

Non-technical summary
Cell volume regulation is an essential function involved not only in homeostasis, but also in cell migration, fission and programmed cell death. The volume-sensitive outwardly rectifying (VSOR) anion channel provides the main pathway for anion transport across the cell membrane during the regulation. We previously demonstrated that an inflammatory mediator, bradykinin, activates the VSOR channels in the major glial cells, astrocytes, in the brain derived from mice and the channels send signals to adjacent neurons through the release of glutamate. Here we demonstrate that this activation is controlled in the immediate vicinity of Ca2+-permeable channel proteins in the astrocytes via high concentrations of intracellular Ca2+, so-called ‘Ca2+ nanodomains’. This mechanism would provide a basis for responding quickly and certainly to even a minute amount of bradykinin released from surrounding tissues (e.g. slightly damaged blood vessel walls) with local control of cell shape changes and signal transmission by astrocytes.
Abstract
Abstract
Volume-sensitive outwardly rectifying (VSOR) anion channels play a key role in a variety of essential cell functions including cell volume regulation, cell death induction and intercellular communications. We previously demonstrated that, in cultured mouse cortical astrocytes, VSOR channels are activated in response to an inflammatory mediator, bradykinin, even without an increase in cell volume. Here we report that this VSOR channel activation must be mediated firstly by ‘nanodomains’ of high [Ca2+]i generated at the sites of both Ca2+ release from intracellular Ca2+ stores and Ca2+ entry at the plasma membrane. Bradykinin elicited a [Ca2+]i rise, initially caused by Ca2+ release and then by Ca2+ entry. Suppression of the [Ca2+]i rise by removal of extracellular Ca2+ and by depletion of Ca2+ stores suppressed the VSOR channel activation in a graded manner. Quantitative RT-PCR and suppression of gene expression with small interfering RNAs indicated that Orai1, TRPC1 and TRPC3 channels are involved in the Ca2+ entry and especially the entry through TRPC1 channels is strongly involved in the bradykinin-induced activation of VSOR channels. Moreover, Ca2+-dependent protein kinases Cα and β were found to mediate the activation after the [Ca2+]i rise through inducing generation of reactive oxygen species. Intracellular application of a slow Ca2+ chelator, EGTA, at 10 mm or a fast chelator, BAPTA, at 1 mm, however, had little effect on the VSOR channel activation. Application of BAPTA at 10 mm suppressed significantly the activation to one-third. These suggest that the VSOR channel activation induced by bradykinin is regulated by Ca2+ in the vicinity of individual Ca2+ release and entry channels, providing a basis for local control of cell volume regulation and intercellular communications.
Introduction
The volume-sensitive outwardly rectifying (VSOR) anion channel is the most influential regulator of cell volume in most types of animal cells. The channel is typically activated by an increase in cell volume, and opening of the channel provides a pathway for outflux of intracellular anions, especially Cl− ions. This outflux of anions, usually accompanied by an outflux of K+ ions, facilitates regulatory volume decrease (RVD) of the cells. The molecular identity of VSOR channel is, however, not determined yet (reviewed by Okada et al. 2009).
The VSOR channel is permeable significantly to amino acids, like glutamate or aspartate. Thus the conditions for opening VSOR channels may also induce intercellular signal transmission mediated by the excitatory amino acid. We previously demonstrated that an inflammatory mediator, bradykinin, activates VSOR channels in cultured mouse cortical astrocytes and the glutamate released via VSOR channels evokes [Ca2+]i rises in adjacent co-cultured neurons through NMDA receptor activation (Liu et al. 2009). In injured brains, the amount of excitatory amino acids released via astrocytic VSOR channels may become very massive and it may cause severe excitotoxic neuronal damage. The damage can indeed be alleviated by application of a specific blocker of VSOR channels, 4-(2-butyl-6, 7-dichloro-2-cyclopentyl - indan - 1 - on - 5 - yl) oxybutyric acid (DCPIB; Zhang et al. 2008).
Activation of VSOR channels is not necessarily accompanied by an increase in cell volume. The bradykinin-induced activation of VSOR channels in our previous study was, in fact, not associated with volume increases. Such a volume-independent activation of VSOR channels may be mediated by generation of reactive oxygen species (ROS) through NADPH oxidases (NOX), as reported in human cervix HeLa cells (Shimizu et al. 2004), rat hepatoma cells (Varela et al. 2004), rat microglia (Harrigan et al. 2008) and rabbit cardiomyocytes (Browe & Baumgarten, 2004; Deng et al. 2010), although it is not a universal mechanism as demonstrated in mouse Ehrlich ascites tumour cells (Lambert et al. 2009). The bradykinin-induced VSOR channel activation in astrocytes was found to be mediated by ROS (Liu et al. 2009). The prototypical NOX, NOX2/gp91phox, is reported to be expressed and driven by activation of protein kinase C (PKC) or [Ca2+]i rises in astrocytes (Pawate et al. 2004; Abramov et al. 2005). The bradykinin action on VSOR channel activation is mediated by the bradykinin B2 receptor coupled to Gαq proteins (Liu et al. 2009). Thus the receptor activation should elicit intracellular Ca2+ release through InsP3 receptors and generation of a PKC activator, diacylglycerol. Large amplitudes of [Ca2+]i rises due to Ca2+ release are indeed generated in astrocytes in response to bradykinin. How the [Ca2+]i rises and PKC activation are linked to VSOR channel activation is, however, yet to be determined. Many reports have long suggested the Ca2+ independence of VSOR channel activation induced by cell swelling (reviewed by Okada, 1997 and Hoffmann et al. 2009). Indeed, the swelling-induced VSOR channel activation is not necessarily accompanied by detectable [Ca2+]i rises in some types of cell (reviewed by Hoffmann et al. 2009), suggesting non-essential contribution of Ca2+ to the swelling-induced activation. In our previous study, suppression of observable bradykinin-induced [Ca2+]i rises by treatment with the membrane-permeable acetoxymethyl (AM) ester form of BAPTA had in fact no effects on VSOR channel activation (Liu et al. 2009). However, a more thorough examination is required because Ca2+ action may be limited within very narrow regions, narrower than the resolution limit of conventional optical microscopy, around the orifices of open Ca2+ channels, so called ‘Ca2+ nanodomains’. In the nanodomains, more than 10 μm of high [Ca2+]i rises proportional to the amplitudes of single-channel Ca2+ currents are generated and only a fast Ca2+ chelator, BAPTA, at very high concentrations can have access to them (Neher, 1998b; Augustine et al. 2003). In the case of the swelling-induced activation of VSOR channels, it has been reported that large amplitudes of the current through VSOR channels are still maintained even in the presence of intracellular BAPTA at 5–10 mm (Kubo & Okada, 1992; Pedersen et al. 1998). However, a different mechanism may regulate the activation mediated by Gαq-coupled receptors without cell swelling. Moreover, since the [Ca2+]i rises within 20 nm of Ca2+ channel pores must be less affected even by BAPTA (Neher, 1998b), it is necessary to examine the effect of thorough removal of Ca2+ both in the extracellular solution and in the intracellular Ca2+ stores and also to examine the dose dependence of the activation on the intracellular Ca2+ chelator, in order to elucidate exactly whether the Ca2+ in the nanodomains may participate partly or not and to determine the distance range of Ca2+ action in the process if it is involved. Ca2+ nanodomain-mediated regulation is well known in the exocytosis of neurotransmitters (Neher, 1998b; Augustine et al. 2003) and at the excitation–contraction coupling in cardiac muscle cells (Cheng & Lederer, 2008). The nanodomain mediation confers fast, accurate and localized regulation of Ca2+-dependent processes on a single cell. Furthermore, Ca2+-sensing molecules involved in the same cellular function but with different Ca2+ sensitivities in a single cell may be distributed at different distances from the centre of a nanodomain, modulating the function over different time scales, as seen in the modulation of neuronal membrane excitability (Akita & Kuba, 2000) and in the regulation of intracellular Ca2+ release itself (Akita & Kuba, 2008). Thus, estimating the distance range of Ca2+ action is very important to realize the significance of a Ca2+-dependent process. It is quite conceivable that Ca2+ nanodomain-mediated regulation would play a role also in local cell volume regulation during cell shape changes, migration or fission, although the evidence is not elucidated yet.
In this study we provide the first evidence for Ca2+ nanodomain-mediated regulation of the cell volume regulator, VSOR channels. First we demonstrate that the bradykinin-induced VSOR channel activation in mouse astrocytes is indeed Ca2+ dependent; the activation was suppressed in parallel with the suppression of intracellular Ca2+ release and store- or receptor-operated Ca2+ entry, and Ca2+-dependent types of PKC were indeed involved in the process. By introducing different types of Ca2+ chelators at given concentrations directly into the cells via whole-cell patch pipettes, we found that only BAPTA at tens of millimolar concentrations could interfere significantly with VSOR channel activation; that is, the Ca2+ action should take place within the nanodomains.
A preliminary report of this work has been made in a meeting (Akita & Okada, 2010).
Methods
Ethical approval
All procedures in this study were performed according to the Guidelines for Care and Use of Laboratory Animals of the Physiological Society of Japan. Experimental protocols were reviewed and approved in advance by the Institutional Animal Care and Use Committee of the National Institutes of Natural Sciences. The authors have read and the experiments comply with the policies and regulations of The Journal of Physiology given by Drummond (2009).
Cell preparation
Astrocytes in primary culture were prepared from the cerebral cortices of 2-day-old ddY mouse pups, as described previously (Liu et al. 2009). The pups (obtained from 44 mother mice in this study) were killed by cervical dislocation, and cerebral cortices were obtained by removing olfactory bulbs, cerebellums, brainstems, basal ganglia, hippocampi and meninges from the brains. The cortices were minced and digested with papain (Sigma-Aldrich), and the cells were dissociated by gentle pipetting with Pasteur pipettes. The cells were plated and cultured in flasks at 37°C in a humidified 5% CO2–95% air environment with minimum essential medium supplemented with 10% fetal bovine serum, 40 U ml−1 penicillin and 100 μg ml−1 streptomycin. The cells were used up within 1 month after the beginning of cultivation. For experiments, the cells in confluent cultures were first trypsinized for 1–2 min with gentle rocking. After discarding the trypsin solution, the cells were detached from the flask bottoms by gentle pipetting with the culture medium, then transferred to polyethyleneimine-coated glass coverslips at a density of 5 × 105 cells cm−2 and cultured overnight before use. Immunocytochemistry revealed that >90% of the cells were glial fibrillary acidic protein-positive, an astrocyte marker, under this condition. The cells with relatively flat and rectangular cell bodies, suggesting protoplasmic type-1 astrocytes, were selected for experiments.
Solutions
The extracellular bathing solution used for whole-cell patch-clamp recordings contained (in mm): 135 N-methyl-d-glucamine (NMDG)-Cl, 2 CaCl2, 1 MgCl2, 10 Hepes and 5 glucose (pH 7.4, 280 mosmol (kg H2O)−1). The standard whole-cell patch pipette solution contained (in mm): 135 NMDGCl, 1 MgCl2, 3 Na2ATP, 5 Hepes and 1 EGTA with 0.2 CaCl2 (estimated free [Ca2+]≈ 15 nm; pH 7.4, 275 mosmol (kg H2O)−1). When high concentrations of EGTA or BAPTA were added to the pipette solution, the Cl− concentration was reduced to keep the same osmolality as that of the standard solution and CaCl2 was added to maintain the same free [Ca2+]. For [Ca2+]i and ROS measurements, we used Ringer solution containing (in mm): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 NaHepes, 6 Hepes and 5 glucose (pH 7.4, 285 mosmol (kg H2O)−1). When Ca2+ was removed from the bathing solutions, equimolar Mg2+ (with or without 0.1 mm EGTA, as mentioned in text) was added to the solutions.
Patch-clamp studies
Whole-cell currents were measured with the whole-cell patch-clamp technique, as described previously (Liu et al. 2009). Patch pipettes were fabricated from borosilicate glass capillaries and had a resistance of 2–3 MΩ when filled with the pipette solution. The currents were recorded with an EPC10 amplifier controlled via Patchmaster software (HEKA Elektronik, Lambrecht/Pfalz, Germany). The current signals were filtered at 1 kHz and sampled at 5 kHz. Liquid junction potentials were calculated with JPCalc in pCLAMP10 software (Molecular Devices, Sunnyvale, CA, USA) and corrected on-line. The series resistance during the recording was kept at <10 MΩ and compensated for by 70%. All the experiments were performed at 25–30°C.
Calcium imaging
[Ca2+]i changes were measured at 25–30°C as the changes in the ratio of fura-2 fluorescence in the cytoplasm. Cells were loaded with the indicator by incubation in the culture medium containing 0.5 μm fura-2 AM (Biotium) for 30 min at 37°C, then washed with Ringer solution for 5 min in the recording chamber mounted on the stage of an Olympus IX71 inverted microscope (Olympus Japan). The cytoplasmic fura-2 was excited at 340 nm and 380 nm (14 nm bandwidth) with a monochromator (Polychrome IV, TILL Photonics, Gräfelfing, Germany), and detected at 510 nm (80 nm bandwidth). The fluorescence images were obtained through a 40× water immersion objective (UApo40XW340, NA1.15, Olympus Japan) with a Roper NTE (Roper Scientific, Trenton, NJ, USA) or a Hamamatsu ORCA-R2 (Hamamatsu Photonics) cooled CCD camera controlled via MetaFluor software (Molecular Devices). The image pairs obtained with 340 nm light and 380 nm light were recorded at 2 s intervals. Image analysis was performed off-line with ImageJ software (National Institutes of Health). The region of interest was defined to include most of the cytoplasm for each cell. The ratios of the fluorescence intensities obtained with 340 nm light (F340) to those with 380 nm light (F380) in the same regions were calculated after background subtraction. The cells showing oscillatory [Ca2+]i changes before bradykinin application were discarded from analysis.
Quantitative RT-PCR
The total RNA in the cells was extracted with an RNeasy Mini kit (Qiagen, Hilden, Germany), and its content and purity were assessed with a NanoDrop 1000 spectrophotometer (Thermo Fischer Scientific). The total RNA of >10 μg with a purity of >2.0 in the absorbance ratio (260 nm/280 nm) was constantly obtained from a confluent culture in a 25 cm2 flask. Reverse transcription with elimination of genomic DNA was made by using a QuantiTect kit (Qiagen) with a GeneAmp 9700 thermal cycler (Applied Biosystems). Quantitative real-time PCR was performed in an Applied Biosystems 7300 system with TaqMan Gene Expression Assay kits (Applied Biosystems) for mouse Orai1 (assay ID: Mm00774349_m1), TRPC1 (Mm00441975_m1), TRPC2 (Mm00441984_m1), TRPC3 (Mm00444690_m1), TRPC4 (Mm00444284_m1), TRPC5 (Mm00437183_m1), TRPC6 (Mm01176083_m1), TRPC7 (Mm00442606_m1), TRPM7 (Mm00457998_m1), PKCα (Mm00440858_m1), PKCβ (Mm00435749_m1), PKCγ (Mm00440861_m1) and β-actin (Mm00607939_s1) as an endogenous reference. PCR was made in a 50 μl reaction volume containing 50 ng of cDNA in each well of 96-well plates, and four replicate wells were taken for each target gene. Cycling conditions consisted of the initial enzyme activation at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15 s and annealing at 60°C for 1 min. No amplification signals when reverse transcription was omitted were detected for all probes (data not shown). mRNA expression levels were compared by the comparative ΔΔCT method, using the level of TRPC1 mRNA as a calibrator. All measurements were repeated at least three times using the mRNA preparations isolated from different cultures.
Immunocytochemistry
Cells were fixed with ice-cold methanol for 10 min at −20°C, and permeabilized with 0.5% saponin (Sigma-Aldrich) and 0.025% Triton X-100 (Sigma-Aldrich) dissolved in phosphate-buffered saline (PBS) for 30 min at room temperature. Antibody reactions were performed at room temperature for 2 h for primary antibodies and for 1 h for secondary antibodies. Non-specific binding of antibodies was blocked by adding 4% bovine serum albumin (Sigma-Aldrich) throughout the permeabilization and the antibody reactions. The staining was observed by epifluorescence illumination with the Polychrome IV monochromator through the Olympus 40× water immersion objective (NA 1.15) after mounting the coverslips with SlowFade Gold (Molecular Probes). The primary antibodies used were rabbit anti-Orai1 (1:100; Abcam, Cambridge, UK), rabbit anti-TRPC1 (1:200; Osenses, Keswick, SA, Australia), rabbit anti-TRPC3 (1:200; Alomone Labs, Jerusalem, Israel) and rabbit anti-TRPM7 (1:100; Alomone Labs). The secondary antibody was Alexa Fluor 546 goat anti-rabbit IgG (1:1000; Molecular Probes). Consistency of staining patterns was confirmed by comparing the staining results with the antibodies at three different concentrations (1:100, 1:200, 1:500) for all the primary antibodies. Negative controls were obtained by omission of primary antibodies.
Small interfering RNA transfection
For gene knockdown studies with small interfering RNA (siRNA), we used Dharmacon ON-TARGETplus SMARTpool siRNA reagents (Thermo Fischer Scientific) designed against mouse Orai1 (catalogue no. L-056431-02), TRPC1 (L-043863-00), TRPC3 (L-049680-00), TRPM7 (L-040716-00), PKCα (L-040348-00) and PKCβ (L-048412-00). This reagent contains a mixture of four different siRNAs targeting the same gene. We also used an ON-TARGETplus Non-Targeting Pool reagent (D-001810-10), containing four different siRNAs but not targeting any known genes, for negative control. Cells at 30–50% confluency were transfected with the siRNA mixture at 1 nm using Lipofectamine RNAiMAX (Invitrogen) in Opti-MEM I Reduced Serum Medium (Invitrogen) without serum and antibiotics at 37°C in a humidified 5% CO2–95% air environment. The transfection medium was replaced with the culture medium after 8–10 h. The effect of knockdown was first assessed by quantitative RT-PCR 3 days after the transfection. Selective suppression of the mRNA level to 10–20% was confirmed for all target genes, as shown in Supplemental Fig. S1. Maximal functional suppression was determined by measuring bradykinin-induced [Ca2+]i rises for channel knockdown and VSOR currents for PKC knockdown 3–5 days after the transfection and comparing them with those in non-targeting siRNA-transfected cells. The maximum was seen at 3 days for knockdown of Orai1, PKCα and PKCβ, and at 4–5 days for TRPC1 and TRPC3.
ROS imaging
Intracellular ROS production was tracked at 25–30°C from the increase in fluorescence of 5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein (CM-DCF) in the cytoplasm. Cells were loaded with the indicator by incubation in Ringer solution containing the membrane-permeant (acetylated) reduced form of CM-DCF (CM-H2DCFDA; Molecular Probes) at 10 μm for 30 min at 37°C, then washed with Ringer solution for 5 min in the recording chamber. The indicator was excited at 490 nm (8 nm bandwidth) with the Polychrome IV monochromator, and detected at 540 nm (50 nm bandwidth). Great care was taken to reduce photo-oxidation of the indicator by using weak excitation light with a short exposure time. The images were obtained through a 20× dry objective (UPlanSApo, NA0.75, Olympus Japan) with the ORCA-R2 camera controlled via MetaFluor software at 10 s intervals. Image analysis was performed off-line with ImageJ. The region of interest was defined to include most of the cytoplasm for each cell. The fluorescence increase was detected as the increase in normalized fluorescence intensity relative to the initial level (F/F0). The initial level was determined as the average over 1 min after the beginning of measurement in the same region. The intensity was measured after background subtraction.
Drugs
All drugs were made up as 1000–10,000× stock solutions, stored frozen and then thawed and diluted with the bathing solution on the day of use. Bradykinin, DCPIB, 2-aminoethoxydiphenyl borate (2-APB), thapsigargin, BAPTA, phorbol 12-myristate 13-acetate (PMA) and Gö6976 were purchased from Sigma-Aldrich. EGTA was from Dojindo (Kumamoto, Japan).
Statistics
Data are expressed as means ± SEM, and error bars in graphs indicate SEM. Statistical multiple comparisons were made by Peritz's closed testing procedure with the F statistic after ANOVA tests. Differences were judged as significant when the P of making at least one incorrect assertion in one set of multiple comparisons, called the familywise error rate, was <0.05. The effect of a treatment on the same sample set was assessed by Student's paired t test.
Results
Bradykinin-induced [Ca2+]i rises are produced by intracellular Ca2+ release and store- or receptor-operated Ca2+ entry
We previously demonstrated that application of 1 μm bradykinin under isotonic conditions activates VSOR channels through bradykinin B2 receptor activation and this is accompanied by prominent [Ca2+]i rises in cultured mouse cortical astrocytes (Liu et al. 2009). To elucidate their Ca2+ sources, we examined the effects of removing extracelluar Ca2+ and the Ca2+ in intracellular Ca2+ stores on the time course of the [Ca2+]i rises. The bradykinin-induced [Ca2+]i rise consisted of two rising phases: the initial large transient phase followed by a small sustained phase (Fig. 1Aa). The initial transient subsided within 1 min after the beginning of bradykinin application, whereas the second sustained phase continued over >15 min in the presence of bradykinin. Removal of extracellular Ca2+ completely abolished the sustained phase and reduced weakly the amplitude of the initial transient (from 0.37 ± 0.02 in the fura-2 ratio (ΔF340/F380) to 0.26 ± 0.02, Fig. 1Ab). Pretreatment of the cells with a blocker of Ca2+ pumps on the Ca2+ stores, thapsigargin (1 μm), in addition to the removal of extracellular Ca2+ eradicated the initial transient (Fig. 1Ac). Thus most of the initial transient component must be caused by intracellular Ca2+ release from Ca2+ stores, and the second sustained component must be caused exclusively by Ca2+ entry at the plasma membrane. The Ca2+ entry would be generated in response to depletion of Ca2+ stores or to bradykinin receptor activation. Indeed, application of the blocker of store- or receptor-operated Ca2+ entry, Gd3+ (1 μm) or 2-APB (30 μm), in the presence of extracellular Ca2+ selectively suppressed the second component without affecting the initial component (Fig. 1Ad and e). When the extent of suppression was evaluated from the time integral of the second component from 1 to 15 min after stimulation, the effects of Gd3+ and 2-APB were found to be statistically similar to that of extracellular Ca2+ removal (Fig. 1B). Thus Ca2+-permeable channels sensitive to both Gd3+ and 2-APB must be involved in the Ca2+ entry responsible for the second component.
Figure 1. Effects of Ca2+ depletion and blockers of Ca2+ entry on bradykinin-induced [Ca2+]i rises in cultured mouse astrocytes.
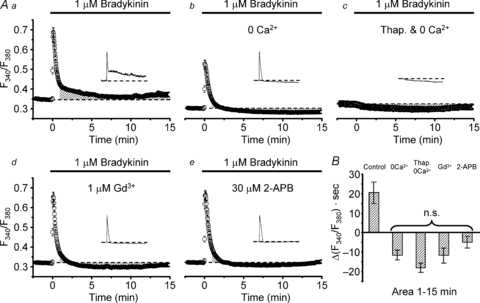
Aa, bradykinin-induced rise in fura-2 ratio (F340/F380) in the cytoplasm averaged over 78 cells. Ab, bradykinin-induced F340/F380 rise in the absence of extracellular Ca2+ (n = 75). EGTA was not added in the bathing solution. Ac, bradykinin response in the absence of extracellular Ca2+ observed after pretreatment with 1 μm thapsigargin (Thap.) for 30 min (n = 69). Thapsigargin was included in the loading solution but not in the bathing solution. Ad, response in the presence of 1 μm Gd3+ (n = 90). Ae, response in the presence of 30 μm 2-APB (n = 85). Cells were preincubated in the Gd3+ or 2-APB containing solution for 10 min at room temperature before the measurements. Insets in Aa–e show typical F340/F380 traces observed in single cells for 15 min. All traces in this figure were recorded with a Roper NTE camera. B, comparison of the time integrals of net F340/F380 rises (areas in Δ(F340/F380) × seconds) from basal levels (indicated by dashed lines in Aa–e) over 1–15 min after the beginning of bradykinin application. The areas for comparison are indicated as shaded regions in Aa–e. All the areas except that in the control conditions (Aa) had negative values. This was presumably due to a temporary facilitating effect of bradykinin on Ca2+ extrusion (Ac). There are no statistical differences between these negative values (judged by multiple comparisons).
Bradykinin-induced activation of VSOR anion channels depends both on Ca2+ release and on Ca2+ entry
Then we examined the effects of removal of extracellular Ca2+ and depletion of Ca2+ stores on VSOR channel activation. The bradykinin application induced development of an anion current, showing mild outward rectification and weak inactivation at high voltages, over 15–20 min under whole-cell voltage clamp conditions (Fig. 2A). We previously checked that this current was blocked by anion channel blockers DIDS, phloretin and DCPIB (the latter two should preferentially and specifically block VSOR channels, respectively), and that the current generation required intracellular ATP and had sensitivity to extracellular hypertonicity but not to Gd3+, consistent with the properties of VSOR channels (Liu et al. 2009). Although the inactivation of the current in this preparation is much weaker than that of the typical swelling-induced VSOR current as seen in epithelial cells (see review by Okada, 1997), the current is quite similar to the swelling-induced currents in this preparation (see the supplemental figure of Liu et al. 2009) and in neurons (Leaney et al. 1997; Inoue et al. 2005), microglia (Harrigan et al. 2008; Schlichter et al. 2011) and cardiomyocytes (Baumgarten & Clemo, 2003).
Figure 2. Effects of Ca2+ depletion on bradykinin-induced whole-cell currents through VSOR anion channels.
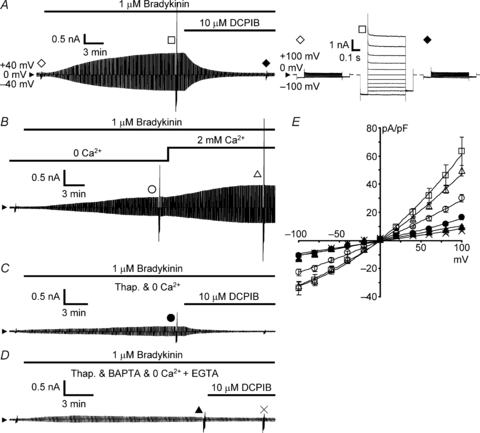
A, time course of an increase in the whole-cell current through VSOR channels during application of 1 μm bradykinin and its suppression by 10 μm DCPIB. Bradykinin and DCPIB were applied through bath perfusion under isotonic conditions. The time course was monitored by repetitively applying a pair of alternating step voltage pulses to ±40 mV (0.5 s duration) from the holding potential of 0 mV every 5 s. The arrowhead indicates the 0 current level. To examine the changes in voltage dependency of the current, test voltage pulses were applied before application of bradykinin (open diamond), at the maximum plateau of the bradykinin-induced current enhancement (open square) and after suppression to the minimum caused by DCPIB (filled diamond). The corresponding current responses are shown in the right panel on an expanded time scale. Test voltage pulses consisted of the pulses (0.5 s duration) ranging from −100 mV to +100 mV in 20 mV increments with fixed pre-pulses (0.1 s duration) to −100 mV and post-pulses (0.1 s duration) to −80 mV. B, bradykinin-induced VSOR current in the absence of extracellular Ca2+ and its enhancement by restoration of extracellular Ca2+ to 2 mm. C, bradykinin-induced VSOR current in the absence of extracellular Ca2+ observed after pretreatment with 1 μm thapsigargin and its suppression by 10 μm DCPIB. D, the bradykinin-induced current observed after thapsigargin treatment and adding 10 mm BAPTA in the pipette solution and 0.1 mm EGTA in the Ca2+-free bathing solution, and its very weak suppression by 10 μm DCPIB. The current responses indicated by the symbols in B–D are those elicited by test voltage pulses as in A. E, current–voltage (I–V) relationships of the whole-cell currents under the conditions shown in A–D. Symbols represent the means ± SEM of the peak current amplitudes elicited by test voltage pulses in current density (pA pF−1). The peak amplitude was determined at 20 ms after the beginning of the pulse. The numbers of cells analysed were 17 for control in the presence of extracellular Ca2+ as in A (open squares), 15 for removal of extracellular Ca2+ (open circles) and Ca2+ restoration (open triangles) as in B, 14 for thapsigargin pretreatment (filled circles) as in C, and 16 for thorough removal of Ca2+ (filled triangles) and DCPIB treatment (crosses) as in D. The mean membrane capacitance of the cells used here was 51.3 ± 3.3 pF.
The maximum amplitude of the VSOR current was suppressed by half in the absence of extracellular Ca2+ (nominally Ca2+ free). The peak amplitude of the current (in current density) elicited by depolarization to +100 mV was 30.0 ± 2.6 pA pF−1 (open circles in Fig. 2B and E), whereas it was 63.4 ± 10.0 pA pF−1 in the presence of Ca2+ (P < 0.05 by multiple comparisons; open squares in Fig. 2A and E). Indeed, restoration of extracellular Ca2+ restored the amplitude to a similar level (49.6 ± 3.8 pA pF−1, open triangles in Fig. 2B and E). Pretreatment with thapsigargin in addition to the removal of extracellular Ca2+ further suppressed the amplitude to one-quarter (16.3 ± 1.7 pA pF−1, P < 0.05 by multiple comparisons; filled circles in Fig. 2C and E). When the residual Ca2+ was thoroughly removed by adding Ca2+ chelators both on the extracellular (0.1 mm EGTA) and intracellular (10 mm BAPTA included in the pipette solution) sides, the amplitude was further reduced (9.9 ± 1.0 pA pF−1, P < 0.05 by multiple comparisons; filled triangles in Fig. 2D and E) and the DCPIB-sensitive current component became very small although it was still statistically significant (suppression to 6.9 ± 0.6 pA pF−1, P < 0.01 by paired t test, crosses in Fig. 2D and E). Thus the results strongly suggest that bradykinin-induced activation of VSOR anion channels depends on intracellular [Ca2+]i rises originating both from intracellular Ca2+ release and from Ca2+ entry.
Orai1, TRPC1 and TRPC3 channels are involved in store- or receptor-operated Ca2+ entry
As Gd3+- and 2-APB-sensitive channels responsible for store- or receptor-operated Ca2+ entry, Orai1 and the canonical type of TRP (TRPC) channels are known in many types of cells (Birnbaumer, 2009). So we checked their expressions in our astrocytes and examined their roles in bradykinin-induced [Ca2+]i rises and VSOR channel activation. In addition, we also checked the expression of TRPM7 channels, which are also Ca2+ permeable and Gd3+ and 2-APB sensitive, because our group previously demonstrated that the Ca2+ entry through these channels assists the RVD process driven by VSOR channel activation in response to hypotonic stimuli in HeLa cells (Numata et al. 2007b).
Our quantitative RT-PCR study indicated that the mRNAs of Orai1, TRPC1, TRPC3 and TRPM7 were expressed abundantly, whereas others were scanty or absent in our astrocytes (Fig. 3A). Immunostaining with the antibodies against Orai1, TRPC1, TRPC3 and TRPM7 proteins indeed suggested actual expression of these channels (Fig. 3B). To examine the contribution of each type of channel to the bradykinin-induced [Ca2+]i rise, we transfected the cells with the siRNA targeting the channel and examined its effects on the [Ca2+]i rise.
Figure 3. Effects of siRNA-mediated knockdown of Orai1, TRPC1, TRPC3 and TRPM7 channels on bradykinin-induced [Ca2+]i rises.
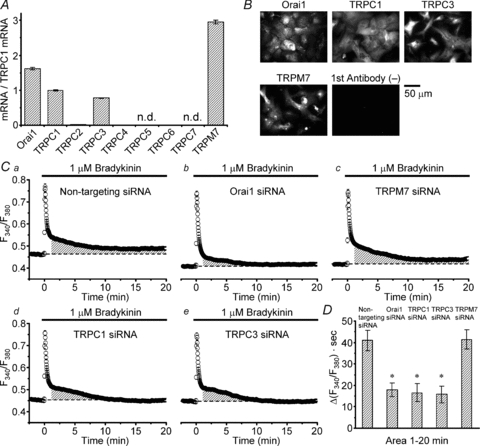
A, comparison of the mRNA expression levels of Gd3+- and 2-APB-sensitive Ca2+ entry channels by quantitative RT-PCR. The expression levels are normalized to that of TRPC1. The levels of TRPC2 (0.013), TRPC4 (0.008) and TRPC6 (0.005) were substantially low and those of TRPC5 and TRPC7 were not detected. The results were obtained from a total of 30 pups born from 3 mother mice. B, immunostaining of the cells with the antibodies against the channels showing high mRNA expression levels. The staining only with secondary antibodies is shown as a negative control. C, bradykinin-induced F340/F380 rise in the cells transfected with non-targeting siRNA as controls (a, averaged over 240 cells) and the rises in the cells transfected with Orai1 siRNA (b, n = 253), TRPM7 siRNA (c, n = 211), TRPC1 siRNA (d, n = 262) and TRPC3 siRNA (e, n = 242). The traces in this figure were recorded with a Hamamatsu ORCA-R2 camera. D, comparison of the areas of net F340/F380 rises over 1–20 min (indicated by shaded regions in Ca–e). The areas in the cells transfected with the siRNAs for Orai1, TRPC1 and TRPC3 were significantly suppressed (marked with asterisks) compared to those in the others (P < 0.05, judged by multiple comparisons). There are no significant differences between the areas in these 3 types of transfected cell. The area in TRPM7 siRNA-transfected cells is not statistically different from that in non-targeting siRNA-transfected cells.
The transfection with the siRNA for Orai1, TRPC1 or TRPC3 channels significantly suppressed selectively the second sustained component of the [Ca2+]i rise (Fig. 3Cb, d and evs. Ca; P < 0.05 judged by multiple comparisons, marked with asterisks in Fig. 3D) without affecting the initial transient component, whereas the transfection with TRPM7 siRNA had effects on neither the initial nor the second components (Fig. 3Ccvs. Ca; Fig. 3D). Thus Orai1, TRPC1 and TRPC3 channels must be involved in the Ca2+ entry after intracellular Ca2+ release. No involvement of TRPM7 channels would be explained by the absence of large changes in cell volume during bradykinin application (Liu et al. 2009), because the channels must be activated through direct mechanical stress (Numata et al. 2007a). In our conditions of siRNA transfection (see Methods and Supplemental Fig. S1A for details), the effects of siRNAs for Orai1, TRPC1 and TRPC3 channels were similar when they were assessed from the decreases in time integral of the [Ca2+]i rise over 1–20 min after the beginning of the rise (Fig. 3D).
Ca2+ entry through TRPC1 channels is strongly involved in bradykinin-induced activation of VSOR anion channels
Then we examined the effects of suppression of Orai1, TRPC1 and TRPC3 channels on bradykinin-induced activation of VSOR channels. Transfection with TRPC1 siRNA significantly suppressed the VSOR current activation when it was compared at 20 min after the beginning of bradykinin application (11.1 ± 1.4 pA pF−1 at +100 mV; open circles in Fig. 4B and C) with that in the control non-targeting siRNA-transfected cells (18.9 ± 2.1 pA pF−1; open squares in Fig. 4A and C; P < 0.05 judged by multiple comparisons, marked with the asterisk in Fig. 4D). A notable thing was that in 70% of the TRPC1 knockdown cells (22 of 31 cells) the current development ceased within 9 min (9.2 ± 0.8 min, average over 22 cells) after the beginning of bradykinin application and then the current decreased gradually over >10 min. An example is shown in Fig. 4B. Such a waxing and waning pattern of development was seen only in 2 of 29 non-targeting siRNA-transfected cells; even in these two cells the waning phase was seen >15 min after the beginning of bradykinin application. This suggests that the Ca2+ entry through TRPC1 channels participates in VSOR channel activation especially in the later phase of the current development. This seemed consistent with the suppression of especially the later phase of the [Ca2+]i rise in TRPC1 siRNA-transfected cells (Fig. 3Cd). In further support of this, a similar pattern of the current decrease could be induced when the extracelluar Ca2+ was removed during the course of the current development (Fig. 4E).
Figure 4. Effects of TRPC1 knockdown on bradykinin-induced VSOR currents.
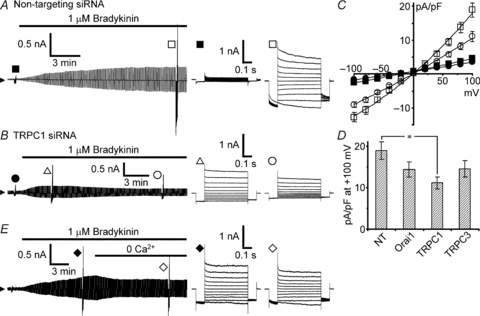
Bradykinin-induced VSOR currents were monitored during application of voltage pulses as in Fig. 2A. A, bradykinin-induced VSOR current in non-targeting siRNA-transfected cells. B, an example showing a waxing and waning pattern of current development induced by bradykinin in TRPC1 siRNA-transfected cells. This pattern was seen in the majority of the cells (22 of 31 cells). C, I–V relationships measured 20 min after the beginning of bradykinin application in these transfected cells. Filled symbols indicate the mean values before application of bradykinin, and open symbols indicate those after 20 min. The numbers of cells analysed were 29 for non-targeting siRNA (squares) and 31 for TRPC1 siRNA (circles). D, comparison of the peak amplitudes of the currents evoked by voltage pulses to +100 mV between the cells transfected with the siRNAs for Orai1 (n = 32), TRPC1 and TRPC3 (n = 27) and the cells with non-targeting (NT) siRNA. Only the amplitude in TRPC1 knockdown cells was significantly suppressed (asterisk, P < 0.05 by multiple comparisons). The examples of current traces and the I–V relationships in Orai1-knockdown and TRPC3-knockdown cells are shown in Supplemental Fig. S2. E, effect of extracellular Ca2+ removal during the course of bradykinin-induced current development. The trace is a representative of three separate experiments.
In Orai1 siRNA-transfected cells, such a pattern of the current decrease was seen in 40% (13 of 32 cells); thus the majority showed a monotonic increase in current amplitude during bradykinin application, as shown in Supplemental Fig. S2A. In TRPC3 siRNA-transfected cells, just half of the cells showed the waxing and waning pattern (Supplemental Fig. S2Ba and b). On average, however, when the amplitudes were compared at 20 min after the beginning of bradykinin application, significant suppression was seen only in TRPC1 siRNA-transfected cells (Fig. 4D). Thus, considering the similar suppression of the [Ca2+]i rises in these three types of transfected cell (Fig. 3D), the results suggest that the Ca2+ entry through TRPC1 channels would be preferentially involved in the VSOR channel activation in the majority of the cells.
Bradykinin-induced activation of VSOR anion channels is mediated by Ca2+-dependent PKCα and β
The foregoing results strongly support the dependence of bradykinin-induced VSOR channel activation on [Ca2+]i rises. However, the time course of the current development was evidently different from that of the [Ca2+]i rise; the current developed gradually and reached a plateau 15–20 min after the beginning of bradykinin action, whereas the [Ca2+]i rise reached a peak immediately after the bradykinin action and decayed rapidly to a small sustained level. This difference implies that Ca2+ would not act directly on VSOR channels. A plausible mediator is evidently the Ca2+-dependent types of PKC. So we explored this possibility.
Application of a PKC activator, PMA (1 μm), instead of bradykinin, indeed induced development of a current showing mild outward rectification and weak inactivation at high voltages (open circles in Fig. 5A and C), which was blocked by DCPIB (crosses in Fig. 5A and C). Thus PMA mimicked the bradykinin action on VSOR channel activation. Furthermore, the bradykinin-induced VSOR current activation was strongly suppressed in the presence of a specific inhibitor of Ca2+-dependent types of PKC, Gö6976 (1 μm; open triangles in Fig. 5B and C), whereas prominent [Ca2+]i rises were still preserved (Fig. 5D).
Figure 5. Involvement of Ca2+-dependent PKC in bradykinin-induced activation of VSOR currents.
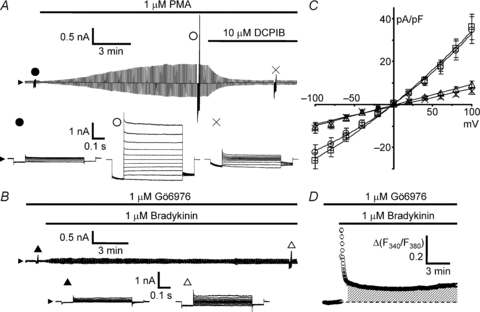
Development and suppression of VSOR currents were monitored during application of voltage pulses as in Fig. 2A. A, development of DCPIB-sensitive whole-cell current induced by 1 μm PMA under isotonic conditions in the absence of bradykinin. B, inhibition of bradykinin-induced VSOR current in the presence of a specific blocker of Ca2+-dependent PKCs, Gö6976 (1 μm). Cells were preincubated with Gö6976 for 30 min at 37°C, and the responses were measured in the presence of Gö6976 at 25–30°C in the bathing solution. C, I–V relationships of the currents shown in A and B. PMA and Gö6976 were dissolved in the bathing solution with 0.01% DMSO. Thus the control bradykinin response was taken in the presence of 0.01% DMSO (open squares; n = 17). The current response induced by PMA (open circles; n = 14) was similar to the control and blocked by 10 μm DCPIB (crosses; n = 14). The response induced by bradykinin was greatly suppressed in the presence of Gö6976 (P < 0.001 by ANOVA, open triangles; n = 21). D, bradykinin-induced [Ca2+]i rise in the presence of Gö6976 averaged over 140 cells. The amplitude and the area of the [Ca2+]i rise over 1–15 min (shaded region) were significantly larger than those of the control (P < 0.05 by ANOVA). The traces were recorded with a Roper NTE camera.
To consolidate this pharmacological result, we also examined the effects of transfection with the siRNAs for Ca2+-dependent PKCs on VSOR current activation. Quantitative RT-PCR indicated that, among three types (α, β and γ) of Ca2+-dependent PKC, PKCα was found to be the major type in our astrocytes (Fig. 6A). The expression of PKCβ mRNA was much lower than that of PKCα, but its level was comparable to that of TRPC1 mRNA. The expression of PKCγ mRNA was at an almost negligible level. Thus we examined the effects of knockdown of PKCα and PKCβ one by one. In our transfection conditions, both the siRNAs for PKCα and PKCβ suppressed selectively the mRNA levels of targeted PKCs to 10% (Supplemental Fig. S1B). Compared with the bradykinin-induced current development in non-targeting siRNA-transfected cells (Fig. 6B), 60% of both the PKCα knockdown cells and the PKCβ knockdown cells exhibited a very mild waxing and waning pattern of current development in response to bradykinin; the current reached a plateau within 8–9 min and it was sustained or declined very slowly over 10 min (17 of 29 PKCα knockdown cells as in Fig. 6C, and 20 of 31 PKCβ knockdown cells as in Fig. 6Da). The rest of the cells showed monotonic current development. On average, when the currents at +100 mV were compared at 20 min after the beginning of bradykinin application, both the amplitude in PKCα knockdown cells (10.8 ± 1.3 pA pF−1; open circles in Fig. 6C and E) and that in PKCβ knockdown cells (12.6 ± 1.4 pA pF−1; open triangles in Fig. 6Da and E) were significantly smaller than that in non-targeting siRNA-transfected cells (18.9 ± 2.1 pA pF−1; open squares in Fig. 6B and E; P < 0.05 by multiple comparisons). However, the typical mild outward rectification of the VSOR current characteristic became obscured especially in PKCβ knockdown cells (open triangles in Fig. 6Db and E), suggesting that contamination of other types of current became noticeable. By extracting the VSOR current component with DCPIB (crosses in Fig. 6Dc), we found that the contaminating current was contained only in the negative potential range, suggesting its inwardly rectifying property. (The asterisks in Fig. 6Db indicate the significant differences in amplitude between the currents before bradykinin application (filled triangles) and after DCPIB treatment (filled diamonds); P < 0.01 by paired t test.) Since no significant shift in reversal potential was seen after the DCPIB treatment, a current through Cl−-permeable ClC-2-like channels bearing a similar inwardly rectifying property (Makara et al. 2003) would probably be involved to some extent. In any case, since the bradykinin-induced current at positive potentials was found to be purely the VSOR current, the reductions in amplitude at +100 mV (Fig. 6E) indicate that both PKCα and β must be involved in the VSOR channel activation. Thus, these data confirm that the VSOR channel activation induced by bradykinin is mediated by Ca2+-dependent PKCα and PKCβ, not by the direct interaction of Ca2+ with the channel.
Figure 6. Effects of siRNAs for PKCα and PKCβ on bradykinin-induced VSOR currents.
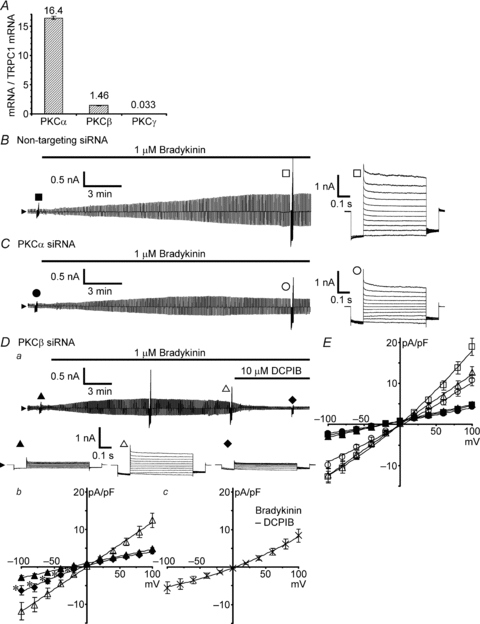
A, mRNA expression levels of Ca2+-dependent PKCα, β and γ, determined by quantitative RT-PCR. The levels are normalized to that of TRPC1 mRNA. The results were obtained from total 30 pups born from 3 mother mice. B and C, bradykinin-induced VSOR currents in non-targeting siRNA-transfected cells and in PKCα siRNA-transfected cells, respectively. The currents were monitored during application of voltage pulses as in Fig. 2A. Da, bradykinin-induced current and its suppression by 10 μm DCPIB in PKCβ siRNA-transfected cells. Db, comparison of the I–V relationships before (filled triangles) and 20 min after the beginning of bradykinin application (open triangles), and after suppression by DCPIB (filled diamonds). Data were from 19 cells. Asterisks indicate the significant differences between the currents before bradykinin application (filled triangles) and after bradykinin application with DCPIB treatment (filled diamonds) at P < 0.01 by paired t test. Dc, I–V relationship of the DCPIB-sensitive current component. Crosses indicate the mean ± SEM of the differences between the currents just before and after the DCPIB treatment in individual cells shown in Db. E, I–V relationships of bradykinin-induced VSOR currents in the siRNA-transfected cells shown in B–D. Filled symbols indicate the mean ± SEM of the amplitudes before application of bradykinin, and open symbols indicate those after 20 min. The numbers of cells analysed were 29 for non-targeting siRNA-transfected cells (squares), 29 for PKCα siRNA-transfected cells (circles) and 31 for PKCβ siRNA-transfected cells (triangles).
Bradykinin-induced ROS generation is mediated by Ca2+-dependent PKC
Moreover, we also examined whether the PKC activation is linked to ROS generation. Bradykinin-induced ROS generation detected with CM-DCF fluorescence (Fig. 7A) was strongly suppressed in the presence of Gö6976, whereas the responsiveness of CM-DCF to exogenous H2O2 was not affected (Fig. 7B). We also checked that the ROS generation was abolished in the presence of a NOX inhibitor, diphenyleneiodonium (DPI; 10 μm, Fig. 7C) as we showed previously (Liu et al. 2009), and also in the absence of extracellular Ca2+ after pretreatment with thapsigargin (Fig. 7D). Similar ROS generation was also produced by PMA instead of bradykinin (Fig. 7E). Since we previously demonstrated that the bradykinin-induced VSOR current and associated glutamate release were prevented in the presence of ROS scavengers and several NOX inhibitors (Liu et al. 2009), the result confirms that PKC activation precedes and is linked serially to ROS generation through NOX for activation of VSOR channels.
Figure 7. Bradykinin-induced ROS generation through NOX and its sensitivity to a blocker of Ca2+-dependent PKC and to Ca2+ depletion.
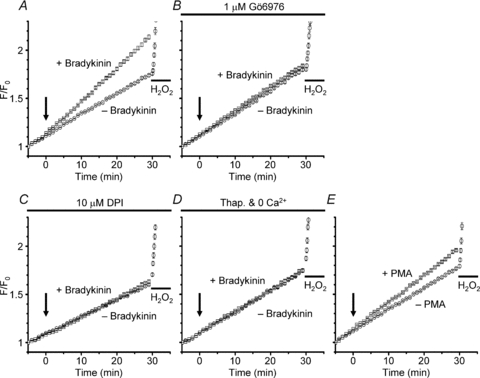
A, comparison of the time courses of increases in normalized fluorescence (F/F0) of CM-DCF with and without application of bradykinin. Bradykinin at 1 μm (+ Bradykinin) or a blank bathing solution (– Bradykinin) was applied 5 min after the beginning of observation (at time 0, indicated by the arrow). The fluorescence was measured at 10 s intervals, and each point in the plot represents the averaged F/F0 value over 1 min. To check the responsiveness of CM-DCF to exogenous ROS, 1 mm H2O2 was applied at 30 min in the ‘− Bradykinin’ condition. The traces are the averages over 148 cells in the ‘+ Bradykinin’ condition (squares) and 164 cells in the ‘− Bradykinin’ condition (circles), respectively. The larger F/F0 rise at 30 min in the ‘+ Bradykinin’ condition is significant at P < 0.01 by ANOVA. The F/F0 rise in the ‘− Bradykinin’ condition would be mostly caused by the photo-oxidation of CM-DCF, since similar amplitudes of the rises were generated even in the presence of a NOX blocker, DPI, as shown below in C. B, suppression of bradykinin-induced F/F0 rise in the presence of 1 μm Gö6976. Gö6976 was included also during dye loading. The traces are from 154 cells in the ‘+ Bradykinin’ condition (squares) and 139 cells in the ‘− Bradykinin’ condition (circles). C, suppression of the F/F0 rise in the presence of 10 μm DPI. The traces are the averages over 227 cells in the ‘+ Bradykinin’ condition (squares) and 230 cells in the ‘− Bradykinin’ condition (circles). D, suppression of the F/F0 rise in the absence of extracellular Ca2+ observed after pretreatment with thapsigargin. Thapsigargin at 1 μm was included in the loading solution but not in the bathing solution. EGTA at 0.1 mm was added to the 0 Ca2+ bathing solution. The traces are from 202 cells in the ‘+ Bradykinin’ condition (squares) and 195 cells in the ‘− Bradykinin’ condition (circles). E, F/F0 rise induced by 1 μm PMA. PMA or a blank solution (– PMA) was applied at time 0 (indicated by the arrow). The traces are from 205 cells in the ‘+ PMA’ condition (squares) and 181 cells in the ‘− PMA’ condition (circles). The larger F/F0 rise at 30 min in the ‘+ PMA’ condition is significant at P < 0.01 by ANOVA.
High concentrations of intracellular BAPTA are required to inhibit bradykinin-induced activation of VSOR anion channels
Ca2+-dependent processes must be inhibited if the Ca2+ binding to its sensing sites is interrupted sufficiently by exogenously applied Ca2+ chelators. However, if the distance between the sensing sites and Ca2+ sources is shorter than 100 nm, higher concentrations of the chelators with faster Ca2+-binding kinetics are required for interruption, because the amount of Ca2+ to be captured around the sensing sites is very high and because the chelators pass through the narrow space between the sensing sites and Ca2+ sources so quickly that Ca2+ is less likely to be captured (Neher, 1998b). Based on the failure of BAPTA-AM treatment to suppress the VSOR channel activation in our previous study (Liu et al. 2009), we examined the effects of applying Ca2+ chelators at given concentrations directly into the cells through patch pipettes on the VSOR channel activation.
By default, we included 1 mm EGTA into the pipette solution for VSOR current recordings (see Methods for details), whereas we had included 10 mm EGTA in our previous study (Liu et al. 2009). However, this difference did not yield a significant difference in the maximum amplitude of VSOR current induced by bradykinin. The amplitude at +100 mV in the presence of intracellular 10 mm EGTA was 49.3 ± 6.4 pA pF−1 in this study (open circles in Fig. 8A and D), whereas it was 63.4 ± 10.0 pA pF−1 in the presence of 1 mm EGTA (n.s. by multiple comparisons; open squares in Fig. 8D). The amplitude was still not significantly affected, when BAPTA, whose Ca2+-binding speed is 200 times faster than EGTA, at 1 mm was included in the pipette solution (44.3 ± 8.5 pA pF−1; filled circles in Fig. 8B and D). When the concentration of BAPTA was increased to 10 mm, the amplitude was suppressed significantly to about one-third (23.1 ± 2.7 pA pF−1, P < 0.05 by multiple comparisons; filled squares in Fig. 8C and D). Thus the results should indicate that Ca2+-sensing sites of the molecules responsible for VSOR channel activation are located or moved so close to Ca2+ sources upon bradykinin receptor activation that only the fast Ca2+ chelator BAPTA at >10 mm can interfere with Ca2+ binding to the sensing sites. The extracellularly applied BAPTA-AM at 50 μm in our previous study (Liu et al. 2009) would have not been concentrated in the cytoplasm at >1 mm.
Figure 8. Effects of intracellular EGTA and BAPTA on bradykinin-induced VSOR currents.
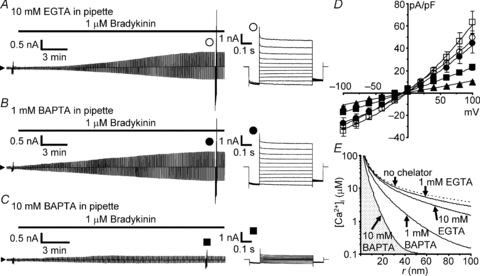
A–C, bradykinin-induced VSOR currents in the presence of intracellular EGTA at 10 mm (A), BAPTA at 1 mm (B) and BAPTA at 10 mm (C). Voltage pulse protocols were the same as those in Fig. 2A. The Ca2+ chelators were applied via patch pipettes. D, I–V relationships at different concentrations of intracellular Ca2+ chelators. The numbers of the cells analysed were 17 for 1 mm EGTA (open squares), 15 for 10 mm EGTA (open circles), 13 for 1 mm BAPTA (filled circles), 14 for 10 mm BAPTA (filled squares) and 16 for 10 mm BAPTA observed after pretreatment with 1 μm thapsigargin and removal of extracellular Ca2+ (filled triangles). Significant suppression in the amplitude at +100 mV was seen only in the presence of 10 mm BAPTA (filled squares and triangles; P < 0.05 by multiple comparisons). E, model of steady-state spatial [Ca2+]i profiles of a single ‘Ca2+ nanodomain’ in the presence of Ca2+ chelators at different concentrations. The profiles were drawn based on the equation proposed by Neher (1998a): 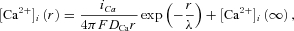 where
where 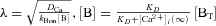 , r is the distance from a Ca2+ channel pore, iCa is the single-channel Ca2+ current (0.1 pA, assuming that iCa through a TRPC channel is slightly smaller than that through a voltage-gated Ca2+ channel), F is the Faraday constant, DCa is the diffusion coefficient of free Ca2+ (223 μm2 s−1; Allbritton et al. 1992), λ is the length constant of the profile, kBon is the Ca2+ binding rate constant of the chelator (2.5 × 106m−1 s−1 for EGTA, 5 × 108m−1 s−1 for BAPTA; Naraghi, 1997), [B] is the concentration of the Ca2+-free chelator, [BT] is the total concentration of the chelator, and KD is the dissociation constant of the chelator (0.438 μm, based on the in vivo calibration of a BAPTA-type Ca2+ indicator by Akita & Kuba (2000); the KD values of EGTA and BAPTA are assumed to be the same). [Ca2+]i(∞) must be the basal [Ca2+]i in the bulk cytoplasm (0.1 μm). Note that [Ca2+]i is plotted on a logarithmic scale. The calculated λ values were 331 nm, 105 nm, 23.4 nm and 7.4 nm in the presence of 1 mm EGTA, 10 mm EGTA, 1 mm BAPTA and 10 mm BAPTA, respectively. The profile in the absence of chelators, indicated by the dotted line, was calculated by putting λ at infinity, i.e. making the exponential term unity.
, r is the distance from a Ca2+ channel pore, iCa is the single-channel Ca2+ current (0.1 pA, assuming that iCa through a TRPC channel is slightly smaller than that through a voltage-gated Ca2+ channel), F is the Faraday constant, DCa is the diffusion coefficient of free Ca2+ (223 μm2 s−1; Allbritton et al. 1992), λ is the length constant of the profile, kBon is the Ca2+ binding rate constant of the chelator (2.5 × 106m−1 s−1 for EGTA, 5 × 108m−1 s−1 for BAPTA; Naraghi, 1997), [B] is the concentration of the Ca2+-free chelator, [BT] is the total concentration of the chelator, and KD is the dissociation constant of the chelator (0.438 μm, based on the in vivo calibration of a BAPTA-type Ca2+ indicator by Akita & Kuba (2000); the KD values of EGTA and BAPTA are assumed to be the same). [Ca2+]i(∞) must be the basal [Ca2+]i in the bulk cytoplasm (0.1 μm). Note that [Ca2+]i is plotted on a logarithmic scale. The calculated λ values were 331 nm, 105 nm, 23.4 nm and 7.4 nm in the presence of 1 mm EGTA, 10 mm EGTA, 1 mm BAPTA and 10 mm BAPTA, respectively. The profile in the absence of chelators, indicated by the dotted line, was calculated by putting λ at infinity, i.e. making the exponential term unity.
Discussion
This study elucidated the dependence of bradykinin-induced VSOR channel activation on Ca2+ nanodomains under isotonic conditions. The Ca2+ involved in the VSOR channel activation was provided initially by Ca2+ release from intracellular Ca2+ stores and then by store- or receptor-operated Ca2+ entry at the plasma membrane. In the Ca2+ entry, Orai1, TRPC1 and TRPC3 channels were found to be involved in our preparations, and the entry through TRPC1 channels seemed to be preferentially used for the VSOR channel activation. The Ca2+ must be utilized at least in part for activation of Ca2+-dependent PKCα and β, and these PKCs induced ROS generation through NOX. The ROS must be involved in VSOR channel activation, although the mechanism is not yet known.
Our study further indicated that the sites of Ca2+ action must be very close to the Ca2+ source channels. How close the distance is can be roughly estimated by using a theoretical model of Ca2+ nanodomains (Neher, 1998a). The model predicts that no significant [Ca2+]i rises would be generated at the sites >40 nm away from individual open channel pores in the presence of 10 mm BAPTA (Fig. 8E). Assuming that the Ca2+ sensors respond to a [Ca2+]i in the range of >1 μm as those of PKCs (Keranen & Newton, 1997), and considering that low concentrations of BAPTA should not affect drastically the VSOR channel activation, the Ca2+ sensors would probably sense the [Ca2+]i rises within ∼20 nm of the channel pores. The sensors involved would include those of phospholipase C and PKC. Activation of these enzymes necessitates their binding to the plasma membrane, because not only Ca2+ but also the activated G proteins, the substrate phosphatidylinositol 4,5-bisphosphate in the membrane and the resultant diacylglycerol are required for their activation (Drin & Scarlata, 2007; Gallegos & Newton, 2008). Thus it is evidently reasonable that these enzymes rely on the Ca2+ in the nanodomains generated just adjacent to the activated G protein-coupled receptors.
It is well known that the ROS generator NOX can be activated by PKC through phosphorylation of its subunit p47phox (Bedard & Krause, 2007), and Ca2+ may enhance synergistically the NOX activity with the phosphorylation (Berthier et al. 2003). Furthermore, although how ROS activate VSOR channels is totally unknown, the ROS signalling involved in normal cellular responses tends to be specifically coupled to its targets, and unwanted spread of ROS is immediately quenched by a wealth of scavengers, like superoxide dismutase and glutathione, in the cytoplasm (Terada, 2006). Therefore, our results strongly suggest that the bradykinin-induced VSOR channel activation is regulated in the immediate vicinity of individual Ca2+ release and entry channels.
It must be noted, however, that this mechanism is different from the principal mechanism of VSOR channel activation induced by osmotic cell swelling in response to hypotonic stimuli. The swelling-induced VSOR current is known to be relatively resistant to intracellular BAPTA at 5–10 mm (Kubo & Okada, 1992; Pedersen et al. 1998), as mentioned in Introduction. This is in contrast to the ∼70% suppression of bradykinin-induced current by BAPTA at 10 mm (Fig. 8C and D). Furthermore, the Ca2+-dependent enzyme cascade should not work unless the [Ca2+]i rise is accompanied by generation of the membrane-associated components, as described above. In this sense, the cascade depends not only on Ca2+ but also on the receptor activation per se. By contrast, the VSOR channel activation induced by hypotonic stimuli was shown to be never abolished by interruption of any known types of signalling cascade (Okada et al. 2009), and the activation correlates directly with the increase in surface area of the cell as a result of cell swelling (Okada, 1997), although the correlation may be modulated more or less by some signalling cascades. Thus the mechanism of hypotonicity-induced VSOR channel activation seems to depend much more on the event associated with intrinsic structural changes of the cell. However, the Ca2+-dependent cascade may contribute to a part of the VSOR channel activity during osmotic swelling. For example, the chemical transmitters released during hypotonic stimuli, like ATP, may contribute to the activity through their binding to Gq-coupled receptors (Wang et al. 1996; Darby et al. 2003; Mongin & Kimelberg, 2005). As another example, the δ-type of phospholipase C is reported to be activated directly by Ca2+ without the help of G-proteins (Kim et al. 1999; Mogami et al. 2003), so this might generate diacylglycerol sufficiently to induce the PKC-NOX signalling in response to the [Ca2+]i rises caused by hypotonic stimuli. These may explain the partial dependence of swelling-induced VSOR channel activation on PKC (Verdon et al. 1995; Du & Sorota, 1999; Gong et al. 2004) or ROS (Varela et al. 2004; Ren et al. 2008) reported in some types of cell, although the dependency on PKC or ROS is known to be quite variable between cell types (reviewed by Okada, 1997; Nilius et al. 1997a; Hoffmann et al. 2009).
The Ca2+ nanodomain-mediated control of the enzyme cascade produced slow but prolonged activation of large VSOR currents, even when the [Ca2+]i rise in the bulk cytoplasm had almost disappeared. This activation pattern is entirely different from that of the rapidly responsive so-called Ca2+-activated Cl− channels. The prototypical Ca2+-activated Cl− current is activated immediately in response to the beginning of a [Ca2+]i rise, and reaches a peak and decays rapidly in parallel with the [Ca2+]i peak and decay (Nilius et al. 1997b; Machaca & Hartzell, 1999). A similar activation pattern of a Ca2+-activated Cl− current was also reported in mouse astrocytes (Park et al. 2009). In our recording conditions, however, we have never seen such a rapid current enhancement just after the beginning of bradykinin application when Ca2+ release began. This would be due to the difference in the mode of current recording or in the amount of Ca2+ chelators in the pipette solution. Park et al. (2009) recorded the currents in a perforated patch clamp mode, whereas we recorded the currents in the conventional whole-cell patch clamp mode with the pipette solution containing 1 mm EGTA by default to fix the basal Ca2+ concentration to ∼15 nm (see Methods for details). Therefore, it is possible that the rapidly activated current component would have been eliminated by intracellular dialysis with 1 mm EGTA in our conditions. In other words, the activation of rapidly responsive Ca2+-activated Cl− channels, if any, would be much more loosely coupled to Ca2+ sources than the VSOR channel activation is in astrocytes.
The characteristics of the current through VSOR channels, especially the outward rectification and inactivation at high voltages, are known to have some variations between cell types, but whether these are due to some modifications on the same channel molecule or others is totally unknown (see reviews by Okada, 1997; Nilius et al. 1997a; Hoffmann et al. 2009). As mentioned in Results, the VSOR current in astrocytes is similar to that in cardiomyocytes, where ClC-3 has been proposed as a candidate for VSOR channels (Duan et al. 1997) although this has been challenged by many reports including those from our groups (Gong et al. 2004; Wang et al. 2005). Since the ClC-3 current may exhibit similar characteristics to the VSOR current, one may argue that the bradykinin-induced current in astrocytes might be caused by the activation of ClC-3. However, the same group that proposed ClC-3 as the candidate (Duan et al. 1999) clearly demonstrated that a site in the amino terminus of ClC-3 can be directly phosphorylated by PKC and this causes suppression of ClC-3 activity. Evidently, this does not match with our case, where PKC activation causes ROS generation for VSOR channel activation. Therefore, the bradykinin-induced current would be unlikely to be caused by ClC-3.
With its properties of slow and sustained activation, the VSOR channel is specialized for cell volume regulation in response to hypotonic exposure and to apoptotic stimuli, both of which follow relatively long time courses of >10 min and >1 h, respectively (reviewed by Okada et al. 2001). Thus the Ca2+ nanodomain-mediated regulation of VSOR channels would provide a firm basis for local control of cell volume regulation, e.g. retraction of a part of a cell causing cell shape change or migration (Schneider et al. 2008), and of intercellular communications on a long time scale, even when Ca2+ signals are transient and confined to a subcellular fine structure level (Koizumi et al. 1999; Conklin et al. 2005).
Bradykinin is widely distributed throughout the brain (Kariya et al. 1985) and known to be released during trauma, stroke and inflammation in the brain (Ellis et al. 1989; Kamiya et al. 1993; Gröger et al. 2005). We speculate that one possible in situ role for local regulation of the VSOR channel in the brain in response to bradykinin might be as follows. A group of astrocytes in the brain is known to form part of the blood–brain barrier, surrounding capillary endothelial cells with their fine endfeet processes (Abbott et al. 2006). Bradykinin is known to be produced easily when the endothelial cell walls are damaged by for example hypertension. Thus, when damage occurs, a small amount of bradykinin may leak out and act on a part of an astrocyte endfoot. If this small amount of bradykinin induces a minute amount of InsP3 production in the cell, the [Ca2+]i rise, if it occurs, may be limited as a local transient event (Callamaras et al. 1998; Koizumi et al. 1999), and may not propagate as Ca2+ waves throughout the astrocyte. Even in this case, however, a number of VSOR channels around the local [Ca2+]i rise would likely be activated based on the nanodomain regulation of VSOR channels. And not only Cl− ions but also excitatory amino acids are released focally through the VSOR channels. Furthermore, because of its slow kinetics, VSOR channel activation would still be maintained for a while even after the [Ca2+]i rise diminishes, and this would facilitate a local retraction of the endfoot process, providing a space for the repair of the injured site by microglia. To elucidate such a role for VSOR channel, fine changes in cell structure and in cell volume induced by bradykinin have to be examined carefully, which remains for future studies.
In conclusion, this study demonstrated for the first time that VSOR channel activation may be mediated by Ca2+ nanodomains, especially when their generation is associated with the activation of Gq-coupled bradykinin receptors. Therefore, it would also be intriguing to see whether activation of other types of Gq-coupled receptors involved in normal cellular functions, like those for ATP or glutamate, may induce similar VSOR channel activation and local cell volume regulation.
Acknowledgments
We thank all members in our department for fruitful discussions and suggestions on this work. This work was supported by Grants-in-Aid for Scientific Research (21790216 to T.A. and 18077008 to Y.O.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Glossary
Abbreviations
- 2-APB
2-aminoethoxydiphenyl borate
- CM-DCF
5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein
- DCPIB
4-(2-butyl-6,7-dichloro-2-cyclopentyl-indan-1-on-5-yl) oxybutyric acid
- DPI
diphenyleneiodonium
- NMDG
N-methyl-d-glucamine
- NOX
NADPH oxidase
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- ROS
reactive oxygen species
- RVD
regulatory volume decrease
- siRNA
small interfering RNA
- VSOR
volume-sensitive outwardly rectifying
Author contributions
The experiments were performed in our Department of Cell Physiology in the National Institute for Physiological Sciences. Conception and design of the experiments were made by T.A. and Y.O. Data collection and analysis were done by T.A., and interpretation was made by T.A. and Y.O. The manuscript was drafted and revised by T.A. and Y.O. Both authors approved the final version of the manuscript.
Supplementary material
Supplemental Figure S1
Supplemental Figure S2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci. 2005;25:9176–9184. doi: 10.1523/JNEUROSCI.1632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, Kuba K. Ca2+-dependent inactivation of Ca2+-induced Ca2+ release in bullfrog sympathetic neurons. J Physiol. 2008;586:3365–3384. doi: 10.1113/jphysiol.2008.153833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, Kuba K. Functional triads consisting of ryanodine receptors, Ca2+ channels, and Ca2+-activated K+ channels in bullfrog sympathetic neurons: Plastic modulation of action potential. J Gen Physiol. 2000;116:697–720. doi: 10.1085/jgp.116.5.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, Okada Y. Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying (VSOR) anion channels via ‘Ca2+ nanodomains’ in mouse cortical astrocytes. J Physiol Sci. 2010;60:S66. doi: 10.1113/jphysiol.2011.208173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Baumgarten CM, Clemo HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog Biophys Mol Biol. 2003;82:25–42. doi: 10.1016/s0079-6107(03)00003-8. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Berthier S, Paclet MH, Lerouge S, Roux F, Vergnaud S, Coleman AW, Morel F. Changing the conformation state of cytochrome b558 initiates NADPH oxidase activation: MRP8/MRP14 regulation. J Biol Chem. 2003;278:25499–25508. doi: 10.1074/jbc.M209755200. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by β1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callamaras N, Marchant JS, Sun XP, Parker I. Activation and co-ordination of InsP3-mediated elementary Ca2+ events during global Ca2+ signals in Xenopus oocytes. J Physiol. 1998;509:81–91. doi: 10.1111/j.1469-7793.1998.081bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Conklin MW, Lin MS, Spitzer NC. Local calcium transients contribute to disappearance of pFAK, focal complex removal and deadhesion of neuronal growth cones and fibroblasts. Dev Biol. 2005;287:201–212. doi: 10.1016/j.ydbio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Darby M, Kuzmiski JB, Panenka W, Feighan D, MacVicar BA. ATP released from astrocytes during swelling activates chloride channels. J Neurophysiol. 2003;89:1870–1877. doi: 10.1152/jn.00510.2002. [DOI] [PubMed] [Google Scholar]
- Deng W, Baki L, Baumgarten CM. Endothelin signalling regulates volume-sensitive Cl− current via NADPH oxidase and mitochondrial reactive oxygen species. Cardiovasc Res. 2010;88:93–100. doi: 10.1093/cvr/cvq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drin G, Scarlata S. Stimulation of phospholipase Cβ by membrane interactions, interdomain movement, and G protein binding – how many ways can you activate an enzyme? Cell Signal. 2007;19:1383–1392. doi: 10.1016/j.cellsig.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XY, Sorota S. Protein kinase C stimulates swelling-induced chloride current in canine atrial cells. Pflugers Arch. 1999;437:227–234. doi: 10.1007/s004240050773. [DOI] [PubMed] [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J Gen Physiol. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Chao J, Heizer ML. Brain kininogen following experimental brain injury: evidence for a secondary event. J Neurosurg. 1989;71:437–442. doi: 10.3171/jns.1989.71.3.0437. [DOI] [PubMed] [Google Scholar]
- Gallegos LL, Newton AC. Spatiotemporal dynamics of lipid signaling: protein kinase C as a paradigm. IUBMB Life. 2008;60:782–789. doi: 10.1002/iub.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Xu H, Shimizu T, Morishima S, Tanabe S, Tachibe T, Uchida S, Sasaki S, Okada Y. ClC-3-independent, PKC-dependent activity of volume-sensitive Cl− channel in mouse ventricular cardiomyocytes. Cell Physiol Biochem. 2004;14:213–224. doi: 10.1159/000080330. [DOI] [PubMed] [Google Scholar]
- Gröger M, Lebesgue D, Pruneau D, Relton J, Kim SW, Nussberger J, Plesnila N. Release of bradykinin and expression of kinin B2 receptors in the brain: role for cell death and brain edema formation after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:978–989. doi: 10.1038/sj.jcbfm.9600096. [DOI] [PubMed] [Google Scholar]
- Harrigan TJ, Abdullaev IF, Jourd'heuil D, Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem. 2008;106:2449–2462. doi: 10.1111/j.1471-4159.2008.05553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Inoue H, Mori S, Morishima S, Okada Y. Volume-sensitive chloride channels in mouse cortical neurons: characterization and role in volume regulation. Eur J Neurosci. 2005;21:1648–1658. doi: 10.1111/j.1460-9568.2005.04006.x. [DOI] [PubMed] [Google Scholar]
- Kamiya T, Katayama Y, Kashiwagi F, Terashi A. The role of bradykinin in mediating ischemic brain edema in rats. Stroke. 1993;24:571–575. doi: 10.1161/01.str.24.4.571. [DOI] [PubMed] [Google Scholar]
- Kariya K, Yamauchi A, Sasaki T. Regional distribution and characterization of kinin in the CNS of the rat. J Neurochem. 1985;44:1892–1897. doi: 10.1111/j.1471-4159.1985.tb07185.x. [DOI] [PubMed] [Google Scholar]
- Keranen LM, Newton AC. Ca2+ differentially regulates conventional protein kinase Cs’ membrane interaction and activation. J Biol Chem. 1997;272:25959–25967. doi: 10.1074/jbc.272.41.25959. [DOI] [PubMed] [Google Scholar]
- Kim YH, Park TJ, Lee YH, Baek KJ, Suh PG, Ryu SH, Kim KT. Phospholipase C-δ1 is activated by capacitative calcium entry that follows phospholipase C-β activation upon bradykinin stimulation. J Biol Chem. 1999;274:26127–26134. doi: 10.1074/jbc.274.37.26127. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Bootman MD, Bobanovic LK, Schell MJ, Berridge MJ, Lipp P. Characterization of elementary Ca2+ release signals in NGF-differentiated PC12 cells and hippocampal neurons. Neuron. 1999;22:125–137. doi: 10.1016/s0896-6273(00)80684-4. [DOI] [PubMed] [Google Scholar]
- Kubo M, Okada Y. Volume-regulatory Cl− channel currents in cultured human epithelial cells. J Physiol. 1992;456:351–371. doi: 10.1113/jphysiol.1992.sp019340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert IH, Klausen TK, Bergdahl A, Hougaard C, Hoffmann EK. ROS activate KCl cotransport in nonadherent Ehrlich ascites cells but K+ and Cl− channels in adherent Ehrlich Lettre and NIH3T3 cells. Am J Physiol Cell Physiol. 2009;297:C198–C206. doi: 10.1152/ajpcell.00613.2008. [DOI] [PubMed] [Google Scholar]
- Leaney JL, Marsh SJ, Brown DA. A swelling-activated chloride current in rat sympathetic neurones. J Physiol. 1997;501:555–564. doi: 10.1111/j.1469-7793.1997.555bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HT, Akita T, Shimizu T, Sabirov RZ, Okada Y. Bradykinin-induced astrocyte-neuron signalling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J Physiol. 2009;587:2197–2209. doi: 10.1113/jphysiol.2008.165084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K, Hartzell HC. Reversible Ca gradients between the subplasmalemma and cytosol differentially activate Ca-dependent Cl currents. J Gen Physiol. 1999;113:249–266. doi: 10.1085/jgp.113.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Rappert A, Matthias K, Steinhäuser C, Spät A, Kettenmann H. Astrocytes from mouse brain slices express ClC-2-mediated Cl− currents regulated during development and after injury. Mol Cell Neurosci. 2003;23:521–530. doi: 10.1016/s1044-7431(03)00080-0. [DOI] [PubMed] [Google Scholar]
- Mogami H, Zhang H, Suzuki Y, Urano T, Saito N, Kojima I, Petersen OH. Decoding of short-lived Ca2+ influx signals into long term substrate phosphorylation through activation of two distinct classes of protein kinase C. J Biol Chem. 2003;278:9896–9904. doi: 10.1074/jbc.M210653200. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. ATP regulates anion channel-mediated organic osmolyte release from cultured rat astrocytes via multiple Ca2+-sensitive mechanisms. Am J Physiol Cell Physiol. 2005;288:C204–C213. doi: 10.1152/ajpcell.00330.2004. [DOI] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium. 1998a;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998b;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G. Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol. 1997a;68:69–119. doi: 10.1016/s0079-6107(97)00021-7. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Szucs G, Wei L, Tanzi F, Voets T, Droogmans G. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J Physiol. 1997b;498:381–396. doi: 10.1113/jphysiol.1997.sp021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata T, Shimizu T, Okada Y. Direct mechano-stress sensitivity of TRPM7 channel. Cell Physiol Biochem. 2007a;19:1–8. doi: 10.1159/000099187. [DOI] [PubMed] [Google Scholar]
- Numata T, Shimizu T, Okada Y. TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol. 2007b;292:C460–C467. doi: 10.1152/ajpcell.00367.2006. [DOI] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am J Physiol Cell Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) J Physiol. 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Sato K, Numata T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J Physiol. 2009;587:2141–2149. doi: 10.1113/jphysiol.2008.165076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, Prenen J, Droogmans G, Hoffmann EK, Nilius B. Separate swelling- and Ca2+-activated anion currents in Ehrlich ascites tumor cells. J Membr Biol. 1998;163:97–110. doi: 10.1007/s002329900374. [DOI] [PubMed] [Google Scholar]
- Park H, Oh SJ, Han KS, Woo DH, Park H, Mannaioni G, Traynelis SF, Lee CJ. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J Neurosci. 2009;29:13063–13073. doi: 10.1523/JNEUROSCI.3193-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Ren Z, Raucci FJ, Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl− current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichter LC, Mertens T, Liu B. Swelling activated Cl− channels in microglia: Biophysics, pharmacology and role in glutamate release. Channels (Austin) 2011;5:128–137. doi: 10.4161/chan.5.2.14310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Klausen TK, Stock C, Mally S, Christensen ST, Pedersen SF, Hoffmann EK, Schwab A. H-ras transformation sensitizes volume-activated anion channels and increases migratory activity of NIH3T3 fibroblasts. Pflugers Arch. 2008;455:1055–1062. doi: 10.1007/s00424-007-0367-3. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Numata T, Okada Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl− channel. Proc Natl Acad Sci U S A. 2004;101:6770–6773. doi: 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada LS. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol. 2006;174:615–623. doi: 10.1083/jcb.200605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela D, Simon F, Riveros A, Jorgensen F, Stutzin A. NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem. 2004;279:13301–13304. doi: 10.1074/jbc.C400020200. [DOI] [PubMed] [Google Scholar]
- Verdon B, Winpenny JP, Whitfield KJ, Argent BE, Gray MA. Volume-activated chloride currents in pancreatic duct cells. J Membr Biol. 1995;147:173–183. doi: 10.1007/BF00233545. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu H, Morishima S, Tanabe S, Jishage K, Uchida S, Sasaki S, Okada Y, Shimizu T. Single-channel properties of volume-sensitive Cl− channel in ClC-3-deficient cardiomyocytes. Jpn J Physiol. 2005;55:379–383. doi: 10.2170/jjphysiol.S655. [DOI] [PubMed] [Google Scholar]
- Wang Y, Roman R, Lidofsky SD, Fitz JG. Autocrine signaling through ATP release represents a novel mechanism for cell volume regulation. Proc Natl Acad Sci U S A. 1996;93:12020–12025. doi: 10.1073/pnas.93.21.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ, Kimelberg HK. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol. 2008;210:514–520. doi: 10.1016/j.expneurol.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.