereas the percentage of IL-10-positive cells in the HspB-pulsed HMDCs was threefold higher than in the Com1-pulsed HMDCs (Figure 3).
Figure 3.
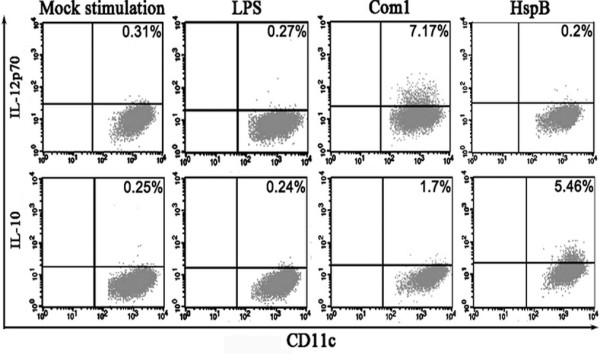
Production of IL-12p70 and IL-10 in antigen-treated HMDCs. After 24 h of stimulation with C. burnetii antigens, IL-12p70 and IL-10 expression in CD11c+ HMDCs were analyzed by intracellular cytokine staining. The percentages of double-positive cells in HMDCs are indicated in the top-right corner, and the results are representative of three independent experiments, each using cells derived from a different donor.
Early T-cell activation induced by antigen-pulsed HMDCs
To evaluate the direct effect of the antigen-pulsed HMDCs on T-cell activation, CD69 expression in both CD4+ and CD8+ cells was determined by flow cytometry after 12 h of interaction of T cells with antigen-pulsed HMDCs (Figure 4). As a result, the percentages of CD4-CD69 and CD8-CD69 double-positive cells among T cells cocultured with Com1-pulsed HMDCs were 8.9- and 14.2-fold higher, respectively, than those cocultured with mock-pulsed HMDCs; the percentages of CD4-CD69 and CD8-CD69 double-positive cells among T cells cocultured with HspB-pulsed HMDCs were only 2.7- and 5.6-fold higher, respectively, than those cocultured with mock-pulsed HMDCs (Figure 4). The percentages of CD4-CD69 and CD8-CD69 double-positive cells among T-cells cocultured with Com1-pulsed HMDCs were 3.3- and 2.5-fold higher, respectively, than those cocultured with HspB-pulsed HMDCs (Figure 4).
Figure 4.
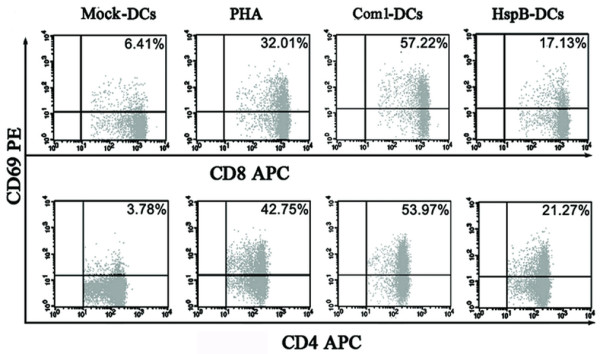
CD69 expression in T cells interacted with antigen-pulsed HMDCs. After coculture with antigen-pulsed HMDCs (Com1-DCs/HspB-DCs), mock-pulsed HMDCs (Mock-DCs), or PHA (positive control), CD69 expression on the surfaces of CD4+ and CD8+ cells were determined in T cells. The percentages of double-positive cells among the T cells are indicated in the top-right corner, and the results are representative of three independent experiments, each using cells derived from a different donor.
Cytokine production of T cells elicited by antigen-pulsed HMDCs
To determine the differentiation orientation of T cells after their interaction with antigen-pulsed HMDCs, the production of cytokines (IFN-γ, TNF-α, and IL-10) was determined in CD4+ and CD8+ cells. The percentages of CD4-IFN-γ and CD8-IFN-γ double-positive cells among T-cells cocultured with Com1-pulsed HMDCs were 2.2- and 15.1-fold higher, respectively, than those cocultured with HspB-pulsed HMDCs (Figure 5). Meanwhile, the percentages of CD4-TNF-α and CD8-TNF-α double-positive cells among T cells cocultured with Com1-pulsed HMDCs were 24.1- and 2.9-fold higher, respectively, than those cocultured with HspB-pulsed HMDCs (Figure 6). Additionally, IL-10 expression of CD4+ and CD8+ cells among T cells treated with Com1-pulsed HMDCs or HspB-pulsed HMDCs remained at baseline levels (data not shown).
Figure 5.
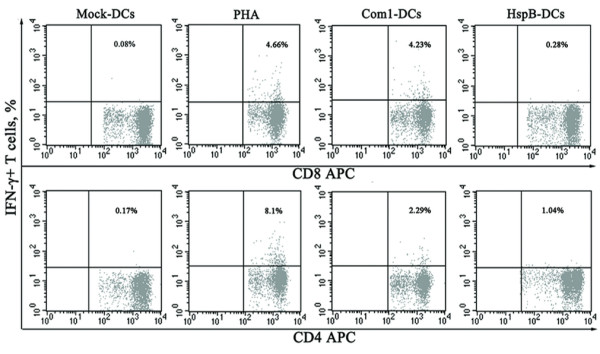
IFN-γ expression in CD4+ and CD8+ cells induced with antigen-pulsed HMDCs. IFN-γ expression in CD4+ and CD8+ cells was analyzed in T cells cocultured with antigen-pulsed HMDCs (Com1-DCs/HspB-DCs), mock-pulsed HMDCs (mock-DCs), or PHA (positive control) by intracellular IFN-γ staining. The percentages of double-positive cells among the T cells are indicated in the top-right corner, and the results are representative of three independent experiments, each using cells derived from a different donor.
Figure 6.
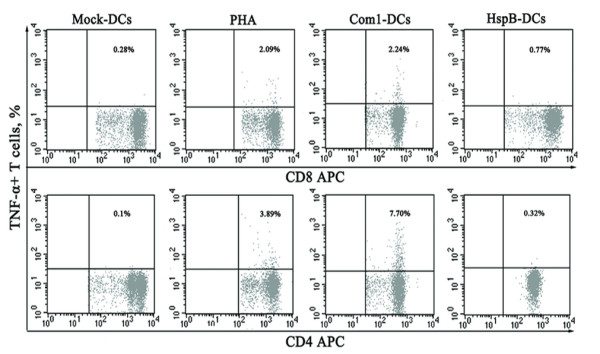
TNF-α expression in CD4+ and CD8+ cells induced with antigen-pulsed HMDCs. TNF-α expression on CD4+ and CD8+ cells was analyzed in T cells cocultured with antigen-pulsed HMDCs (Com1-DCs/HspB-DCs), mock-pulsed HMDCs (mock-DCs), or PHA (positive control) by intracellular TNF-α staining. The percentages of double-positive cells among the T cells are indicated in the top-right corner, and the results are representative of three independent experiments, each using cells derived from a different donor.
T-cell proliferation induced by antigen-pulsed HMDCs
T-cell proliferation induced by Com1- or HspB-pulsed HMDCs was evaluated by carboxyfluorescein diacetate succinimidyl ester (CFSE)-based proliferation assay after 96 h of interaction of T cells with antigen-pulsed HMDCs. The proliferation rates of CD4+ and CD8+ cells among T-cells cocultured with Com1-pulsed HMDCs were 4.1- and 7.1-fold higher, respectively, than those cocultured with HspB-pulsed HMDCs (Figure 7).
Figure 7.
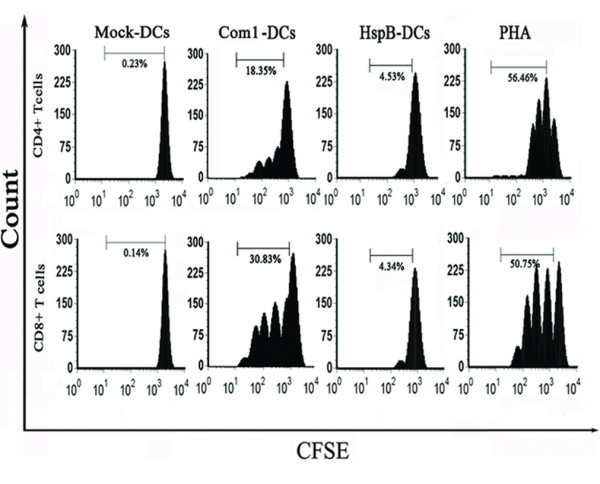
Proliferation of CD4+ and CD8+ cells in response to antigen-treated HMDCs. The CFSE-stained T cells were stimulated with antigen-treated HMDCs (Com1-DCs/HspB-DCs), mock-pulsed HMDCs (Mock-DCs), or PHA (positive control) for 96 h. The results are representative of three independent experiments, each using cells derived from a different donor.
Immune protective analysis of Com1- and HspB-pulsed BMDCs
Compared with mice receiving mock-pulsed BMDCs (control), animals receiving Com1-pulsed BMDCs exhibited significantly lower coxiella burden; mice receiving HspB-pulsed BMDCs displayed similar high levels of coxiella burden after challenge with C. burnetii (Figure 8). Coxiella burden in mice receiving HspB-pulsed BMDCs was significantly higher than in those receiving Com1-pulsed BMDCs; coxiella burden in animals receiving I Ag-pulsed BMDCs was much lower than in mice receiving Com1-pulsed BMDCs (Figure 8).
Figure 8.
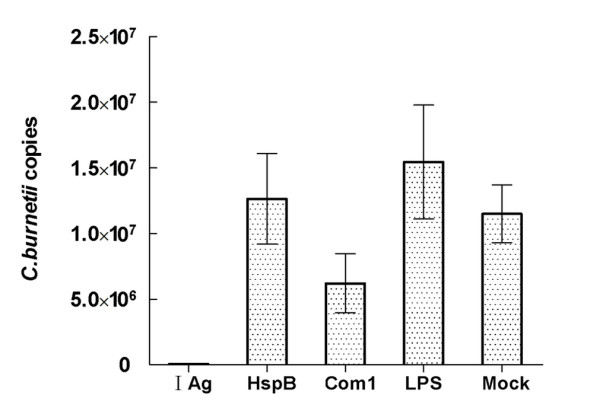
Estimation of coxiella burden in recipient mice of antigen-pulsed BMDCs. Coxiella DNA copies were determined in five mouse samples per group by quantitative PCR. Data are expressed as the average copy number of five samples, and error bars indicate the standard deviation. Mice receiving phase I Ag-pulsed BMDCs (I Ag) and mock-pulsed BMDCs (Mock) were used as positive and negative controls, respectively. In quantitative PCR analysis, the copy number per mouse was obtained with 1% of the DNA sample extracted from 10 mg spleen tissue. Coxiella burden in mice receiving Com1-pulsed BMDCs was significantly lower than in mice receiving HspB-pulsed BMDCs (P<0.05). This result is from one independent experiment.
Discussion
Com1 was first recognized as an immunoreactive protein associated with the outer membrane of C. burnetii by Hendrix and colleagues [19], and it was identified as a major immunodominant antigen of C. burnetii in other studies [20,21]. HspB was similarly recognized as a major immunodominant antigen of C. burnetii [20] and was suggested as being a surface-associated protein [24]. However, it is unclear whether Com1 and HspB are able to mount specific immune responses, particularly a cell-mediated immune response against C. burnetii in humans.
In the present study, Com1 and HspB were applied to stimulate HMDCs, and the antigen-pulsed HMDCs were arranged to stimulate isogenic T cells. To explore a possible mechanism of the immune response induced by Com1 or HspB, the immune effector functions (including T-cell activation-associated cytokines expressed in the antigen-pulsed HMDCs and the cytokines associated with intracellular bactericidal activities in the T cells activated by the antigen-pulsed HMDCs) were measured by flow cytometry.
DCs are present in most tissues in an immature status, and they undergo maturation upon antigen capture, whereupon they migrate to the lymph nodes to elicit adaptive immune responses [1]. In the present study, the Com1-pulsed HMDCs exhibited increased expression of surface molecules, including the DC-maturation marker (CD83), costimulatory molecules (CD40, CD80, and CD86), and adhesion molecules (CD54 and CD58). In addition, CD83, CD80, and CD86 are DC-maturation markers, and thus their increased expression in Com1-pulsed HMDCs suggests that Com1 has the ability to induce efficient maturation of HMDCs.
These surface molecules expressed on mature HMDCs may efficiently initiate the primary adaptive immune response. During this response, naive T cells polarize toward the antigen-presenting DCs, and a specific large-scale molecular complex is built at the DC-T-cell interface [26]. T-cell activation is regulated primarily by signalling events that derive from the interaction between the T-cell receptor (TCR) and the presented antigens on DCs and the interaction between T-cell surface molecules (CD28, CD154, CD2, and CD11a) and costimulatory (CD40, CD80, and CD86) or adhesion molecules (CD54 and CD58) on DCs [27]. The CD80/CD86-CD28 interaction provides important signals for T-cell activation and survival, and the CD40-CD154 interaction is crucial for the development of CD4+ T-cell-dependent effector functions; the interactions of both CD54-CD11a and CD58-CD2 act as a central part in the clustering of DCs and CD4+ T cells [27]. Therefore, the increased expression of costimulatory and adhesion molecules on Com1-activated HMDCs strongly indicates that Com1-activated HMDCs possess the ability to induce efficient proliferation and activation of naive T cells.
T-cell proliferation induced by antigen-treated HMDCs was measured by flow cytometry of CFSE dye dilution. Our results showed that the proliferation levels of CD4+ and CD8+ cells in T cells cocultured with Com1-activated HMDCs were significantly higher than those cocultured with mock-pulsed HMDCs. This suggests that Com1-activated HMDCs efficiently stimulated naive T cells to expand the populations of both CD4+ and CD8+ subsets before acquiring T-cell effector function.
It is well established that antigen-activated DCs can regulate the balance of Th1 and Th2 by differential expression between IL-12 and IL-10. If IL-12 is much higher than IL-10, the naive T cells will be skewed toward the Th1 phenotype; conversely, if IL-10 is much higher than IL-12, the naive T cells will differentiate toward the Th2 phenotype. In the present study, high IL-12p70 and low IL-10 expression were found in Com1-pulsed HMDCs, which suggests that Com1-activated HMDCs possess the ability to drive T cells toward Th1 differentiation through the Th1-specific cytokine IL-12.
To demonstrate the capability of Com1-pulsed HMDCs to activate T cells, the T-cell activation marker CD69 on CD4+ and CD8+ cells was determined after T-cell interaction with Com1-pulsed HMDCs. Our results showed that the percentages of CD4-CD69 and CD8-CD69 double-positive cells among T cells that interacted with Com1-pulsed HMDCs were much greater than those that interacted with mock-pulsed HMDCs. This firmly suggests that both CD4+ and CD8+ cells are efficiently activated through strong accessory signals derived from the surface-molecule interactions between Com1-activated HMDCs and T cells.
Laboratory studies have demonstrated that protection against C. burnetii infection is conferred by a Th1-oriented immune response, which depends on the Th1-specific cytokine IFN-γ and inflammatory cytokine TNF-α since both cytokines have the ability to upregulate the bactericidal activity of macrophages [28,29]. In the present study, Th1-specific cytokine IFN-γ and Th2-specific cytokine IL-10 as well as inflammatory cytokine TNF-α were determined in T cells after exposure to Com1-activated HMDCs. Our results showed that the expression of IFN-γ and TNF-α in CD4+ and CD8+ cells was dramatically increased after T-cell interaction with Com1-pulsed HMDCs. However, the IL-10 expression in CD4+ and CD8+ cells in this T-cell population remained at baseline levels. These results strongly imply that Com1-pulsed HMDCs have the ability to drive differentiation of naive CD4+ and CD8+ cells toward, respectively, CD4+ Th1 and CD8+ Tcl cells and that HMDC-activated CD4+ and CD8+ cells may produce IFN-γ and TNF-α to enhance the intracellular bactericidal activity of the host cells (macrophages).
To demonstrate the immunoprotective ability of Com1, Com1-pulsed BMDCs were adaptively transferred to naive mice. Our results showed that mice receiving Com1-pulsed BMDCs exhibited significantly lower coxiella burden than control animals after challenge with virulent C. burnetii. However, the coxiella burden in mice receiving HspB-pulsed BMDCs was significantly higher than in those receiving Com1-pulsed BMDCs. This suggests that HspB is not a protective antigen.
We found that HspB was unable to induce efficient maturation and activation of HMDCs. The expressions of maturation marker (CD83), costimulatory (CD40, CD80, and CD86), and adhesion molecules (CD54 and CD58) of HspB-pulsed HMDCs were much lower than those of Com1-pulsed HMDCs. Moreover, HspB-pulsed HMDCs produced much lower levels of IL-12, but much higher levels of IL-10, than Com1-pulsed HMDCs. The increased expression of IL-10 in DCs may inhibit T-cell proliferation [30,31]. Similarly, Motta et al. [32] showed that Mycobacterium tuberculosis Hsp70 inhibited T-cell proliferation in vitro, since it impaired DC maturation and induced high production of IL-10 in DCs; this suggests that Hsp70 does not have inflammatory potential but rather immunosuppressive properties.
In this study, the percentages of CD8-CD69 and CD4-CD69 double-positive cells among T cells cocultured with HspB-pulsed HMDCs were much lower than those cocultured with Com1-pulsed HMDCs. HspB-pulsed HMDCs caused a negligible proliferation of CD8+ and CD4+ cells, which suggests that the low-level expression of costimulatory molecules and adherence molecules of HspB-pulsed HMDCs was unable to activate the T cells.
Generally, the interaction between the innate/adaptive immune system and invading C. burnetii is sufficient to eradicate the organisms, which elicit asymptomatic or mild, self-limiting, flu-like symptoms in most human C. burnetii infections. The bactericidal immune response is mainly based on inflammatory cytokines IFN-γ and TNF-α [33,34]. However, a variety of pathogens can take advantage of the anti-inflammatory properties of IL-10 (which is crucial for maintaining the subtle balance between immunity against pathogens and a pathology that can arise when an immune response is left unchecked) as a mean of avoiding sterilizing immunity and establishing a persistent infection [35-37]. In C. burnetii infections, IL-10 is believed to have the ability to disarm bactericidal responses and contribute to chronic development of C. burnetii infection. Previous studies [38,39] showed that IL-10 promoted replication of C. burnetii in human monocytes and C. burnetii established a more robust infection in IL-10-overexpressing transgenic mice owing to the macrophages failing to kill the bacteria. Another study revealed that IL-10 overproduction was involved in establishing persistent C. burnetii infections and was related to Q fever endocarditis development [40]. Clearly, IL-10 plays an important role in persistent C. burnetii infection. Therefore, the high level of anti-inflammatory cytokine IL-10 generated in HspB-pulsed HMDCs may be an important factor in inhibiting proliferation and activation of T cells.
In our recent study, adaptive transfer of mouse BMDCs stimulated with coxiella antigens into BABL/c mice showed that the coxiella burden in animals receiving Com1-pulsed BMDCs was significantly lower than in mice receiving EnhA-pulsed BMDCs [41]. Although both Com1 and EnhA had the ability to activate BMDCs to express MHC-II and costimulatory molecules, CD4+ T cells from mice receiving Com1-pulsed BMDCs exhibited significantly higher CD69 expression than animals receiving EnhA-pulsed BMDCs after coculture with homologous antigen-pulsed BMDCs. In addition, the percentages of CD4+ and IFN-γ double-positive cells in mice receiving Com1-pulsed BMDCs were significantly higher than in animals receiving EnhA-pulsed BMDCs; the percentages of CD4+ and IL-17 double-positive cells in mice receiving Com1-BMDCs were significantly higher than in mice receiving EnhA-pulsed BMDCs [41]. However, the percentages of regulatory T lymphocytes in mice receiving Com1-pulsed BMDCs were substantially lower than in animals receiving EnhA-pulsed BMDCs [41]. Our results suggest that the protection offered by Com1-pulsed BMDCs is correlated with the increased proliferation of Th1 CD4+ T cells, preferential development of Th17 cells, and impaired expansion of regulatory T lymphocytes [41].
Conclusions
Our results demonstrate that Com1 is able to induce complete maturation and activation of HMDCs that drive T cells toward Th1 and Tc1 polarization; however, HspB-induced incompletely mature HMDCs are unable to induce efficient T-cell proliferation and activation. Unlike HspB, Com1 is a potent protective antigen of C. burnetii, which has been demonstrated by adoptive transfers of Com1-pulsed and HspB-pulsed BMDCs into naive BALB/c mice.
Methods
Preparation of whole-cell antigen and recombinant proteins
C. burnetii Xinqiao strain (phase I) isolated from ticks in China [42] was propagated in embryonated eggs, inactivated with formalin, and purified by renografin density centrifugation, as described previously [16]. The purified coxiella cells were suspended in phosphate-buffered saline (PBS) buffer as a phase I antigen (I Ag). Recombinant proteins, Com1 and HspB, were prepared in our laboratory according to described methods [20]. Briefly, the com1 gene was amplified from C. burnetii (Xinqiao strain) genomic DNA with a pair of primers: primer 1 (5'-ATCGGATCCTTAGCCGGAACCTTGACC-3') with the BamHI restriction site; and primer 2 (5'-GCCCTCGAGTAACGCTTTATTACCAATGACG-3') with the XhoI restriction site. The amplified gene fragment was cloned into a prokaryotic expression plasmid, pET-32a, by the restriction sites. E. coli BL21 cells were transformed by com1-recombined pET-32a. The hspB gene was amplified from C. burnetii genomic DNA with the other primer pair: primer 1 (5'-GCCGAGCTCGTGACGTTGGGACCAAAAGG-3') with the SacI restriction site; and primer 2 (5'-ATACTGCAGCCGCCCATTCCTCCCATGCC-3') with the PstI restriction site. The amplified gene fragment was cloned into a prokaryotic-expression plasmid pQE30 by the restriction sites.
E. coli M15 cells were transformed by hspB-recombined pQE30. The transformed bacterial cells were grown to an optical density (OD600 = 0.6) at 37°C in Luria-Bertani broth, containing 50 μg/ml ampicillin and then induced for 4 h with 0.5 mM isopropyl β-D-thiogalactoside (IPTG, Sigma-Aldrich, St Louis, MO, USA). The recombinant proteins containing six consecutive histidine residues were purified from the cellular debris of the transformed E. coli cells using the nickel-nitrilotriacetic resin affinity chromatography kit (QIAexpressionist, Qiagen GmbH, Germany) according to the manufacturer's instructions. The recombinant proteins Com1 and HspB were identified in SDS-PAGE (Figure 1A) and recognized using the sera from BALB/c mice experimentally infected with C. burnetii in an immunoblot assay (Figure 1B).
The purified recombinant proteins were treated with Triton X-114 (Amresco, Solon, OH, USA) to remove LPS, as described previously [43]. In brief, 5 μl Triton X-114 was added to 500 μl recombinant protein solution (1 μg/ml), and then the mixture was vigorously shaken in a vortex mixer for several seconds. The mixture was kept on ice for 5 min and then incubated at 37°C for 5 min. Finally, the mixture was briefly centrifuged, and the supernatant was collected. This procedure was repeated three times. Each LPS-removed protein was concentrated with a concentrator (VivaScience, Hannover, Germany), and the concentrated protein was diluted with the elution buffer in the affinity chromatography kit to make a protein solution (400 μg/ml). The recombinant proteins were used only when the LPS level was below 0.005 endotoxin units (EU)/ml, determined using the Limulus amoebocyte assay (Sigma-Aldrich, St Louis, MO, USA).
Isolation and culture of cells
The human blood samples in this study were obtained from three healthy donors in our research group who clearly knew the purpose of this study, according to the Blood Donation Law of the People's Republic of China. This study was approved by the ethics committee of the Beijing Institute of Microbiology and Epidemiology.
HMDCs were generated from human peripheral blood mononuclear cells (PBMCs), as described previously [44]. In brief, PBMCs were isolated from blood buffy coats of healthy donors by Ficoll-Paque Plus (Haoyang Biotech, Beijing, China) density-gradient centrifugation, and the monocyte and T-cell fractions were obtained from PBMCs by Percoll density-gradient centrifugation (Haoyang Biotech, Beijing, China), according to the manufacturer's protocols. Residual lymphocytes were removed from the monocyte fraction by plastic adherence in 12-well plates (Costar, Corning, NY, USA), and the purity of the collected monocytes (CD14+) was greater than 90%, as determined by flow cytometry. The isogeneic T cells were collected from the supernatants, and more than 90% of the collected cells were CD3-positive in flow cytometric analysis.
The adherence monocytes were cultured with RPMI 1640 medium (Hyclone, Beijing, China) supplemented with 10% fetal bovine serum (FBS; Hyclone, Beijing, China), 50 ng/ml human IL-4 (PeproTech, Rocky Hill, NJ, USA), and 50 ng/ml human granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech) at 37°C and 5% CO2. IL-4 and GM-CSF were added to the cultured cells every other day. After 5 days of culture, more than 95% of the cells had converted to immature HMDCs (iHMDCs), with a phenotype of CD11c+, CD54 low, CD83 low, CD40 low, CD58 low, CD86low, and CD80low, as determined by flow cytometry.
Approximately 1 × 106 iHMDCs was added to each well of a six-well plate; they were then treated with Com1 (20 μg/ml), HspB (20 μg/ml), E. coli LPS (6 μg/ml; Sigma-Aldrich, St Louis, MO, USA), and 50 μl elution buffer (mock pulse) for 24 h at 37°C. After 24 h of antigen stimulation, the HMDCs were harvested by centrifugation at 500 × g for 5 min, and their maturation and activation status were determined by flow cytometry, as described below.
Flow cytometric assay
The harvested HMDCs were suspended in PBS containing 5% FBS, and the following monoclonal antibodies (mAbs) were used to characterize the cell phenotypes (BD Pharmingen, San Jose, CA, USA): anti-CD11c-APC, anti-CD58-FITC, anti-CD80-PE, anti-CD40-FITC, anti-CD83-PE, anti-CD54-PE, and anti-CD86-PE. In parallel, HMDCs were immunostained with the isotype-matched control mAb. Approximately 1 × 106 HMDCs were immunostained with three-color antibodies in 50 μl PBS containing 5% FBS. The HMDCs were incubated with different antibodies at room temperature for 15 min. After washing, 1% paraformaldehyde was added to fix the stained HMDCs. Samples were run on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed using CellQuest software (BD Biosciences, San Jose, CA, USA).
Intracellular cytokine detection by flow cytometry
After 20 h of antigen stimulation, HMDCs were incubated for 4 h with brefeldin A solution (1 μl/106 cells), and then the HMDCs were harvested and washed twice in PBS containing 1% bovine serum albumin (BSA). The brefeldin-treated HMDCs were stained with anti-CD11c-APC for 15 min at room temperature. After one fixing step with FACS lysing solution, HMDCs were permeabilized and immunostained with PE-conjugated anti-cytokine antibody (anti-IFN-α, anti-IL-12p70, or anti-IL-10) or isotype-matched control mAb for 30 min at room temperature.
After 20 h of incubation with the antigen-pulsed HMDCs, the T cells were incubated with brefeldin A for 4 h. After washing, the T cells were stained with anti-CD3-PerCP, anti-CD4-APC/anti-CD8-APC, anti-IFN-γ-FITC, anti-TNF-α-PE, or anti-IL-10-PE by the method described above. After washing, the stained cells were collected and suspended in PBS, and the different cytokine expression in HMDCs, CD4+, or CD8+ cells was determined by flow cytometry.
Measurement of early T-cell activation
After 12 h of incubation with the antigen-treated HMDCs, the T cells were stained with anti-CD3-PerCP, anti-CD4-APC/anti-CD8-APC, and anti-CD69-PE in 50 μl PBS containing 5% FBS at room temperature for 15 min. After washing, 1% paraformaldehyde was added to fix the stained cells, and then the CD69 expression on the CD4+ or CD8+ cells was determined by flow cytometry. In parallel, the cells were stained with isotype-matched control mAb.
T-cell proliferation assay
Approximately 5 × 107 T cells in 1 ml PBS was mixed with 10 μM CFSE (Invitrogen, Eugene, OR, USA), and then the cells were incubated at 37°C for 10 min in the dark. After incubation, 10 ml PBS containing 5% FBS was added to each sample, followed by 5 min of incubation on ice. After three washes in PBS, the cells were harvested by centrifugation at 400 × g for 5 min. Approximately 1 × 105 CFSE-labelled responder cells was incubated with 1 × 104 antigen-pulsed HMDCs at 37°C and 5% CO2 for 96 h. The CFSE-labelled responder cells were stimulated with PHA (10 μg/ml) for 96 h as a positive control. After incubation, the cells were harvested and stained with anti-CD3-PerCP and anti-CD4-APC/anti-CD8-APC. CFSE-labelled responder cell proliferation in CD4+ or CD8+ cells was determined by flow cytometry, and the proliferation percentages of the T-cell fractions were calculated by ModFit software (BD Biosciences, San Jose, CA, USA).
Immunoprotection analysis of Com1- and HspB-pulsed BMDCs
Mouse bone marrow dendritic cells (BMDCs, CD11c+) were isolated from the bone marrow of BALB/c mice, according to the protocol described previously [45]. Briefly, a single-cell suspension from the bone marrow was prepared from mouse femurs and cultured in a complete 1640 medium, containing 10% FBS, 100 μg/ml streptomycin sulfate, and 100 U/ml penicillin at 37°C and 5% CO2. Approximately 1 × 106 cells was added to each well of a six-well plate, and mouse GM-CSF (20 ng/ml; Peprotech) and IL-4 (10 ng/ml; Peprotech) was added to the culture medium every other day. After 6 days of culture, BMDCs were stimulated with C. burnetii I Ag (10 μg/ml), Com1 (10 μg/ml), HspB (10 μg/ml), E. coli LPS (2 μg/ml), or 25 μl elution buffer (mock pulse) for 24 h at 37°C and 5% CO2.
After antigen stimulation, the antigen-pulsed BMDCs were intraperitoneally (i.p.) injected into BALB/c mice (6 weeks age, female; 5 × 105 cells/mouse). Fourteen days after the BMDCs injection, mice were i.p. challenged with C. burnetii Xinqiao strain (1 × 106 cells/mouse) in Biosafety Level 3 Laboratory. Mice were sacrificed on day 7 post-challenge, and their spleens were harvested for detection of C. burnetii burden by a quantitative polymerase chain reaction (PCR) analysis [41,46]. For each mouse, 10 mg of spleen tissue was used to extract DNA with DNeasy Blood &Tissue kit (Qiagen, Germany), and the purified DNA was eluted from the column with 200 μl elution buffer. The data analysis was performed using SPSS software (version 10.0; SPSS). Group comparison was performed with the one-way analysis of variance (ANOVA) test. Values were expressed as means with standard deviations (SDs). P values of <0.05 were considered significant.
Authors' contributions
YW carried out the analyses of HMDCs pulsed with antigens and T cells interacted with antigen-pulsed HMDCs and drafted the manuscript. XX performed the immune protective analysis of antigen-pulsed BMDCs against C. burnetii and drafted the manuscript. DW assisted the analyses of HMDCs and T-cells, and LW assisted the immune protective analysis of antigen-pulsed BMDCs. All authors read and approved the final manuscript.
Contributor Information
Ying Wang, Email: wcyli@sina.com.
Xiaolu Xiong, Email: xiongxiaolu624@sohu.com.
Deping Wu, Email: wudeping520@yahoo.com.cn.
Xile Wang, Email: wangxile80@163.com.
Bohai Wen, Email: bohaiwen@sohu.com.
Acknowledgements
This research was supported by grants (30670101 and 30901369) from the National Natural Science Foundation of China and a grant (2010CB530200/2010CB530205) from the National Basic Research Program of China.
References
- Banchereau J, Steinman R. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Steinman RM. Some interfaces of dendritic cell biology. Acta Pathol Microbiol Immunol Scand. 2003;111:675–697. doi: 10.1034/j.1600-0463.2003.11107802.x. [DOI] [PubMed] [Google Scholar]
- Sundquist M, Johansson C, Wick MJ. Dendritic cells as inducers of antimicrobial immunity in vivo. Acta Pathol Microbiol Immunol Scand. 2003;111:715–724. doi: 10.1034/j.1600-0463.2003.11107804.x. [DOI] [PubMed] [Google Scholar]
- Chung NP, Chen Y, Chan VS, Tam PK, Lin CL. Dendritic cells: sentinels against pathogens. Histol Histopathol. 2004;19:317–24. doi: 10.14670/HH-19.317. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Akira S. TLR signaling and the function of dendritic cells. Chem Immunol Allergy. 2005;86:120–135. doi: 10.1159/000086657. [DOI] [PubMed] [Google Scholar]
- Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/S0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S, Galan JE, Mellman I. Developmental Control of Endocytosis in Dendritic Cells by Cdc42. Cell. 2000;102:325–34. doi: 10.1016/S0092-8674(00)00038-6. [DOI] [PubMed] [Google Scholar]
- Berón W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–21. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Hackstadt T, Samuel JE. Developmental biology of Coxiella burnetii. Trends Microbiol. 1999;7:149–54. doi: 10.1016/S0966-842X(99)01475-4. [DOI] [PubMed] [Google Scholar]
- Maurin M, Raoult D. Q fever. Clin Microbiol Rev. 1999;12:518–553. doi: 10.1128/cmr.12.4.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander L. Q fever epidemiology and pathogenesis. Microbes Infect. 2000;2:417–424. doi: 10.1016/S1286-4579(00)00325-7. [DOI] [PubMed] [Google Scholar]
- Marrie TJ. Q fever pneumonia. Curr Opin Infect Dis. 2004;17:137–142. doi: 10.1097/00001432-200404000-00012. [DOI] [PubMed] [Google Scholar]
- Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci USA. 2003;100:5455–60. doi: 10.1073/pnas.0931379100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waag DM, England MJ, Tammariello RF, Byrne WR, Gibbs P, Banfield CM, Pitt MLM. Comparative efficacy and immunogenicity of Q fever chloroform:methanol residue (CMR) and phase I cellular (Q-Vax) vaccines in cynomolgus monkeys challenged by aerosol. Vaccine. 2002;20:2623–34. doi: 10.1016/S0264-410X(02)00176-7. [DOI] [PubMed] [Google Scholar]
- Williams JC, Peacock MG, Waag DM, Kent G, England MJ, Nelson G, Stephenson EH. Vaccines against coxiellosis and Q fever. Development of a chloroform:methanol residue subunit of phase I Coxiella burnetii for the immunization of animals. Ann N Y Acad Sci. 1992;653:88–111. doi: 10.1111/j.1749-6632.1992.tb19633.x. [DOI] [PubMed] [Google Scholar]
- Waag DM, England MJ, Bolt CR, Williams JC. Phase I clinical trial of CMR Q fever vaccine: low-dose priming enhances humoral and cellular immune responses to Coxiella burnetii. Clin Vaccine Immunol. 2008;15:1505–12. doi: 10.1128/CVI.00119-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GQ, To H, Yamaguchi T, Fukushi H, Hirai K. Differentiation of Coxiella burnetii by sequence analysis of the gene (com1) encoding a 27-kDa outer membrane protein. Microbiol Immunol. 1997;41:871–7. doi: 10.1111/j.1348-0421.1997.tb01943.x. [DOI] [PubMed] [Google Scholar]
- Hendrix LR, Mallavia LP, Samuel JE. Cloning and sequencing of Coxiella burnetii outer membrane protein gene com1. Infect Immun. 1993;61:470–7. doi: 10.1128/iai.61.2.470-477.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Kiss K, Seshadri R, Hendrix LR, Samuel JE. Identification and cloning of immunodominant antigens of Coxiella burnetii. Infect Immun. 2004;72:844–52. doi: 10.1128/IAI.72.2.844-852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Chen C, Bouman T, Pablo J, Unal B, Cockrell DC, Brown WC, Barbian KD, Porcella SF, Samuel JE, Felgner PL, Heinzen RA. Candidate antigens for Q fever serodiagnosis revealed by immunoscreening of a Coxiella burnetii protein microarray. Clin Vaccine Immunol. 2008;15:1771–9. doi: 10.1128/CVI.00300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekeyová Z, Kowalczewska M, Decloquement P, Pelletier N, Spitalská E, Raoult D. Identification of protein candidates for the serodiagnosis of Q fever endocarditis by an immunoproteomic approach. Eur J Clin Microbiol Infect Dis. 2009;28:287–95. doi: 10.1007/s10096-008-0621-4. [DOI] [PubMed] [Google Scholar]
- Fernandez RC, Logan SM, Lee SH, Hoffman PS. Elevated levels of Legionella pneumophila stress protein Hsp60 early in infection of human monocytes and L929 cells correlate with virulence. Infect Immun. 1996;64:1968–76. doi: 10.1128/iai.64.6.1968-1976.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garduño RA, Garduño E, Hoffman PS. Surface-associated Hsp60 chaperonin of Legionella pneumophila mediates invasion in a HeLa Cell Model. Infect Immun. 1998;66:4602–10. doi: 10.1128/iai.66.10.4602-4610.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waag DM. Coxiella burnetii: Host and bacterial responses to infection. Vaccine. 2007;25:7288–7295. doi: 10.1016/j.vaccine.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Rothoeft T, Gonschorek A, Bartz H, Anhenn O, Schauer U. Antigen dose, type of antigen-presenting cell and time of differentiation contribute to the T helper 1/T helper 2 polarization of naive T cells. Immunology. 2003;110:430–9. doi: 10.1111/j.1365-2567.2003.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimanianda V, Haensler J, Lacroix-Desmazes S, Kaveri SV, Bayry J. Novel cellular and molecular mechanisms of induction of immune responses by aluminum adjuvants. Trends Pharmacol Sci. 2009;30:287–95. doi: 10.1016/j.tips.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Honstettre A, Ghigo E, Moynault A, Capo C, Toman R, Akira S, Takeuchi O, Lepidi H, Raoult D, Mege JL. Lipopolysaccharide from Coxiella burnetii is involved in bacterial phagocytosis, filamentous actin reorganization, and inflammatory responses through Toll-like receptor 4. J Immunol. 2004;172:3695–703. doi: 10.4049/jimmunol.172.6.3695. [DOI] [PubMed] [Google Scholar]
- Shannon JG, Heinzen RA. Adaptive immunity to the obligate intracellular pathogen Coxiella burnetii. Immunol Res. 2009;43:138–48. doi: 10.1007/s12026-008-8059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner MA, Kim SH, Bianco NR, Francisco LM, Sharpe AH, Robbins PD. B7-1/2, but not PD-L1/2 molecules, are required on IL-10-treated tolerogenic DC and DC-derived exosomes for in vivo function. Eur J Immunol. 2009;39:3084–90. doi: 10.1002/eji.200939407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez N, Karumuthil-Melethil S, Li R, Prabhakar BS, Holterman MJ, Vasu C. Preferential costimulation by CD80 results in IL-10-dependent TGF-β1(+) -adaptive regulatory T cell generation. J Immunol. 2008;180:6566–76. doi: 10.4049/jimmunol.180.10.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta A, Schmitz C, Rodrigues L, Ribeiro F, Teixeira C, Detanico T, Bonan C, Zwickey H, Bonorino C. Mycobacterium tuberculosis Hsp70 impairs maturation of DCs from bone marrow precursors, induces IL-10 production and inhibits T cell proliferation in vitro. Immunology. 2007;121:462–472. doi: 10.1111/j.1365-2567.2007.02564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun. 2007;75:3245–55. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigo E, Capo C, Tung CH, Raoult D, Gorvel JP, Mege JL. Coxiella burnetii survival in THP-1 monocytes involves the impairment of phagosome maturation: IFN-gamma mediates its restoration and bacterial killing. J Immunol. 2002;169:4488–95. doi: 10.4049/jimmunol.169.8.4488. [DOI] [PubMed] [Google Scholar]
- Aliberti J. Host persistence: exploitation of anti-inflammatory pathways by Toxoplasma gondii. Nat Rev Immunol. 2005;5:162–70. doi: 10.1038/nri1547. [DOI] [PubMed] [Google Scholar]
- Nylén S, Sacks D. Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol. 2007;28:378–84. doi: 10.1016/j.it.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Tufariello JM, Chan J, Flynn JL. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect Dis. 2003;3:578–90. doi: 10.1016/S1473-3099(03)00741-2. [DOI] [PubMed] [Google Scholar]
- Ghigo E, Capo C, Raoult D, Mege JL. Interleukin-10 stimulates Coxiella burnetii replication in human monocytes through tumor necrosis factor down-modulation: role in microbicidal defect of Q fever. Infect Immun. 2001;69:2345–52. doi: 10.1128/IAI.69.4.2345-2352.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghari S, Bechah Y, Capo C, Lepidi H, Raoult D, Murray PJ, Mege JL. Persistent Coxiella burnetii infection in mice overexpressing IL-10: an efficient model for chronic Q fever pathogenesis. PLoS Pathog. 2008;4:23–31. doi: 10.1371/journal.ppat.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honstettre A, Imbert G, Ghigo E, Gouriet F, Capo C, Raoult D, Mege JL. Dysregulation of cytokines in acute Q fever: role of interleukin-10 and tumor necrosis factor in chronic evolution of Q fever. J Infect Dis. 2003;187:956–62. doi: 10.1086/368129. [DOI] [PubMed] [Google Scholar]
- Wei Y, Wang X, Xiong X, Wen B. Coxiella burnetii antigen-stimulated dendritic cells mediated protection against Coxiella burnetii in BALB/c mice. J Infect Dis. 2011;203:283–291. doi: 10.1093/infdis/jiq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Yu S, Yu G, Li Q, Zhang X. Analysis of proteins and lipopolysaccharide from Chinese isolates of Coxiella burnetii with monoclonal antibodies. Acta Virol. 1991;35:538–44. [PubMed] [Google Scholar]
- Aida Y, Pabst MJ. Removal of endotoxin from protein solutions by phase separation using Triton X-114. J Immunol Methods. 1990;132:191–5. doi: 10.1016/0022-1759(90)90029-U. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophages colony- stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wen B, Chen M, Zhang J, Niu D. Balb/c mouse model and real-time quantitative polymerase chain reaction for evaluation of the immunoprotectivity against Q fever. Ann N Y Acad Sci. 2005;1063:171–5. doi: 10.1196/annals.1355.027. [DOI] [PubMed] [Google Scholar]