

Non-technical summary
Heat stress reduces brain blood flow and impairs orthostatic tolerance. Brain blood flow is largely controlled by the partial pressure of arterial 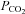 . Indeed, hyperthermia-induced over-breathing and related reductions in arterial
. Indeed, hyperthermia-induced over-breathing and related reductions in arterial 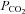 account for ∼50% of the reduction in brain blood flow. This investigation tested the unique hypothesis that the distribution of cardiac output during heat stress (challenged by thermoregulatory increases in skin blood flow and sweat loss) contributes to the remaining 50%. We show that cardiac output is not related to brain blood flow, but rather arterial
account for ∼50% of the reduction in brain blood flow. This investigation tested the unique hypothesis that the distribution of cardiac output during heat stress (challenged by thermoregulatory increases in skin blood flow and sweat loss) contributes to the remaining 50%. We show that cardiac output is not related to brain blood flow, but rather arterial 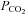 plays a much larger role than previously suggested. These findings help us understand the mechanisms relating heat stress with an increased likelihood of fainting, and are also relevant to pathological conditions that are accompanied by elevations in body temperature.
plays a much larger role than previously suggested. These findings help us understand the mechanisms relating heat stress with an increased likelihood of fainting, and are also relevant to pathological conditions that are accompanied by elevations in body temperature.
Abstract
Abstract
Cerebral blood flow (CBF) is reduced during passive heat stress, with 50% of this reduction associated with hyperventilatory-induced hypocapnia and subsequent cerebral vasoconstriction. It remains unknown, however, what other factors may contribute to the remaining 50%. We tested the hypothesis that the distribution of cardiac output plays an important role in maintaining cerebral perfusion during mild and severe heat stress. Middle cerebral artery and posterior cerebral artery blood flow velocity (MCAv and PCAv; transcranial Doppler) and left ventricular end-diastolic and end-systolic volumes (2-D echocardiography) were measured under conditions of normothermia and mild and severe passive heat stress (core temperature +0.8 ± 0.1°C (Protocol I; n = 10) and 1.8 ± 0.1°C (Protocol II; n = 8) above baseline). Venous return was manipulated by passive tilt table positioning (30 deg head-down tilt (HDT) and 30 deg head-up tilt (HUT)). Measurements were made under poikilocapnic and isocapnic conditions. Protocol I consisted of mild heat stress which resulted in small reductions in end-tidal CO2 (−5.6 ± 3.5%), MCAv/PCAv (−7.3 ± 2.3% and −10.3 ± 2.9%, respectively) and stroke volume (−8.5 ± 4.2%); while end-diastolic volume was significantly reduced (−16.9 ± 4.0%) and cardiac output augmented (17.2 ± 7.4%). During mild heat stress, CBF was related to left ventricular end-diastolic volume (MCAv, r2 = 0.81; PCAv, r2 = 0.83; P < 0.05) and stroke volume (MCAv, r2 = 0.38; PCAv, r2 = 0.43), but not with cardiac output. Protocol II consisted of severe heat stress which resulted in much greater reductions in end-tidal CO2 (−87.5 ± 31.5%) and CBF (MCAv, −36.4 ± 6.1%; PCAv, −30.1 ± 4.8%; P < 0.01 for all variables), while end-diastolic volume and stroke volume decreased to a similar extent as for mild heat stress. Importantly, isocapnia restored MCAv and PCAv back to normothermic baseline. This investigation therefore produced two novel findings: first, that venous return and stroke volume are related to CBF during mild heat stress; and second, that hyperventilatory hypocapnia has a major influence on CBF during severe passive heat stress.
Introduction
Heat stress reduces orthostatic tolerance compared to normothermic conditions (Lind et al. 1968; Allan & Crossley, 1972; Johnson et al. 1973; Shvartz et al. 1975; Wilson et al. 2002). While the mechanism(s) remains unclear, it is well established that cerebral blood flow (CBF) is reduced during passive heat stress (Wilson et al. 2006; Brothers et al. 2009). Indeed, any condition that jeopardizes CBF will ultimately lead to syncope (Van Lieshout et al. 2003). As CBF is highly sensitive to arterial hypocapnia (Fan et al. 2008), hyperthermia-induced hyperventilation and consequent hypocapnia (Wilson et al. 2006; Brothers et al. 2009) severely reduces CBF. While this mechanism has been suggested to account for approximately 50% of the reduction in cerebral vascular conductance (Brothers et al. 2009), it currently remains unknown what accounts for the remaining 50%. Although the classical notion is that CBF is largely controlled by the partial pressure of arterial 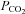 (Ide et al. 2003; Fan et al. 2008) and blood pressure (Lucas et al. 2010), CBF may also be influenced by the distribution of cardiac output (reviewed in Ogoh & Ainslie, 2009).
(Ide et al. 2003; Fan et al. 2008) and blood pressure (Lucas et al. 2010), CBF may also be influenced by the distribution of cardiac output (reviewed in Ogoh & Ainslie, 2009).
During passive heat stress, to support the large increase in cutaneous blood flow (up to 7 l min−1), cardiac output can more than double, reaching values as high as 13 l min−1 (Rowell et al. 1969). Therefore, it is possible that the peripheral distribution of cardiac output during heat stress does not favour adequate cerebral perfusion. In support of this, it has recently been reported (Fan et al. 2008) that hyperthermia-induced elevations in cardiac output were related to declines in CBF velocity. In that study, in contrast to Brothers et al. (2009), reductions in CBF were fully accounted for by heat stress-induced reductions in end-tidal 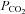 . However, limitations of this study were the indirect assessment of cardiac output (finger photoplethysmography) and the lack of manipulation of cardiac output or end-tidal
. However, limitations of this study were the indirect assessment of cardiac output (finger photoplethysmography) and the lack of manipulation of cardiac output or end-tidal 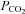 to determine the influence of these variables, if any, on CBF.
to determine the influence of these variables, if any, on CBF.
Therefore, based on this background, the purpose of the present study was to investigate the potential contributions of cardiac output and hypocapnia on CBF in heat-stressed humans. We hypothesized that alterations in venous return and cardiac output would be related to changes in CBF, especially during heat stress and related changes in vascular compliance. To address this hypothesis, we combined mild and severe heat stress with and without clamping of end-tidal 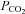 and concurrent measurements of CBF velocity (transcranial Doppler) and cardiac volumes (echocardiography) during supine rest and orthostatically manipulated cardiac output (using head-up and head-down tilt).
and concurrent measurements of CBF velocity (transcranial Doppler) and cardiac volumes (echocardiography) during supine rest and orthostatically manipulated cardiac output (using head-up and head-down tilt).
The clinical relevance of passive heat stress studies are numerous and include, but are not limited to, exploring the pathophysiology of heat-induced syncope, and furthering our understanding of heat-induced cardiovascular stress in a range of occupational settings (e.g. fire-fighting, desert military operations, operating machinery, etc.). The study of passive heat stress is also relevant to the myriad of pathological conditions that are accompanied by elevations in body temperature.
Methods
Ten healthy male subjects participated in this experiment (mean ± SD): age, 24.9 ± 4.7 years; height, 175.8 ± 6.0 cm; and weight, 78.6 ± 11.7 kg. Females were excluded in order to avoid known conflicts with the menstrual cycle (Charkoudian & Joyner, 2004), as this was beyond the scope of the present investigation. All subjects were free of cardiovascular, metabolic or neurological disease and were not taking any medications. Subjects were informed of the purpose and risks of the study prior to providing written informed consent. The study was approved by the University of Alberta Health Research Ethics Board. Subjects refrained from alcohol, caffeine and exercise for 24 h prior to the study, and were asked to arrive at the laboratory well nourished and fully hydrated.
Instrumentation and measurements
Upon arrival at the laboratory, each subject swallowed a telemetry pill for the measurement of core temperature (Jonah ingestible capsule, VitalSense, Mini Mitter Co. Inc., Bend, OR, USA). Mean skin temperature was measured from the weighted average of four biomedical ceramic chip thermocouples (MA 100, 10K0 negative coefficient, Thermometrics, NJ, USA) affixed to the left side of the body (Ramanathan, 1964). Subjects were dressed in a water-perfused tube-lined suit (Med-Eng, Ottawa, Canada) that covered the entire body except for the head, hands and feet. Core and skin temperature could therefore be controlled by adjusting the temperature of water perfusing the suit. Sweat loss was estimated as a change in nude body mass, measured immediately before and after the experimental protocol.
Beat-by-beat heart rate and arterial blood pressure were continuously obtained from R-wave detection on an electrocardiogram (lead II, Dual Bio Amp, ADInstruments) and finger photoplethysmography (Finometer model 2, Finapres, Amsterdam, the Netherlands), respectively. An automated blood pressure cuff, worn on the opposite arm, was used to verify and calibrate finger photoplethysmography. Mean arterial blood pressure was averaged across each condition, calculated as 1/3 pulse pressure plus diastolic blood pressure.
Cerebral blood flow velocity (mean) was measured through the temporal window in the middle cerebral artery (MCAv; 1 cm distal to the MCA–anterior cerebral artery bifurcation) and P1-segment of the posterior cerebral artery (PCAv; confirmed by velocity increase during ipsilateral carotid occlusion and by response to visual stimuli) using a 2 MHz pulsed Doppler ultrasound system (Spencer Technologies, Seattle WA, USA). An optimal signal for each vessel was identified, as described in detail elsewhere (Willie et al. 2011). The probe was then held in place using a headband fixation device. Cerebral blood velocity in the PCA was measured in addition to MCA because each artery perfuses distinct regions of the brain and are fed by distinct conduit vessels – the MCA by the internal carotid arteries and the PCA by the vertebral arteries, which contribute approximately 75% and 25%, respectively, to total cerebral blood flow. The MCA provides blood to a larger total brain mass, principally the frontal and temporal cortices and anterior portions of the occipital cortex, whereas the PCA carries a substantially smaller blood flow supplying the occipital cortex only. Measuring both MCAv and PCAv therefore provides an opportunity to determine regional differences in brain perfusion during whole body heating. Moreover, there is recent evidence to suggest that flow regulation differs between the anterior and posterior circulations during exercise (Willie et al.; Sato & Sadamoto, 2010; Sato et al. 2011); the role of body temperature in these data, however, is unknown. Thus, the combined quantification of cerebral blood velocity in the major arteries of the anterior and posterior cerebral conduit vessels provides insight into regional brain blood distribution during poikilocapnic and isocapnic whole body heating. Cerebrovascular conductance index (CVC) could therefore be estimated for the MCA (CVCM) and PCA (CVCP), calculated by dividing mean cerebral blood flow velocity (MCAv and PCAv) by MAP (Lucas et al. 2011).
Subjects breathed through a mouthpiece with the nose occluded. Inspired gas was humidified (HC 150; Fisher and Paykel Healthcare, Auckland, New Zealand) and delivered continuously using a flow-through system to prevent re-breathing of expired gas. Ventilation was measured by a pneumotachometer (3700 series; Hans Rudolph, Kansas City, MO, USA). Expired CO2 and O2 (mmHg) were measured (CD-3A and S-3A; AEI Technologies, Naperville, IL, USA) continuously from a small sample port off the mouthpiece. End-tidal CO2 was maintained at baseline values by adding medical grade CO2 to the inspired gas.
Two-dimensional transthoracic echocardiography was performed using a commercially available ultrasound machine (VividQ or Vivid7, GE, VingMed, Horten, Norway). Parasternal short axis images (level of the papillary muscles) and apical four chamber images were acquired in order to calculate left ventricular end-diastolic and end-systolic volumes, according to the area–length formula (Lang et al. 2005). Stroke volume was therefore calculated as the difference between left ventricular end-diastolic and end-systolic volumes, and cardiac output was calculated as the product of stroke volume and heart rate.
Experimental protocol
Protocol 1 – mild heat stress
To evaluate the influence of venous return, cardiac output and hypocapnia on CBF, 10 subjects underwent haemodynamic and thermal testing in the supine position, and during 30 deg head-up tilt (HUT) and 30 deg head-down tilt (HDT) while normothermic, and during passive heat stress (Fig. 1A). During heat stress, cerebral vascular measurements were obtained under poikilocapnic and isocapnic conditions. Due to the prevalence of orthostatic intolerance during heat stress, and the linear reduction in CBF with incremental heat stress (Fan et al. 2008), a mild level of passive heat stress (∼0.8°C above baseline) was chosen for this protocol. During normothermia, thermoneutral water (34°C) was circulated through the tube-lined suit, in order to establish a physiological baseline. To elevate core temperature, 50°C water was circulated throughout the suit for 45 min, as previously reported (Nelson et al. 2010a,b). After 45 min of passive heat stress, the temperature of water circulating the suit was reduced to 47°C to attenuate the rate of rise in core temperature. Each tilt condition was held for 5 min before thermal and haemodynamic data were collected for a maximum of 10 min.
Figure 1. Schematic representation of the experimental protocols (I and II).
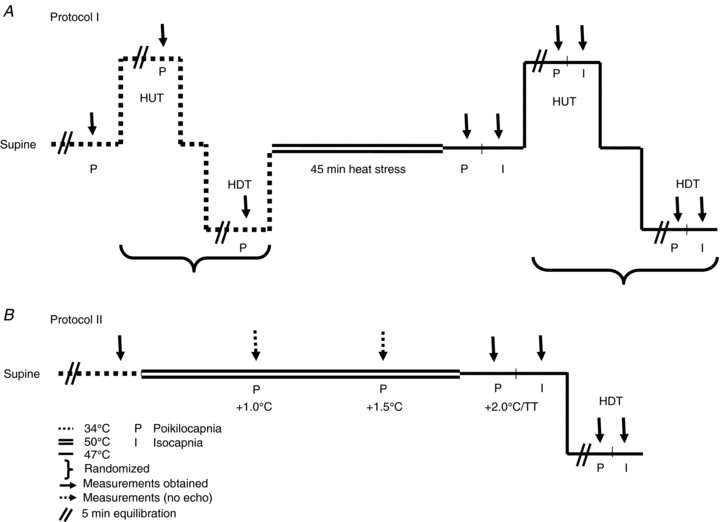
Dashed lines indicate normothermia (34°C water perfusing throughout the tube-lined suit), double continuous lines indicate the initial heating period (50°C water), while single continuous lines indicate a reduction in the circulating water temperature (47°C). The orientation of the lines represents changes in posture (supine, head-up tilt (HUT) and head-down tilt (HDT)). Measurements were obtained after 5 min at each posture (indicated by //), followed by 10 min of data collection (↓). Heat stress was performed for 45 min during protocol I, in order to elevate core temperature ∼0.8°C, whereas during protocol II, 93 ± 16 min of heat stress was required to elevate core temperature 2.0°C above baseline (or to reach thermal tolerance). Measurements were obtained under poikilocapnic (P) and isocapnic (I) conditions.
Protocol II – severe heat stress
To revisit the influence of hypocapnia on CBF during passive heat stress, eight subjects returned to the laboratory on a separate day (>1 month apart). Following instrumentation, subjects rested in the supine position and underwent normothermic baseline thermal and haemodynamic testing. Subjects were then passively heated (as described in Protocol I), until core temperature rose ∼2°C above baseline. This level of heat stress has previously been shown to reduce CBF (indexed from MCAv) and end-tidal 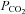 by 35% and 63%, respectively (Fan et al. 2008). Once the target core temperature was achieved, the water temperature circulating the suit was reduced, as described above, to attenuate the rate of rise in core temperature. Following thermal and haemodynamic measurements, the subjects remained on the mouth-piece while end-tidal
by 35% and 63%, respectively (Fan et al. 2008). Once the target core temperature was achieved, the water temperature circulating the suit was reduced, as described above, to attenuate the rate of rise in core temperature. Following thermal and haemodynamic measurements, the subjects remained on the mouth-piece while end-tidal 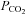 was returned to the normothermic level (isocapnia). Similar to Protocol I, 30 deg HDT was then performed with and without end-tidal CO2 clamping to evaluate the influence of venous return on CBF during severe heat stress. Fig. 1B provides a detailed schematic of Protocol II. Due to the severity of this protocol, subjects were not tested in the HUT position; furthermore, normothermic tilt table positioning was not repeated. With the exception of left ventricular volumes, measurements were conducted at normothermic baseline, at select time points during the heating process (core temperature +1.0°C, +1.5°C and +2.0°C above baseline), and during isocapnic supine and HDT heat stress. Left ventricular volumes were assessed at normothermic baseline, during severe supine heat stress and during HDT heat stress.
was returned to the normothermic level (isocapnia). Similar to Protocol I, 30 deg HDT was then performed with and without end-tidal CO2 clamping to evaluate the influence of venous return on CBF during severe heat stress. Fig. 1B provides a detailed schematic of Protocol II. Due to the severity of this protocol, subjects were not tested in the HUT position; furthermore, normothermic tilt table positioning was not repeated. With the exception of left ventricular volumes, measurements were conducted at normothermic baseline, at select time points during the heating process (core temperature +1.0°C, +1.5°C and +2.0°C above baseline), and during isocapnic supine and HDT heat stress. Left ventricular volumes were assessed at normothermic baseline, during severe supine heat stress and during HDT heat stress.
Data analysis
Significance was set at P < 0.05. Results are reported as the mean ± SEM, unless otherwise specified. For both normothermia and heat stress, but non-clamped, conditions, data were averaged over the entire 10 min data collection period. During the heat stress plus isocapnia, data were averaged during a minimum of 30 s in which end-tidal CO2 was clamped at normothermic levels. As performed previously (Nelson et al. 2011), all volumetric data (echocardiography) were calculated from the average of three cardiac cycles.
Protocol I
The relationship between heat stress, posture and CBF was analysed using Pearson product-moment correlation coefficients. The effect of heat stress and posture on thermal and haemodynamic measurements was analysed using a two-way repeated measures analysis of variance, with Tukey post hoc analysis where appropriate to identify discrete differences. To evaluate the influence of end-tidal 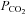 on cerebrovascular measurements, poikilocapnic and isocapnic data were compared to normothermic values using a two-way ANOVA.
on cerebrovascular measurements, poikilocapnic and isocapnic data were compared to normothermic values using a two-way ANOVA.
Protocol II
The influence of severe heat stress (with and without isocapnia) on thermal, haemodynamic and respiratory variables was evaluated using a one-way repeated measures analysis of variance, followed by Tukey post hoc analyses to identify discrete differences. Pearson product-moment correlation coefficients were used to evaluate the relationship between venous return and cardiac output and CBF, as well as to assess the relationship between end-tidal CO2 and CBF over a wide range of body temperatures (normothermic baseline, +1.0°C, +1.5°C and +2.0°C above baseline).
Results
Protocol I
The results from Protocol I are presented in Table 1. As expected, heat stress increased mean skin temperature (3.6 ± 0.2°C), and core temperature (0.8 ± 0.06°C), with posture exhibiting no influence on either variable. On average, participants lost an estimated 1.2 ± 0.1 l of fluid throughout the protocol. Heart rate increased significantly above baseline with head-up tilt (6 ± 2 beats min−1), heat stress (24 ± 4 beats min−1), and during head-up tilt combined with heat stress (38 ± 3 beats min−1), but did not change during head-down tilt. Mean arterial blood pressure was similar between conditions. Tilt table positioning and heat stress manipulated left ventricular volumes. Left ventricular end-diastolic volume decreased (P < 0.05) from baseline with head-up tilt (−27.5 ± 8.4 ml), heat stress (−23.9 ± 5.8 ml), and head-up tilt combined with heat stress (−43.3 ± 10.9); but, end-diastolic volume was not affected by temperature during head-down tilt. Stroke volume was reduced with head-up tilt (independent of thermal condition) but did not change during head-down tilt. Independent of posture, cardiac output increased significantly with heat stress.
Table 1.
Thermal, haemodynamic and cerebrovascular variables during normothermia and passive heat stress tilt table positioning under poikilocapnic and isocapnic conditions
| Normothermic | Heat stress | |||||
|---|---|---|---|---|---|---|
| Supine | HUT | HDT | Supine | HUT | HDT | |
| Thermal | ||||||
| Skin temp. (°C) | 33.4 ± 0.1 | 33.4 ± 0.1 | 33.2 ± 0.1 | 36.9 ± 0.1* | 36.7 ± 0.2* | 36.6 ± 0.1* |
| Core temp. (°C) | 37.0 ± 0.05 | 37.0 ± 0.05 | 37.0 ± 0.07 | 37.9 ± 0.1* | 38.0 ± 0.09* | 37.9 ± 0.08* |
| Haemodynamics | ||||||
| HR (beats min−1) | 61.3 ± 2.9 | 66.4 ± 2.9† | 60.4 ± 2.6 | 86.4 ± 3.0* | 101.6 ± 2.6*† | 83.7 ± 3.0* |
| MAP (mmHg) | 71.9 ± 2.5 | 77.6 ± 2.5 | 75.1 ± 2.5 | 71.6 ± 2.3 | 77.0 ± 3.2 | 76.0 ± 3.7 |
| LV EDV (ml) | 166.7 ± 7.8 | 142.7 ± 7.6† | 166.7 ± 7.1 | 146.2 ± 6.7*‡ | 125.8 ± 6.4*† | 164.7 ± 7.5 |
| SV (ml) | 99.0 ± 6.9 | 78.9 ± 4.4† | 97.8 ± 4.8 | 91.3 ± 4.6 | 81.4 ± 5.3† | 103.5 ± 5.3 |
| CO (l min−1) | 6.0 ± 0.5 | 5.2 ± 0.3 | 5.8 ± 0.3 | 7.9 ± 0.6* | 8.4 ± 0.7* | 8.7 ± 0.6* |
| Cerebrovascular | ||||||
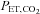 (mmHg) (mmHg) | ||||||
| Poikilocapnic | 41.7 ± 0.9 | 39.7 ± 1.4 | 42.0 ± 1.1 | 39.8 ± 1.8 | 36.6 ± 1.7*† | 39.0 ± 1.4 |
| Isocapnic | — | — | — | 41.2 ± 0.7 | 41.6 ± 0.6 | 41.4 ± 0.8 |
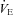 (l min−1) (l min−1) | ||||||
| Poikilocapnic | 9.6 ± 0.4 | 10.3 ± 0.6 | 9.7 ± 0.5 | 11.3 ± 0.8* | 12.6 ± 0.9* | 11.5 ± 0.8* |
| Isocapnic | — | — | — | 11.4 ± 0.8* | 13.0 ± 0.8* | 11.6 ± 0.8 |
| VT (l) | ||||||
| Poikilocapnic | 0.8 ± 0.05 | 0.9 ± 0.06 | 0.9 ± 0.07 | 1.0 ± 0.09* | 1.1 ± 0.1* | 1.0 ± 0.1 |
| Isocapnic | — | — | — | 0.9 ± 0.07* | 1.0 ± 0.08 | 1.0 ± 0.08 |
| RR (breaths min−1) | ||||||
| Poikilocapnic | 12.5 ± 0.7 | 11.3 ± 0.6 | 11.3 ± 1.0 | 12.1 ± 0.9 | 11.6 ± 0.6 | 12.6 ± 1.1 |
| Isocapnic | — | — | — | 12.5 ± 0.9 | 13.3 ± 0.8 | 12.5 ± 0.9 |
| Middle cerebral artery | ||||||
| MCAv (cm s−1) | ||||||
| Poikilocapnic | 62.8 ± 3.7 | 59.3 ± 3.2‡ | 65.6 ± 3.4 | 58.6 ± 3.3 | 50.3 ± 2.5*† | 60.1 ± 3.3 |
| Isocapnic | — | — | — | 60.3 ± 3.0 | 57.1 ± 2.6‡ | 64.0 ± 3.2 |
| CVCM (cm s−1 mmHg−1) | ||||||
| Poikilocapnic | 0.88 ± 0.04 | 0.77 ± 0.03§ | 0.88 ± 0.05 | 0.82 ± 0.05 | 0.66 ± 0.03*† | 0.80 ± 0.05 |
| Isocapnic | — | — | — | 0.84 ± 0.04 | 0.75 ± 0.03‡ | 0.85 ± 0.04 |
| Posterior cerebral artery | ||||||
| PCAv (cm s−1) | ||||||
| Poikilocapnic | 40.6 ± 3.3 | 36.8 ± 2.7§ | 40.1 ± 2.9 | 36.6 ± 2.4* | 32.2 ± 1.7*† | 37.6 ± 2.9 |
| Isocapnic | — | — | — | 37.6 ± 2.0 | 36.9 ± 1.6 | 41.0 ± 2.7 |
| CVCP (cm s−1 mmHg−1) | ||||||
| Poikilocapnic | 0.57 ± 0.04 | 0.48 ± 0.03§ | 0.53 ± 0.03 | 0.51 ± 0.03* | 0.42 ± 0.02*† | 0.50 ± 0.04 |
| Isocapnic | — | — | — | 0.52 ± 0.02 | 0.48 ± 0.02‡ | 0.54 ± 0.03 |
Data reported as mean ± SEM. HR, heart rate; MAP, mean arterial blood pressure; LV EDV, left ventricular end-diastolic volume; ESV, end-systolic volume; SV, stroke volume; CO, cardiac output; 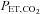 , end-tidal
, end-tidal 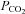 ;
; 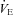 , ventilation; VT, tidal volume; RR, respiratory rate; and CVC, cerebral vascular conductance. Data were collected in the supine position, as well as during 30 deg head-up tilt (HUT) and 30 deg head-down tilt (HDT).
, ventilation; VT, tidal volume; RR, respiratory rate; and CVC, cerebral vascular conductance. Data were collected in the supine position, as well as during 30 deg head-up tilt (HUT) and 30 deg head-down tilt (HDT).
indicates a significant difference between respective normothermic posture
indicates a significant difference from supine and HDT
indicates a significant difference from HDT
indicates a significant difference from supine. P < 0.05.
Mild heat stress tended to reduce end-tidal CO2 (−1.4 ± 1.2 mmHg), MCAv (−4.2 ± 1.4 cm s−1; P = 0.09) and CVCM (0.06 ± 0.03 cm s−1 mmHg−1) from baseline, while PCAv (−4.0 ± 1.2 cm s−1) and CVCP (0.06 ± 0.02 cm s−1 mmHg−1) were significantly reduced and ventilation was augmented (+1.7 ± 0.8 l min−1). Combining HUT with heat stress further reduced (P < 0.01) end-tidal 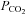 (−5.1 ± 1.0 mmHg), MCAv (−12.5 ± 3.0 cm s−1) and PCAv (−8.4 ± 2.0 cm s−1), while ventilation remained significantly elevated above baseline (1.8 ± 1.2 l min−1). Head-down tilt combined with heat stress returned MCAv and PCAv towards baseline values, while end-tidal
(−5.1 ± 1.0 mmHg), MCAv (−12.5 ± 3.0 cm s−1) and PCAv (−8.4 ± 2.0 cm s−1), while ventilation remained significantly elevated above baseline (1.8 ± 1.2 l min−1). Head-down tilt combined with heat stress returned MCAv and PCAv towards baseline values, while end-tidal 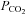 remained depressed (P = 0.07). Similarly, during isocapnic heat stress, MCAv and PCAv were elevated during all three postures.
remained depressed (P = 0.07). Similarly, during isocapnic heat stress, MCAv and PCAv were elevated during all three postures.
As illustrated in Fig. 2, changes in both stroke volume and left ventricular end-diastolic volume were related to both MCAv and PCAv, while cardiac output was not related to CBF regardless of end-tidal 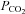 . Individual relationships tended to support this finding, with end-diastolic volume and stroke volume being related to CBF (r2 = 0.26–0.61) and cardiac output sharing virtually no relationship (r2 = 0.03–0.10).
. Individual relationships tended to support this finding, with end-diastolic volume and stroke volume being related to CBF (r2 = 0.26–0.61) and cardiac output sharing virtually no relationship (r2 = 0.03–0.10).
Figure 2. Relationship between the group average middle cerebral artery blood flow velocity (MCAv) or posterior cerebral artery blood flow velocity (PCAv) and the group average stroke volume, cardiac output and left ventricular end-diastolic volume under normothermic and passive heat stress conditions during passive tilt table positioning (supine, head-up tilt and head-down tilt).
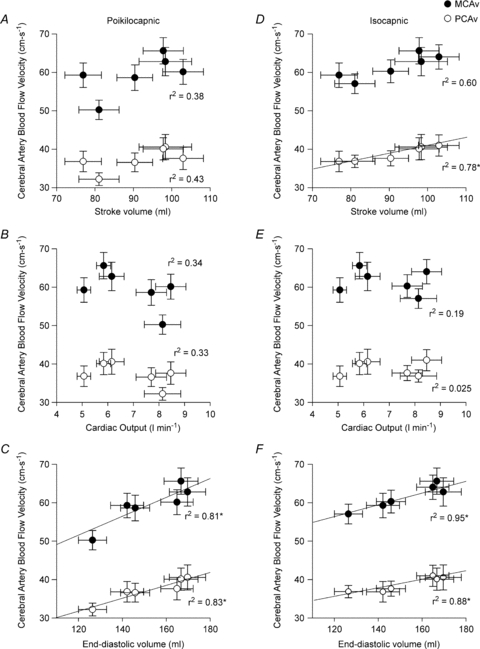
A–C illustrate these relationships during poikilocapnia, while D–F illustrate the isocapnic relationships. * indicates a significant relationship (P < 0.05). n = 10, protocol I.
Protocol II
As shown in Table 2, severe heat stress significantly increased skin and core temperature (5.9 ± 0.2°C and 1.8 ± 0.03°C, respectively), heart rate (43.7 ± 4.7 beats min−1) and cardiac output (2.7 ± 0.5 l min−1), decreased end-tidal 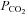 (−14.6 ± 3.4 mmHg), MCAv (−14.7 ± 2.2 cm s−1), PCAv (−9.7 ± 1.2 cm s−1), CVCM (0.19 ± 0.03 cm s−1 mmHg−1), CVCP (0.12 ± 0.03 cm s−1 mmHg−1) and left ventricular end-diastolic volume (−25.9 ± 8.9 ml), while stroke volume and arterial blood pressure remained unchanged. Severe heat stress also resulted in an estimated sweat loss of 1.6 ± 0.3 l. Interestingly, isocapnia completely restored MCAv and PCAv (Fig. 3) to pre-heat stress values, independent of changes in skin and core temperature, heart rate, cardiac output, stroke volume or arterial blood pressure (Table 2). In support of this finding, both MCAv and PCAv were strongly related to end-tidal
(−14.6 ± 3.4 mmHg), MCAv (−14.7 ± 2.2 cm s−1), PCAv (−9.7 ± 1.2 cm s−1), CVCM (0.19 ± 0.03 cm s−1 mmHg−1), CVCP (0.12 ± 0.03 cm s−1 mmHg−1) and left ventricular end-diastolic volume (−25.9 ± 8.9 ml), while stroke volume and arterial blood pressure remained unchanged. Severe heat stress also resulted in an estimated sweat loss of 1.6 ± 0.3 l. Interestingly, isocapnia completely restored MCAv and PCAv (Fig. 3) to pre-heat stress values, independent of changes in skin and core temperature, heart rate, cardiac output, stroke volume or arterial blood pressure (Table 2). In support of this finding, both MCAv and PCAv were strongly related to end-tidal 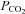 (r2 = 0.97 and r2 = 0.97, respectively) when analysed over a wide range of body temperatures (Fig. 4). Head-down tilt combined with severe heat stress increased end-diastolic volume (+9.3 ± 3.9 ml), while cardiac output, skin and core temperature, heart rate and ventilation remained significantly elevated above normothermic baseline; stroke volume and arterial blood pressure remained unchanged, and MCAv, PCAv and end-tidal
(r2 = 0.97 and r2 = 0.97, respectively) when analysed over a wide range of body temperatures (Fig. 4). Head-down tilt combined with severe heat stress increased end-diastolic volume (+9.3 ± 3.9 ml), while cardiac output, skin and core temperature, heart rate and ventilation remained significantly elevated above normothermic baseline; stroke volume and arterial blood pressure remained unchanged, and MCAv, PCAv and end-tidal 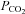 remained significantly depressed (Table 2). Head-down tilt combined with isocapnia, however, completely restored MCAv and PCAv to pre-heat stress values (Table 2). Unlike mild heat stress, during severe heat stress end-diastolic volume and stroke volume were not found to be related to MCAv or PCAv (P = 0.123–0.195).
remained significantly depressed (Table 2). Head-down tilt combined with isocapnia, however, completely restored MCAv and PCAv to pre-heat stress values (Table 2). Unlike mild heat stress, during severe heat stress end-diastolic volume and stroke volume were not found to be related to MCAv or PCAv (P = 0.123–0.195).
Table 2.
Thermal, haemodynamic and respiratory variables during normothermic baseline, severe heat stress, and during severe heat stress combined with head-down tilt and end-tidal 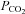 clamping
clamping
| Baseline | Heat stress | Heat stress + 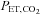 clamping clamping |
Head-down tilt | Head-down tilt + 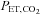 clamping clamping |
|
|---|---|---|---|---|---|
| Thermal | |||||
| Skin temp. (°C) | 33.3 ± 0.2 | 39.1 ± 0.1* | 38.9 ± 0.1* | 38.8 ± 0.2* | 38.8 ± 0.2* |
| Core temp. (°C) | 36.9 ± 0.05 | 38.7 ± 0.05* | 38.8 ± 0.02* | 38.9 ± 0.05* | 38.9 ± 0.05* |
| Haemodynamics | |||||
| Heart rate (beats min−1) | 62 ± 4 | 106 ± 3* | 106 ± 3* | 99 ± 3* | 102 ± 2* |
| Mean arterial pressure (mmHg) | 72 ± 2 | 70 ± 3 | 73 ± 2 | 71 ± 2 | 76 ± 3 |
| End-diastolic volume (ml) | 154.5 ± 8.2 | 128.5 ± 4.8* | — | 137.9 ± 6.5 | — |
| Stroke volume (ml) | 73.1 ± 8.6 | 67.1 ± 5.1 | — | 66.1 ± 4.2 | — |
| Cardiac output (l min−1) | 4.4 ± 0.3 | 7.0 ± 0.4* | — | 6.5 ± 0.4* | — |
| Respiratory and cerebrovascular | |||||
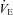 (l min−1) (l min−1) |
9.7 ± 1.1 | 14.7 ± 2.3* | 17.1 ± 2.2* | 14.4 ± 1.3* | 18.3 ± 2.3* |
| VT (l) | 0.8 ± 0.1 | 1.2 ± 0.2 | 1.4 ± 0.2* | 1.1 ± 0.2 | 1.5 ± 0.2* |
| RR (breaths min−1) | 12 ± 1 | 13 ± 1 | 12 ± 1 | 15 ± 1 | 13 ± 2 |
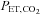 (mmHg) (mmHg) |
39.5 ± 3.0 | 24.9 ± 4.0* | 40.0 ± 2.7 | 25.9 ± 3.6* | 39.7 ± 2.7 |
| MCAv (cm s−1) | 57.5 ± 2.8 | 42.8 ± 3.0* | 52.7 ± 2.5 | 43.8 ± 2.6* | 55.5 ± 2.9 |
| CVCM (cm s−1 mmHg−1) | 0.80 ± 0.03 | 0.61 ± 0.04* | 0.73 ± 0.03 | 0.62 ± 0.04* | 0.74 ± 0.05 |
| PCAv (cm s−1) | 43.1 ± 2.2 | 33.4 ± 2.0* | 41.6 ± 2.1 | 34.3 ± 2.3* | 44.0 ± 3.0 |
| CVCP (cm s−1 mmHg−1) | 0.60 ± 0.03 | 0.48 ± 0.03* | 0.58 ± 0.04 | 0.48 ± 0.03* | 0.59 ± 0.05 |
Date reported as mean ± SEM. n = 8. 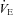 , ventilation; VT, tidal volume, RR, respiratory rate,
, ventilation; VT, tidal volume, RR, respiratory rate, 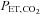 , end-tidal
, end-tidal 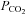 ; MCAv, middle cerebral artery mean blood velocity; PCAv, posterial cerebral artery mean blood velocity; CVC, cerebral vascular conductance.
; MCAv, middle cerebral artery mean blood velocity; PCAv, posterial cerebral artery mean blood velocity; CVC, cerebral vascular conductance.
indicates a significant difference from baseline.
Figure 3. Middle cerebral artery blood flow velocity (MCAv; A) and posterior cerebral blood flow velocity (PCAv; B) during normothermic baseline, severe heat stress and severe heat stress with end-tidal 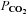 clamping.
clamping.
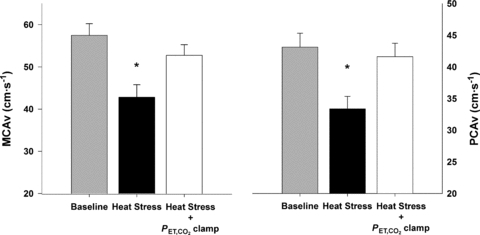
Data reported as mean ± SEM. * indicates a significant difference from baseline. Severe heat stress significantly reduced MCAv and PCAv from baseline (P < 0.001, for both), while isocapnia restored MCAv and PCAv to baseline levels (P = 0.183 and 0.948, respectively). Poikilocapnic head-down tilt did not alter MCAv or PCAv from severe heat stress (P = 0.989 and 0.991, respectively), remaining significantly reduced relative to baseline (P < 0.001, for both). Isocapnic head-down tilt restored MCAv and PCAv to baseline values (P = 0.992 and 0.882, respectively) n = 8, protocol II.
Figure 4. Relationship between the group average middle cerebral artery blood flow velocity (MCAv) and posterior cerebral artery blood flow velocity (PCAv) and the group average end-tidal  over a wide range of body temperatures (from right to left: normothermic baseline, +1.0°C above baseline, +1.5°C above baseline and +1.8°C above baseline).
over a wide range of body temperatures (from right to left: normothermic baseline, +1.0°C above baseline, +1.5°C above baseline and +1.8°C above baseline).
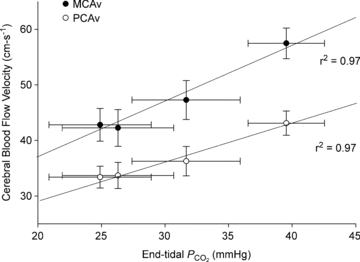
Both relationships were significant (P < 0.05). n = 8, protocol II.
Discussion
This investigation produced two novel findings: First, stroke volume and left ventricular end-diastolic volume, but not cardiac output, were strongly related to changes in cerebral perfusion. Moreover, changes in venous return partly accounted for the variability in cerebral hypoperfusion associated with mild heat stress. Second, restoration of end-tidal  during severe heat stress (1.8°C above baseline) restored global CBF (MCAv and PCAv) to normothermic levels. Collectively, these findings indicate that alterations in venous return and stroke volume are important determinants of CBF during mild heat stress, while during severe heat stress, hyperthermic-induced hyperventilation and related hypocapnia has a much greater influence on CBF than previously reported (Brothers et al. 2009).
during severe heat stress (1.8°C above baseline) restored global CBF (MCAv and PCAv) to normothermic levels. Collectively, these findings indicate that alterations in venous return and stroke volume are important determinants of CBF during mild heat stress, while during severe heat stress, hyperthermic-induced hyperventilation and related hypocapnia has a much greater influence on CBF than previously reported (Brothers et al. 2009).
Heat stress and CBF
It has long been established that CBF, as indexed by MCAv and cerebral vascular conductance, is significantly reduced during passive heat stress (Wilson et al. 2002, 2006; Fan et al. 2008; Low et al. 2008; Brothers et al. 2009). Because of heat-induced hyperventilation, 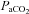 (Crandall et al. 2008) and end-tidal
(Crandall et al. 2008) and end-tidal 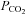 (Fan et al. 2008; Brothers et al. 2011) are also reduced during passive heating. Given the close linear relationship between changes in end-tidal CO2 and CBF (Ide et al. 2003), it follows that changes in CBF during heat stress are at least partially due to reduced
(Fan et al. 2008; Brothers et al. 2011) are also reduced during passive heating. Given the close linear relationship between changes in end-tidal CO2 and CBF (Ide et al. 2003), it follows that changes in CBF during heat stress are at least partially due to reduced 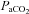 (as assessed by end-tidal
(as assessed by end-tidal 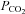 ). In support of this rational, Brothers et al. (2009) recently found that restoration of end-tidal
). In support of this rational, Brothers et al. (2009) recently found that restoration of end-tidal 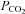 during passive heat stress (+1.3°C above baseline) partially restored CBF to normothermic levels, accounting for approximately 50% of the reduction in MCAv. On the basis of this finding, combined with a related report (Fan et al. 2008), we reasoned that other factors such as re-distribution of cardiac output (measured) and/or sympathetic cerebral vasoconstriction (speculated in Brothers et al. 2009) would account for some of the unexplained differences. These aspects, and considerations of the related divergent findings, are now considered.
during passive heat stress (+1.3°C above baseline) partially restored CBF to normothermic levels, accounting for approximately 50% of the reduction in MCAv. On the basis of this finding, combined with a related report (Fan et al. 2008), we reasoned that other factors such as re-distribution of cardiac output (measured) and/or sympathetic cerebral vasoconstriction (speculated in Brothers et al. 2009) would account for some of the unexplained differences. These aspects, and considerations of the related divergent findings, are now considered.
Mild-heat stress – relationship to venous return
In addition to 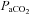 , cerebral perfusion also depends on the arterial perfusion pressure (Lucas et al. 2010). Indeed, cardiac output supports cerebral perfusion at rest and during exercise (Ide et al. 1998), independent of cerebral autoregulation (Ogoh et al. 2005). Our findings suggest that in addition to end-tidal
, cerebral perfusion also depends on the arterial perfusion pressure (Lucas et al. 2010). Indeed, cardiac output supports cerebral perfusion at rest and during exercise (Ide et al. 1998), independent of cerebral autoregulation (Ogoh et al. 2005). Our findings suggest that in addition to end-tidal 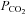 , venous return and stroke volume are strongly related to changes in CBF during passive heat stress. We originally hypothesized that changes in the distribution of cardiac output would account for some of the reduction in CBF. To our surprise, however, cardiac output was not related to CBF in the present investigation. It is important to note, however, that the present experimental conditions (i.e. passive tilt table positioning with and without heat stress) did not produce a wide range of cardiac outputs. Rather, despite significant alterations in venous return, cardiac output was compensated for by changes in heart rate, therefore producing only two distinct data points. Interestingly, the relationship between stroke volume and end-diastolic volume was much stronger (Fig. 2). Considering that stroke volume is the only variable in the heart rate–stroke volume–total peripheral resistance relation that is directly affected by gravity (Levine et al. 2002), and because stroke volume shares a direct, linear function with central blood volume and left ventricular end-diastolic volume (Levine et al. 2002), it is not surprising that changes in stroke volume and end-diastolic volume provide a better relationship with CBF than global measures like cardiac output or arterial pressure (Levine et al. 2002). Others (Levine et al. 2002; Charkoudian et al. 2005; Fu et al. 2005) have also used stroke volume to assess the relationship between cardiac function and muscle sympathetic nerve activity, as stroke volume is known to play a more significant role in baroreflex control than cardiac output per se. We therefore believe that the strong linear relationships between stroke volume/end-diastolic volume and CBF (MCAv and PCAv), found in the present study, provide novel insight into the regulation of CBF in heat-stressed humans.
, venous return and stroke volume are strongly related to changes in CBF during passive heat stress. We originally hypothesized that changes in the distribution of cardiac output would account for some of the reduction in CBF. To our surprise, however, cardiac output was not related to CBF in the present investigation. It is important to note, however, that the present experimental conditions (i.e. passive tilt table positioning with and without heat stress) did not produce a wide range of cardiac outputs. Rather, despite significant alterations in venous return, cardiac output was compensated for by changes in heart rate, therefore producing only two distinct data points. Interestingly, the relationship between stroke volume and end-diastolic volume was much stronger (Fig. 2). Considering that stroke volume is the only variable in the heart rate–stroke volume–total peripheral resistance relation that is directly affected by gravity (Levine et al. 2002), and because stroke volume shares a direct, linear function with central blood volume and left ventricular end-diastolic volume (Levine et al. 2002), it is not surprising that changes in stroke volume and end-diastolic volume provide a better relationship with CBF than global measures like cardiac output or arterial pressure (Levine et al. 2002). Others (Levine et al. 2002; Charkoudian et al. 2005; Fu et al. 2005) have also used stroke volume to assess the relationship between cardiac function and muscle sympathetic nerve activity, as stroke volume is known to play a more significant role in baroreflex control than cardiac output per se. We therefore believe that the strong linear relationships between stroke volume/end-diastolic volume and CBF (MCAv and PCAv), found in the present study, provide novel insight into the regulation of CBF in heat-stressed humans.
Severe heat stress – arterial 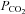 accounts for heat-induced reductions in CBF
accounts for heat-induced reductions in CBF
Our findings that restoration of end-tidal 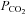 fully accounts for the reduction in CBF with severe heat stress, is in contrast to the aforementioned study (Brothers et al. 2009). Our findings clearly suggest that hyperthermia-induced hyperventilation and associated hypocapnia, is a primary mechanism for reduced CBF during severe passive heat stress (Fig. 3). We extend previous work by providing a global measure of CBF during passive heat stress, by including the PCA (which supplies the occipital cortex). To our knowledge, this is the first study to include an index of CBF during passive heat stress outside of the MCA. In support of our findings, Fan et al. (2008) reported a strong linear relationship (r2 = 0.98) between end-tidal
fully accounts for the reduction in CBF with severe heat stress, is in contrast to the aforementioned study (Brothers et al. 2009). Our findings clearly suggest that hyperthermia-induced hyperventilation and associated hypocapnia, is a primary mechanism for reduced CBF during severe passive heat stress (Fig. 3). We extend previous work by providing a global measure of CBF during passive heat stress, by including the PCA (which supplies the occipital cortex). To our knowledge, this is the first study to include an index of CBF during passive heat stress outside of the MCA. In support of our findings, Fan et al. (2008) reported a strong linear relationship (r2 = 0.98) between end-tidal 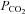 and MCAv. We confirm and extend this finding by showing a similar relationship (Fig. 4) between MCAv and end-tidal
and MCAv. We confirm and extend this finding by showing a similar relationship (Fig. 4) between MCAv and end-tidal 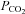 (r2 = 0.97, P = 0.015), and PCAv and end-tidal
(r2 = 0.97, P = 0.015), and PCAv and end-tidal 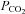 (r2 = 0.97; P = 0.014) under a similar range of core temperatures and end-tidal
(r2 = 0.97; P = 0.014) under a similar range of core temperatures and end-tidal 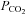 . The discrepancy between these studies is unclear; however, differences in methodological design (e.g. 1.3 vs. 1.8°C increase in core temperature) and severity of hypocapnia (∼6 mmHg vs. 14 mmHg reduction in end-tidal
. The discrepancy between these studies is unclear; however, differences in methodological design (e.g. 1.3 vs. 1.8°C increase in core temperature) and severity of hypocapnia (∼6 mmHg vs. 14 mmHg reduction in end-tidal 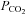 ) may be related. Regardless of the methodological differences, our findings do not support the notion of a major influence of ‘other’ factors (e.g. elevated SNA or cardiac output redistribution) influencing CBF during severe passive heat stress. We acknowledge, however, that heightened tachycardia (reduced diastolic filling time) and greater levels of fluid loss (reduced venous return) may have contributed to the disappearance of a relationship between end-diastolic volume and stroke volume with CBF during severe heat stress. It is also possible that redundant mechanisms, or the magnitude of hypocapnia (possibly overwhelming the system), contributed to the present results.
) may be related. Regardless of the methodological differences, our findings do not support the notion of a major influence of ‘other’ factors (e.g. elevated SNA or cardiac output redistribution) influencing CBF during severe passive heat stress. We acknowledge, however, that heightened tachycardia (reduced diastolic filling time) and greater levels of fluid loss (reduced venous return) may have contributed to the disappearance of a relationship between end-diastolic volume and stroke volume with CBF during severe heat stress. It is also possible that redundant mechanisms, or the magnitude of hypocapnia (possibly overwhelming the system), contributed to the present results.
Limitations
Blood velocity in the MCA and PCA were used as indices of CBF. This methodology is limited to the assumption that cerebral vessel diameter does not change with posture or with passive heat stress. We recognize that changes in diameter will significantly alter blood flow (radius to the fourth power); however, vessel diameter has been directly measured in humans, under a wide range of mean arterial pressures and end-tidal 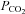 , with diameter remaining relatively stable (Giller et al. 1993; Valdueza et al. 1997; Serrador et al. 2000). Furthermore, changes in MCAv during related changes in
, with diameter remaining relatively stable (Giller et al. 1993; Valdueza et al. 1997; Serrador et al. 2000). Furthermore, changes in MCAv during related changes in 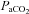 parallel changes in global CBF, as estimated directly based on the Fick principle (reviewed in Ainslie & Duffin, 2009). Thus, it is likely that changes in cerebral artery blood velocity in the present study reflect changes in CBF.
parallel changes in global CBF, as estimated directly based on the Fick principle (reviewed in Ainslie & Duffin, 2009). Thus, it is likely that changes in cerebral artery blood velocity in the present study reflect changes in CBF.
Our findings are based on the assumption that end-tidal 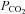 reflects arterial
reflects arterial 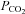 and that the cerebrovascular responsiveness to CO2 is not altered with passive heat stress. In support of the former, estimation of arterial
and that the cerebrovascular responsiveness to CO2 is not altered with passive heat stress. In support of the former, estimation of arterial 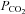 using end-tidal
using end-tidal 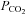 has recently been validated under a relatively wide range of core temperatures and orthostatic stress (Brothers et al. 2011). In support of the latter, we (Fan et al. 2008) and others (Low et al. 2008) have shown that cerebrovascular responsiveness to CO2 is unaltered during passive heat stress.
has recently been validated under a relatively wide range of core temperatures and orthostatic stress (Brothers et al. 2011). In support of the latter, we (Fan et al. 2008) and others (Low et al. 2008) have shown that cerebrovascular responsiveness to CO2 is unaltered during passive heat stress.
Implications
There are a number of implications of our findings for the understanding of the pathophysiology of heat-induced syncope. During mild heat stress, the relationship between venous return and CBF highlights the importance of postural-induced hypotension in the etiology of cerebral hypo-perfusion and related syncope. For example, a steep fall in cardiac output is the main mechanism in the initiation of a vasovagal faint (Verheyden et al. 2008). Since venous return is progressively reduced during heat stress at rest, it seems reasonable to speculate that this is an important mechanism that may not only impact on cardiac output but also CBF (Fig. 2). Although cerebral autoregulation is a critical mechanism that acts to maintain a relatively constant CBF despite changes in blood pressure, it has been previously shown that dynamic cerebral autoregulation is unaltered with mild heat stress (Low et al. 2009). Since MAP was maintained with severe heat stress, we feel that in the conditions of the present study, changes in cerebral autoregulation is not a major factor contributing to our findings. Rather, during more severe heat stress, the marked hypocapnia seems to be a much more important regulator of cerebral hypoperfusion and therefore syncope. Although speculative, validated techniques such as lower body muscle tensing (Krediet et al. 2005) and the regularization of breathing, may represent a simple non-pharmacological means capable of offsetting eminent syncope during heat stress.
Conclusion
This investigation produced two novel findings: first, that changes in CBF appear to be related to stroke volume and end-diastolic volume, suggesting a possible role for carotid and/or pulmonary baroreceptor control over CBF during mild heat stress; and second, that changes in CBF during severe heat stress are primarily related to arterial hypocapnia secondary to hyperthermia-induced hyperventilation.
Acknowledgments
The authors wish to acknowledge the subjects for their participation. We also wish to acknowledge Brad Welch, Timothy Just and Mathew Rieger for their assistance with data collection, as well as Dr Stephen Cheung for the use of his equipment. M.D.N. was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Postgraduate Scholarship), Alberta Scholarship Programs (Ralph Steinhauer Award of Distinction), and the University of Alberta (Andrew Stewart Memorial Graduate Prize). C.K.W. is funded by NSERC of Canada (Doctoral Scholarship). M.K.S. was supported by a Canadian Institutes of Health Research (CIHR) New Investigator Award. P.N.A. is supported by CIHR, NSERC and a Canada Research Chair in Cerebrovascular Physiology.
Glossary
Abbreviations
- CBF
cerebral blood flow
- CVC
cerebral vascular conductance
- HDT
head-down tilt
- HUT
head-up tilt
- MAP
mean arterial blood pressure
- MCA
middle cerebral artery
- PCA
posterior cerebral artery
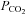
partial pressure of carbon dioxide
Author contributions
Each author was involved in the conception and design of the study, drafting the article and revising it for intellectual content, and has provided final approval of the version of the manuscript to be published.
References
- Ainslie PN, Duffin J. Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: mechanisms of regulation, measurement, and interpretation. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1473–R1495. doi: 10.1152/ajpregu.91008.2008. [DOI] [PubMed] [Google Scholar]
- Allan JR, Crossley RJ. Effect of controlled elevation of body temperature on human tolerance to +G z acceleration. J Appl Physiol. 1972;33:418–420. doi: 10.1152/jappl.1972.33.4.418. [DOI] [PubMed] [Google Scholar]
- Brothers RM, Ganio MS, Hubing KA, Hastings JL, Crandall CG. End-tidal carbon dioxide tension reflects arterial carbon dioxide tension in the heat-stressed human with and without simulated hemorrhage. Am J Physiol Regul Integr Comp Physiol. 2011;300:R978–R983. doi: 10.1152/ajpregu.00784.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothers RM, Wingo JE, Hubing KA, Crandall CG. The effects of reduced end-tidal carbon dioxide tension on cerebral blood flow during heat stress. J Physiol. 2009;587:3921–3927. doi: 10.1113/jphysiol.2009.172023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charkoudian N, Joyner MJ. Physiologic considerations for exercise performance in women. Clin Chest Med. 2004;25:247–255. doi: 10.1016/j.ccm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Charkoudian N, Joyner MJ, Johnson CP, Eisenach JH, Dietz NM, Wallin BG. Balance between cardiac output and sympathetic nerve activity in resting humans: role in arterial pressure regulation. J Physiol. 2005;568:315–321. doi: 10.1113/jphysiol.2005.090076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall CG, Wilson TE, Marving J, Vogelsang TW, Kjaer A, Hesse B, Secher NH. Effects of passive heating on central blood volume and ventricular dimensions in humans. J Physiol. 2008;586:293–301. doi: 10.1113/jphysiol.2007.143057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J-L, Cotter JD, Lucas RAI, Thomas K, Wilson L, Ainslie PN. Human cardiorespiratory and cerebrovascular function during severe passive hyperthermia: effects of mild hypohydration. J Appl Physiol. 2008;105:433–445. doi: 10.1152/japplphysiol.00010.2008. [DOI] [PubMed] [Google Scholar]
- Fu Q, Witkowski S, Okazaki K, Levine BD. Effects of gender and hypovolemia on sympathetic neural responses to orthostatic stress. Am J Physiol Regul Integr Comp Physiol. 2005;289:R109–R116. doi: 10.1152/ajpregu.00013.2005. [DOI] [PubMed] [Google Scholar]
- Giller C, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–741. discussion 741–742. [PubMed] [Google Scholar]
- Ide K, Eliasziw M, Poulin MJ. Relationship between middle cerebral artery blood velocity and end-tidal PCO2 in the hypocapnic-hypercapnic range in humans. J Appl Physiol. 2003;95:129–137. doi: 10.1152/japplphysiol.01186.2002. [DOI] [PubMed] [Google Scholar]
- Ide K, Pott F, Lieshout JJV, Secher NH. Middle cerebral artery blood velocity depends on cardiac output during exercise with a large muscle mass. Acta Physiol Scand. 1998;162:13–20. doi: 10.1046/j.1365-201X.1998.0280f.x. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Niederberger M, Rowell LB, Eisman MM, Brengelmann GL. Competition between cutaneous vasodilator and vasoconstrictor reflexes in man. J Appl Physiol. 1973;35:798–803. doi: 10.1152/jappl.1973.35.6.798. [DOI] [PubMed] [Google Scholar]
- Krediet CTP, de Bruin IGJM, Ganzeboom KS, Linzer M, van Lieshout JJ, Wieling W. Leg crossing, muscle tensing, squatting, and the crash position are effective against vasovagal reactions solely through increases in cardiac output. J Appl Physiol. 2005;99:1697–1703. doi: 10.1152/japplphysiol.01250.2004. [DOI] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Levine BD, Pawelczyk JA, Ertl AC, Cox JF, Zuckerman JH, Diedrich A, et al. Human muscle sympathetic neural and haemodynamic responses to tilt following spaceflight. J Physiol. 2002;538:331–340. doi: 10.1113/jphysiol.2001.012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind AR, Leithead CS, McNicol GW. Cardiovascular changes during syncope induced by tilting men in the heat. J Appl Physiol. 1968;25:268–276. doi: 10.1152/jappl.1968.25.3.268. [DOI] [PubMed] [Google Scholar]
- Low DA, Wingo JE, Keller DM, Davis SL, Cui J, Zhang R, Crandall CG. Dynamic cerebral autoregulation during passive heat stress in humans. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1598–R1605. doi: 10.1152/ajpregu.90900.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low DA, Wingo JE, Keller DM, Davis SL, Zhang R, Crandall CG. Cerebrovascular responsiveness to steady-state changes in end-tidal CO2 during passive heat stress. J Appl Physiol. 2008;104:976–981. doi: 10.1152/japplphysiol.01040.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas SJE, Burgess KR, Thomas KN, Donnelly J, Peebles KC, Lucas RAI, et al. Alterations in cerebral blood flow and cerebrovascular reactivity during 14 days at 5050 m. J Physiol. 2011;589:741–753. doi: 10.1113/jphysiol.2010.192534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas SJE, Tzeng YC, Galvin SD, Thomas KN, Ogoh S, Ainslie PN. Influence of changes in blood pressure on cerebral perfusion and oxygenation. Hypertension. 2010;55:698–705. doi: 10.1161/HYPERTENSIONAHA.109.146290. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Altamirano-Diaz LA, Petersen SR, DeLorey DS, Stickland MK, Thompson RB, Haykowsky MJ. Left ventricular systolic and diastolic function during tilt table positioning and passive heat stress in humans. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00127.2011. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Haykowsky MJ, Petersen SR, DeLorey DS, Cheng-Baron J, Thompson RB. Increased left ventricular twist, untwisting rates, and suction maintain global diastolic function during passive heat stress in humans. Am J Physiol Heart Circ Physiol. 2010a;298:H930–H937. doi: 10.1152/ajpheart.00987.2009. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Haykowsky MJ, Petersen SR, DeLorey DS, Stickland MK, Cheng-Baron J, Thompson RB. Aerobic fitness does not influence the biventricular response to whole-body passive heat stress. J Appl Physiol. 2010b;109:1545–1551. doi: 10.1152/japplphysiol.00769.2010. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Ainslie PN. Cerebral blood flow during exercise: mechanisms of regulation. J Appl Physiol. 2009;107:1370–1380. doi: 10.1152/japplphysiol.00573.2009. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Brothers RM, Barnes Q, Eubank WL, Hawkins MN, Purkayastha S, O-Yurvati A, Raven PB. The effect of changes in cardiac output on middle cerebral artery mean blood velocity at rest and during exercise. J Physiol. 2005;569:697–704. doi: 10.1113/jphysiol.2005.095836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan NL. A new weighting system for mean surface temperature of the human body. J Appl Physiol. 1964;19:531–533. doi: 10.1152/jappl.1964.19.3.531. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Brengelmann GL, Murray JA. Cardiovascular responses to sustained high skin temperature in resting man. J Appl Physiol. 1969;27:673–680. doi: 10.1152/jappl.1969.27.5.673. [DOI] [PubMed] [Google Scholar]
- Sato K, Ogoh S, Hirasawa A, Oue A, Sadamoto T. The distribution of blood flow in the carotid and vertebral arteries during dynamic exercise in humans. J Physiol. 2011;589:2847–2856. doi: 10.1113/jphysiol.2010.204461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Sadamoto T. Different blood flow responses to dynamic exercise between internal carotid and vertebral arteries in women. J Appl Physiol. 2010;109:864–869. doi: 10.1152/japplphysiol.01359.2009. [DOI] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Shvartz E, Strydom NB, Kotze H. Orthostatism and heat acclimation. J Appl Physiol. 1975;39:590–595. doi: 10.1152/jappl.1975.39.4.590. [DOI] [PubMed] [Google Scholar]
- Valdueza J, Balzer J, Villringer A, Vogl T, Kutter R, Einhaupl K. Changes in blood flow velocity and diameter of the middle cerebral artery during hyperventilation: assessment with MR and transcranial Doppler sonography. AJNR Am J Neuroradiol. 1997;18:1929–1934. [PMC free article] [PubMed] [Google Scholar]
- Van Lieshout JJ, Wieling W, Karemaker JM, Secher NH. Syncope, cerebral perfusion, and oxygenation. J Appl Physiol. 2003;94:833–848. doi: 10.1152/japplphysiol.00260.2002. [DOI] [PubMed] [Google Scholar]
- Verheyden B, Liu J, van Dijk N, Westerhof BE, Reybrouck T, Aubert AE, Wieling W. Steep fall in cardiac output is main determinant of hypotension during drug-free and nitroglycerine-induced orthostatic vasovagal syncope. Heart Rhythm. 2008;5:1695–1701. doi: 10.1016/j.hrthm.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Willie CK, Colino FL, Bailey DM, Tzeng YC, Binsted G, Jones LW, et al. Utility of transcranial doppler ultrasound for the integrative assessment of cerebrovascular function. J Neurosci Methods. 2011;196:221–237. doi: 10.1016/j.jneumeth.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Willie CK, Cowan EC, Ainslie PN, Taylor CE, Smith KJ, Sin PYW, Tzeng YC. Neurovascular coupling and distribution of cerebral blood flow during exercise. J Neurosci Methods. 2011;198:270–273. doi: 10.1016/j.jneumeth.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Wilson TE, Cui J, Zhang R, Crandall CG. Heat stress reduces cerebral blood velocity and markedly impairs orthostatic tolerance in humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1443–R1448. doi: 10.1152/ajpregu.00712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Cui J, Zhang R, Witkowski S, Crandall CG. Skin cooling maintains cerebral blood flow velocity and orthostatic tolerance during tilting in heated humans. J Appl Physiol. 2002;93:85–91. doi: 10.1152/japplphysiol.01043.2001. [DOI] [PubMed] [Google Scholar]