

Abstract
Cytotoxic CD8 T lymphocytes (CTLs) play a pivotal role in the control of infection. Activated CTLs, however, often lose effector function during chronic infection. PD-1 receptor and its ligand PD-L1 of the B7/CD28 family function as a T cell coinhibitory pathway and are emerging as major regulators converting effector CTLs into exhausted CTLs during chronic infection with human immunodeficiency virus, hepatitis B virus, hepatitis C virus, and other pathogens capable of establishing chronic infections. Importantly, blockade of the PD-1/PD-L1 pathway is able to restore functional capabilities to exhausted CTLs and early clinical trials have shown promise. Further research will reveal how chronic infection induces upregulation of PD-1 on CTLs and PD-L1 on antigen-presenting cells and other tissue cells and how the PD-1/PD-L1 interaction promotes CTLs exhaustion, which is crucial for developing effective prophylactic and therapeutic vaccination against chronic infections.
1. Introduction
Our immune system is tasked with the formidable challenge of balancing elimination of pathogenic entities and maintaining tolerance to prevent autoimmune disease. T cells are central to preserving this balance, and their proper regulation, primarily coordinated by the B7/CD28 family of molecules, is of utmost importance. Optimal T cell activation requires two signals. The first is an antigen-specific signal generated by T cell receptor (TCR) recognition of peptide-MHC presented by an antigen-presenting cell (APC), while the second, antigen-independent, signal is supplied by binding of one of the classic B7 family members, B7-1 (CD80) or B7-2 (CD86) to CD28 on the T cell surface. It is imperative that both signals are received, as antigen recognition by the TCR without the costimulatory signal can render T cells unresponsive or anergic. The B7/CD28 family has several additional members, which are capable of either costimulating (positive) or coinhibiting (negative) T cells depending on which receptor is ligated [1]. Integration of such positive and negative signals ultimately determines the outcome of T cell activation and subsequent effector phase during response to an invading pathogen.
Chronic viral infections, such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV), are a significant burden on global public health and present a unique and significant challenge to developing vaccination strategies. The role of CTLs in controlling viral infection is one of cardinal significance. Upon recognition of viral antigen, as presented by APC, naive T cells expand and differentiate into effector cells, dispersing throughout the body specifically recognizing and eliminating the foreign threat by targeting virus-infected cells. Following viral clearance effector cells contract, however, a select number remain as memory T cells that are capable of rapid expansion and acquisition of effector functions upon re-exposure to the same virus. The goal of both prophylactic and therapeutic vaccinations is to elicit this type of response to either prevent or treat viral infection, respectively. However, the persistent antigen stimulation during chronic viral infections often renders CTLs “exhausted,” a state of dysfunction defined by the progressive loss of key components of effector function. Recent studies have revealed that programmed death-1 (PD-1), an inhibitory member of the B7-CD28 family, is a master regulator of CTL exhaustion (Figure 1).
Figure 1.
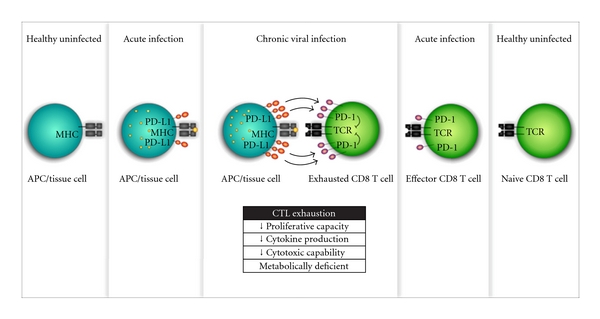
PD-1/PD-L1-mediated cytotoxic T lymphocyte exhaustion during chronic viral infection. During chronic viral infection, the persistent presentation of antigen causes CD8 T cells to highly upregulate PD-1, a T cell coinhibitory receptor. PD-L1, the ligand for PD-1, is also upregulated on APC or resident tissue cells during chronic viral infection. This severe overrepresentation of the inhibitory PD-1/PD-L1 pathway is a major cause of exhaustion in CD8 T cells. Exhausted CD8 T cells are functionally deficient and have decreased proliferative capacity, cytokine production, and cytotoxic capacity and are metabolically deficient. Exhausted CD8 T cells are ineffective at clearing virus and, in turn, the chronic infection persists.
2. The PD-1 Receptor and Its PD-L1 Ligand as a T Cell Coinhibitory Pathway
PD-1 was discovered almost two decades ago on a T cell hybridoma line undergoing activation-induced programmed cell death, hence the name [2]. This original observation led to the hypothesis that PD-1 may function as a cell death inducer, but the expression seen was more likely due to T cell activation, as it is now well established that PD-1 is upregulated on activated T cells, B cells, and monocytes [3–5]. The mechanism of PD-1 expression is not yet well explored, but two different transcription factors, NFATc1 and T-bet, have been implicated as positive and negative regulators of PD-1 expression, respectively [6, 7]. Both transcription factors were found to associate with regulatory elements in the PD-1 gene. Using a T cell line that constitutively expresses PD-1, it was found that blocking NFATc1 by cyclosporine A resulted in a significant decline in PD-1 [6]. Conversely, overexpression of T-bet was associated with a decline in PD-1 as well as other inhibitory receptors [7].
PD-1 has two identified ligands, PD-L1 (B7-H1, CD274) [5, 8] and PD-L2 (B7-DC, CD273) [9, 10], and both are members of the B7 family. While PD-L2 expression tends to be more restricted, found primarily on activated professional APCs [9, 10], PD-L1 has a much more promiscuous expression profile. PD-L1 is expressed on cells of hematopoietic lineage, including activated T cells, B cells, monocytes [5, 8], dendritic cells [11], and macrophages [12]. However, it is also extensively expressed in peripheral nonlymphoid tissues, with intermediate to high expression detected in heart, skeletal muscle, placenta, lung, kidney, and liver [5, 8]. Interferon gamma (IFN-γ) is known to be a major regulator of PD-L1 expression for a wide range of cell types. A study looking at cancer-associated PD-L1 upregulation found that IFN-γ induced the expression of the transcription factor interferon regulatory factor 1 (IRF-1), which binds to regulatory sites in the PD-L1 gene and is largely responsible for the observed increase in PD-L1 [13]. PD-L1 can also be upregulated on macrophages in a TLR-4-dependent manner by LPS [12], which is reliant on Stat-1 signaling [14].
Binding of PD-L1 and PD-L2 to their PD-1 receptor negatively regulates T cells, causing decreased proliferation and production of effector cytokines, such as IL-2 and IFN-γ [5, 11]. Complementary to this, blocking of the PD-1/PD-L pathways results in enhanced proliferative capacity of T cells [15]. While both PD-L1 and PD-L2 have short cytoplasmic tails without known signaling motifs, the cytoplasmic domain of PD-1 has two structural motifs [4, 16–19]. Proximal to the cell membrane (N-terminal) is an immunoreceptor tyrosine-based inhibitory motif (ITIM) [4] and distal to the cell membrane (C-terminal) is an immunoreceptor tyrosine-based switch motif (ITSM) [17]. PD-1 ligation, along with TCR signaling, results in phosphorylation of the cytoplasmic domain tyrosines [16] and recruitment of SHP-2, a Src homology 2-containing tyrosine phosphatase, to the C-terminal tyrosine in the ITSM [16–18]. SHP-2 then dephosphorylates TCR-associated CD-3ζ and ZAP70 [18], resulting in inhibition of downstream signaling. Specifically, activation of phosphatidylinositol 3-kinase (PI3K) is blocked, which affects upregulation of the cell survival gene Bcl-XL and decreases IL-2 production and glucose metabolism [19]. The ITSM, not the ITIM, is the critical motif for this signaling in both T cells [17] and B cells [16]. It has been suggested that PD-L1 also mediates an inhibitory signal by ligating B7-1 [20], although with the short cytoplasmic tails characteristic of B7 family members it is not clear how this signal may be mediated.
The inhibitory signal generated by PD-1 ligation with its ligand PD-L1 along with the unique expression pattern of PD-L1, both on hematopoietic cells and in nonlymphoid tissues, suggests that this pathway may have a significant role in regulating peripheral tolerance. In vivo data supports this hypothesis as PD-1-deficient mice develop spontaneous autoimmune disease. Aged C57BL/6 mice lacking PD-1 develop a lupus-like glomerulonephritis and destructive arthritis [21], while deficient BALB/c mice develop fatal dilated cardiomyopathy, dying as young as five weeks old [22]. Additionally, in a mouse model of adenovirus-induced liver injury PD-1-deficient mice cleared infection faster but died from severe liver-specific immunopathology [23]. Along with loss of T cell coinhibition, another mechanism proposed to explain this breakdown of tolerance involves stop signals induced by TCR ligation. Such signals inhibit T cell mobility, sustaining T-cell-APC interaction for formation of the immunological synapse. It was found that the PD-1/PD-L1 interaction constrains these signals in a model where blocking of the pathway lowered T cell mobility and enhanced T-cell-APC contacts [24]. Another mechanism highlights that PD-L1 promotes CD4 T cell conversion to induced T regulatory cells (iTreg), which are capable of suppressing the response of effector T cells [25].
The PD-1/PD-L1 pathway regulates T cell functional capabilities by inhibiting proliferation and production of associated effector cytokines. In concert with the wide expression of PD-L1 in peripheral nonlymphoid tissues, the negative regulatory function of this pathway makes it important for preventing T-cell-mediated autoimmunity and immunopathology. However, this type of protective regulation can be usurped by pathogens, allowing them to escape immune recognition and establish a chronic infection. Overwhelming evidence shows that this does occur, particularly for chronic viral infections where CTL overexpression of PD-1 renders T cells exhausted.
3. PD-1/PD-L1-Mediated CTL Exhaustion in Chronic LCMV Mouse Model
During acute infection, activated pathogen-specific CTLs proliferate and acquire effector functions, such as cytokine production and cytotoxic capability, which enable them to effectively clear infection. Following clearance, a small pool of pathogen-specific memory T cells remain that have the ability to very rapidly reactivate and acquire their killing functions following re-exposure to the same pathogen. However, during chronic viral infection this does not occur, as pathogen-specific CTLs are found to be functionally deficient and unable to eliminate infection. These exhausted CTLs are defined by their impaired proliferative capacity, cytokine production and loss of cytotoxic capabilities. [26, 27]. Additionally, as compared to normal T cells, exhausted CTLs have metabolic deficiencies and altered expression of genes involved in chemotaxis, adhesion, and migration [28]. This phenomenon was originally defined using a well-established mouse model of chronic viral infection in mice, lymphocytic choriomeningitis virus (LCMV) [26, 27]. The Armstrong strain of LCMV causes an acute infection that is cleared by the immune system, generating a robust CTL memory. On the other hand, the Clone 13 strain of LCMV establishes a chronic infection in mice that renders CTLs exhausted and unable to clear infection.
In a study conducted to reveal mechanisms that lead to exhaustion, the genetic profile of exhausted CTLs from a chronic LMCV infection was compared to that of functional CTLs responding to an acute LCMV infection [29]. The novel finding was that exhausted CTLs have significant overexpression of PD-1, whereas the functional LCMV-specific CTLs had no appreciable expression of PD-1. Expression of PD-1 was found to correlate with the defined functional impairment seen in exhausted T cells and, in turn, higher viral loads. Blocking the PD-1/PD-L1 pathway, with an anti-PD-L1 antibody, in chronically infected mice resulted in enhanced CTL response that caused a decrease in viral loads. PD-1 expression by exhausted CTLs is dependent on persisting antigen-specific stimulation, as loss of presentation of specific epitope during chronic infection leads to functional restoration and decreased PD-1 expression on epitope-specific CTLs [30]. Persistent antigen stimulation during chronic viral infection has a progressive effect on loss of CTL function and correlated increase in PD-1 expression, meaning that more exhausted CTLs (PD-1hi) are less susceptible to functional rescue by PD-1 blocking than others (PD-1int) [31]. Further research will help to reveal how persistent antigen stimulation upregulates PD-1 and PD-L1 expression.
4. PD-1/PD-L1-Mediated CTL Exhaustion in HIV Infection
The reversal of CTL exhaustion by PD-1/PD-L1 blockade in murine chronic LCMV infection, even in absence of CD4 T cell help [29], bade exciting news for HIV research. HIV represents a huge burden on both global health and economics. In 2009, there was a reported 33.3 million people living with HIV and 2.6 million new infections that same year [32]. HIV is characterized by a persisting infection that targets and significantly depletes CD4 T cells, which are important helpers for generation of an optimal CTL response. There has been limited success in development of either a prophylactic or a therapeutic vaccine to combat HIV. Vaccine design faces the significant problem of overcoming the limited functional capacity of CTLs in chronic HIV infection, as these cells are essential for viral control. As with the murine LCMV model, PD-1 was found to be upregulated on virus-specific CTLs from HIV-infected patients [33, 34]. In vitro experiments demonstrated that HIV-specific PD-1-expressing CTLs were exhausted, having decreased proliferative capacity and secretion of effector cytokines, but were capable of functional rescue by blockade of the PD-1/PD-L1 pathway [33, 34]. Interestingly, both patients receiving highly active antiretroviral therapy (HAART) [34] and patients naturally maintaining low viral replication, called long-term nonprogressors (LTNP) [35], had lower levels of PD-1 expression on their CTLs. However, when HAART therapy was discontinued HIV-specific T cells reacquired their elevated levels of PD-1 expression [34]. PD-1 negatively regulates T cells by abrogating positive TCR and costimulatory signaling, which is a likely way the PD-1/PD-L1 pathway impairs CTL function. PD-1 is capable of upregulating a unique set of genes in exhausted CTLs, which includes the AP-1 family transcription factor basic leucine transcription factor (BATF) [36]. Overexpression of BATF resulted in an exhaustion-like T cell dysfunction, while silencing of the BATF gene was able to rescue function in impaired HIV-specific CTLs [36]. Simian immunodeficiency virus (SIV), a nonhuman primate model of HIV, similarly has functionally deficient CTLs during chronic infection. PD-1 expression increased on SIV-specific T cells over time, with higher expression on T cells from lymph nodes or genital mucosa, which are the major sites of viral replication, as compared to those from peripheral blood [37]. High expression of PD-1 on SIV-specific CTLs correlated with the decreased proliferative capacity characteristic of exhausted T cells, which was reversible with blockade of the PD-1/PD-L1 pathway [37, 38].
Virus-specific CD4 T cells from HIV-infected patients also show higher PD-1 expression that correlates with a rescuable exhaustion phenotype [33]. HIV-specific CD4 T cells from the lymph nodes were found to have higher expression of PD-1 than those from the blood [39]. This illustrates that continued antigen recognition is an important causation of increased PD-1 expression and resulting exhaustion. Like HIV-specific CTLs, CD4 T cells from patients receiving HAART had reduced PD-1 expression [39]. Monocytes from HIV-infected patients also show elevated levels of PD-1 expression, which correlates with higher levels of IL-10, a classic anti-inflammatory cytokine, and inhibition of CD4 T cell function [40]. Even before the discovery that PD-1 is expressed on exhausted T cells, the importance of the PD-1/PD-L1 pathway in chronic infections was hinted at by the observation that PD-L1 is upregulated on monocytes and B cells in HIV-infected patients [41]. Higher PD-L1 expression is also seen in dendritic dells from HIV patients, as compared to those from patients receiving HAART or from LTNP [42]. In the same study, ligation of dendritic cell-expressed PD-L1 with PD-1 resulted in decreased maturation and increased apoptosis [42]. In vitro exposure of human monocyte-derived macrophages to replication competent HIV also resulted in an increase of PD-L1 expression [43]. These data show that HIV is a master manipulator of the immune response, usurping the immunoregulatory functions of the PD-1/PD-L1 pathway, on both T cells and APCs, to evade recognition and establish a chronic infection. It appears that the current HAART regimen is capable of some reduction in T cell exhaustion and perhaps clinical application of PD-1/PD-L1 blockade, shown to reanimate exhausted T cells, could enhance this even further.
5. The PD-1/PD-L1 Pathway in Other Chronic Infections
PD-1/PD-L1-pathway-mediated CTL exhaustion is also important for other clinically relevant chronic infections. Hepatitis B virus (HBV) and hepatitis C virus (HCV) are two of the most prevalent chronic viral infections in the United States, with about 1-2% of the population afflicted [44]. Annually there are about 15,000 deaths in the USA from liver diseases caused by chronic infection with HBV or HCV [44]. As with HIV, chronic viral hepatitis causes virus-specific CTLs to become exhausted, allowing virus to escape immune recognition and establish a persistent infection [45, 46]. HBV-specific CTLs from chronically infected patients expressed PD-1, which correlated with exhaustion that was reversed by blocking the PD-1/PD-L1 pathway [47, 48]. Additionally, in comparison to healthy subjects, chronic HBV patients had elevated PD-L1 expression on peripheral blood mononuclear cells [48], specifically dendritic cells [49]. In vitro, these PD-L1hi dendritic cells from chronic HBV patients rendered healthy CTLs and CD4 T cells hyporesponsive [49]. CTLs from patients with chronic HCV infection also have high expression of PD-1 that is associated with an exhausted phenotype [50]. A study following HCV-infected patients throughout the course of their disease found that patients with higher PD-1 expression on T cells during acute infection tended to progress to having a chronic infection [51]. The liver is the major site of infection for HCV, and intrahepatic virus-specific CTLs from chronic patients have higher PD-1 expression [52] and are less susceptible to functional restoration by PD-1/PD-L1 blockade [53], as compared to circulating virus-specific CTLs.
PD-1/PD-L1-mediated T cell exhaustion is most well defined for LCMV, HIV, HBV, and HCV; however, there has been some investigation into this pathway in additional pathogens, including other viruses, bacteria, parasites, and even helminthes. Herpes simplex virus 1 (HSV-1) is a persistent virus that establishes a latent infection with viral reservoirs in neural ganglia [54]. It was found that higher corneal scarring, associated with reactivated HSV-1 infection in a murine model, correlated with higher PD-1 mRNA expression in the cornea and ganglia, suggesting a possible role for PD-1-mediated T cell exhaustion [54]. This is supported by a subsequent study where a higher level of latency, and in turn corneal scarring, correlated with higher numbers of PD-1-expressing CTLs and CD4 T cells [55]. Additionally, mice lacking either PD-1 or PD-L1, but not PD-L2, had reduced HSV-1 latency [56]. HSV-1-specific CTLs activated without the help of CD4 T cells have high PD-1 expression and characteristics of exhaustion that are reversible with blockade of the PD-1/PD-L1 pathway [57]. Friend virus, which establishes a chronic infection and ultimately causes leukemia in mice, also has PD-1-expressing exhausted CTLs that can be rescued by PD-1/PD-L1 blockade, leading to virus control [58]. Helicobacter pylori causes a chronic gastric infection that is not cleared and eventually can cause gastritis, ulcers, and gastric cancer [59, 60]. Following H. pylori chronic infection, PD-L1 was upregulated on dendritic cells [60]. This was also true for gastric epithelial cells (GECs) [59], which are resident nonhematopoietic cells shown to be capable of T cell activation. In vitro, PD-L1hi GEC had reduced capacity to induce proliferation in CD4 T cells, which was reversed by blocking PD-L1 [59]. Furthermore, analysis of gastric biopsies from H. pylori-infected patients found that gastric CD4 T cells had higher levels of PD-1 expression, which correlated with density of the pathogen [61]. In chronic Mycobacterium bovis infection in mice, dendritic cells in chronic granulomas had higher PD-L1 expression than those in acute granulomas, suggesting that these cells may facilitate latent tuberculosis infection by decreasing protective T cell responses [62]. Dendritic cells also upregulate PD-L1 expression in chronic Leishmania donovani infection, and in conjunction with the observed increase in CTL PD-1 it is likely that this pathogen manipulates the PD-1/PD-L1 pathway to avoid immune recognition [63]. In vivo blockade of PD-L1 partially rescued CTL function and decreased parasite burden in the spleens of L. donovani-infected mice [63]. Chronic infection with Toxoplasma gondii, an extremely prevalent parasite, caused a gradated increase in PD-1 expression on exhausted CTLs as disease persisted [64]. Blockade of the PD-1/PD-L1 pathway during chronic T. gondii infection rejuvenated the exhausted CTLs, leading to control of parasite reactivation and prevention of mortality in a mouse model [64]. Finally, PD-L1 on macrophages [65], as well as PD-L2 on dendritic cells [66], were found to be upregulated following infection with the helminth Schistosoma mansoni and are associated with chronic morbidity [66].
6. Blockade of the PD-1/PD-L1 Pathway as an Emerging Immunotherapy
Functionally deficient exhausted CTLs are a significant cause and consequence of chronic viral infection. This presents a major challenge to vaccine designs that aim to either prevent or eliminate such infections by mediating T cell response. Whether developing a prophylactic vaccine to prevent disease in healthy individuals or a therapeutic vaccine to clear disease in infected individuals, it is imperative to keep this considerable hurdle in mind. This is aptly illustrated in a chimpanzee model of vaccination against HCV, where although viremia was initially controlled during the acute phase of infection, ultimately the majority of animals developed a persistent disease [67]. Analysis for PD-1 in the liver revealed that expression was significantly higher in the animals that developed a chronic infection despite vaccination [67]. The critical role that the PD-1/PD-L1 pathway plays in development of CTL exhaustion represents an exciting and promising target for defeating this challenge. Following the original observation of the importance of PD-1 in CTL exhaustion, there has been a deluge of studies in chronic viral infection models that show the remarkable potential of manipulating the PD-1/PD-L1 pathway to enhance viral clearance.
Blockade of the PD-1/PD-L1 pathway is able to restore functional capabilities to exhausted CTLs in the LCMV murine model of chronic viral infection [29, 31] and in ex vivo experiments with CTLs from patients with chronic HIV [33–35] or chronic viral hepatitis [47, 50, 52, 53]. Promising results have been seen in combing PD-1/PD-L1 blockade with prophylactic and therapeutic vaccination. In nonhuman primates that were treated with a SIV-specific prophylactic vaccine in conjunction with antibody to block PD-1, it was found that the antibody had an adjuvant-like quality and induced higher numbers of SIV-specific T cells as compared to vaccine alone [68]. In a mouse model of human papilloma virus (HPV), which can cause cancer, prophylactic vaccination against HPV combined with soluble PD-1 to block the PD-1/PD-L1 interaction resulted in enhanced HPV-specific CTL response and a potent antitumor effect [69]. During an established chronic LCMV infection, therapeutic vaccination along with PD-1 blockade reversed CTL exhaustion and enhanced LCMV-specific CTL response and viral clearance [70]. This effect held true even without CD4 T cell help [70], indicating promise for the likely low CD4 T cell counts in HIV-infected individuals.
The PD-1/PD-L1 pathway is essential for maintaining peripheral tolerance and preventing both spontaneous autoimmunity and immunopathology during T cell response to disease. Therefore, blocking of this pathway to combat chronic viral infection could pose a serious risk for developing such problems. This is illustrated in a study where blocking the PD-1/PD-L1 pathway during chronic LCMV infection in mice caused serious immunopathology directed at PD-L1-expressing splenic fibroblastic reticular cells (FRCs) [71]. This immunopathology was CTL mediated, as eliminating this immune population abrogated destruction of FRCs [71]. This concern is somewhat relieved by the results of an in vivo nonhuman primate study and a clinical trial in humans. Blocking PD-1 in nonhuman primates with chronic SIV infection resulted in rapid expansion of previously exhausted SIV-specific CTLs, in both the gut and peripheral blood, and a significant decrease in viral load [72]. This therapy was shown to be safe, and it ultimately decreased progression to AIDS-like symptoms and prolonged survival [72]. In a phase-1 clinical trail, PD-1 blockade, by means of a humanized anti-PD-1 antibody, was tested in patients with advanced hematologic malignancies [73]. The treatment proved to be safe and well tolerated in all patients, ultimately showing clinical benefit in 33% of those in the trial [73]. One study found that PD-1/PD-L1 blockade was able to enhance expansion of pathogen-specific regulatory T cells (Tregs) [74]. This suggests that blockade may be safe because it expands effector cells that, while apt at controlling infection, have the potential to be immunopathogenic, but it also expands Tregs that are capable of abrogating such destruction. Exhausted CTLs express multiple inhibitory receptors, including PD-1, and several studies have illustrated that combining blockade of PD-1 with other inhibitory molecules has a synergistic effect on reversing CTL exhaustion. In murine chronic LCMV infection coblockade of the PD-1/PD-L1 pathway with IL-10 [75] or LAG3 [76] resulted in better functional restoration of CTLs than blocking any receptor alone. Additionally, in vitro blockade of both PD-1 and CTLA-4 on exhausted CTLs from chronic HCV infection resulted in a collective increase in reacquisition of CTL functional capabilities [53]. In nonhuman primate chronic SIV infection, PD-1/PD-L1 blockade therapy was also shown to increase the number of SIV-specific B cells and, in turn, anti-SIV antibodies [72, 77].
Tuberculosis is one of the most common and lethal opportunistic infections in HIV-infected patients. In 2009, it was a leading cause of death among HIV-infected individuals, causing approximately 380,000 deaths in that year alone [32]. Two independent studies infected PD-1 deficient mice with Mycobacterium tuberculosis (TB), expecting to see the enhanced survival seen in other infection models in these mice. Shockingly, TB-infected PD-1-deficient mice were severely susceptible to infection and died significantly faster than their wild-type counterparts [78, 79]. PD-L1-deficient mice were also more susceptible to infection but less so than the PD-1-deficient mice [79]. HIV is a prominent chronic infection being investigated for PD-1/PD-L1 blocking therapies. However, the high prevalence of TB in HIV-positive individuals and the severe mortality seen in TB-infected mice lacking PD-1 could be a serious impediment to therapeutic strategies blocking this pathway to control HIV infection. It is imperative to see if in vivo blockade of the PD-1/PD-L1 pathway has the same lethal effect as the gene knockout mice as it is possible that transient, rather than complete, elimination of these molecules will have less severe effects in TB infection. If transient blockade of the pathway still has such severe consequences, it would be of utmost importance to eliminate the possibility of TB infection in an HIV-positive individual before commencing any therapy involving PD-1 or PD-L1.
7. Conclusion
The PD-1/PD-L1 T cell coinhibitory pathway has emerged as a potent regulator of CTL exhaustion during chronic viral infection. This was established in the murine LCMV model and was expanded to clinically relevant human viral diseases, such as HIV. Studies have shown that blocking the PD-1/PD-L1 pathway can restore function to exhausted CTLs, enhancing both prophylactic and therapeutic vaccinations in tested models. This is very promising as CTLs are critical to viral clearance and exhaustion presents a major hurdle to treating chronic infections. Continuing work towards better understanding of how this pathway operates is important and will facilitate development of new vaccine strategies that can overcome CTL exhaustion.
Acknowledgments
K. A. Hofmeyer is supported by NIH Training Grant T32DK007513. Research in the Zang lab is supported in part by NIH Type 1 Diabetes Pathfinder Award DP2DK083076 and DOD New Investigator Award PC094137. Albert Einstein Cancer Center and Diabetes Research Center are supported by NIH P30CA013330 and P60DK020541, respectively.
References
- 1.Zang X, Allison JP. The B7 family and cancer therapy: costimulation and coinhibition. Clinical Cancer Research. 2007;13(18, part 1):5271–5279. doi: 10.1158/1078-0432.CCR-07-1030. [DOI] [PubMed] [Google Scholar]
- 2.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO Journal. 1992;11(11):3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agata Y, Kawasaki A, Nishimura H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. International Immunology. 1996;8(5):765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 4.Vibhakar R, Juan G, Traganos F, Darzynkiewicz Z, Finger LR. Activation-induced expression of human programmed death-1 gene in T- lymphocytes. Experimental Cell Research. 1997;232(1):25–28. doi: 10.1006/excr.1997.3493. [DOI] [PubMed] [Google Scholar]
- 5.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of Experimental Medicine. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. The Journal of Immunology. 2008;181(7):4832–4839. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kao C, Oestreich KJ, Paley MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nature Immunology. 2011;12(7):663–671. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nature Medicine. 1999;5(12):1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 9.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature Immunology. 2001;2(3):261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 10.Tseng SY, Otsuji M, Gorski K, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. The Journal of Experimental Medicine. 2001;193(7):839–845. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter LL, Fouser LA, Jussif J, et al. PD-1:PD-L inhibitory pathway affects both CD4+ and CD4+ T cells and is overcome by IL-2. European Journal of Immunology. 2002;32(3):634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. The Journal of Immunology. 2002;169(10):5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 13.Lee SJ, Jang BC, Lee SW, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274) FEBS Letters. 2006;580(3):755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 14.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. The Journal of Immunology. 2003;170(3):1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 16.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13866–13871. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. The Journal of Immunology. 2004;173(2):945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 18.Sheppard KA, Fitz LJ, Lee JM, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Letters. 2004;574(1-3):37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 19.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and Cellular Biology. 2005;25(21):9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity. 2007;27(1):111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 22.Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 23.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. The Journal of Experimental Medicine. 2003;198(1):39–50. doi: 10.1084/jem.20022235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fife BT, Pauken KE, Eagar TN, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nature Immunology. 2009;10(11):1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. The Journal of Experimental Medicine. 2009;206(13):3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zajac AJ, Blattman JN, Murali-Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. The Journal of Experimental Medicine. 1998;188(12):2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallimore A, Glithero A, Godkin A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. The Journal of Experimental Medicine. 1998;187(9):1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 30.Blattman JN, Wherry EJ, Ha SJ, Van Der Most RG, Ahmed R. Impact of epitope escape on PD-1 expression and CD8 T-cell exhaustion during chronic infection. Journal of Virology. 2009;83(9):4386–4394. doi: 10.1128/JVI.02524-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(39):15016–15021. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. UN Joint Programme on HIV/AIDS, Global Report: UNAIDS Report on the Global AIDS Epidemic, December 2010, http://www.unhcr.org/refworld/docid/4cfca9c62.html/
- 33.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 34.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nature Medicine. 2006;12(10):1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 35.Zhang JY, Zhang Z, Wang X, et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood. 2007;109(11):4671–4678. doi: 10.1182/blood-2006-09-044826. [DOI] [PubMed] [Google Scholar]
- 36.Quigley M, Pereyra F, Nilsson B, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nature Medicine. 2010;16(10):1147–1151. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velu V, Kannanganat S, Ibegbu C, et al. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. Journal of Virology. 2007;81(11):5819–5828. doi: 10.1128/JVI.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrovas C, Price DA, Mattapallil J, et al. SIV-specific CD8+ T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood. 2007;110(3):928–936. doi: 10.1182/blood-2007-01-069112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Souza M, Fontenot AP, Mack DG, et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. The Journal of Immunology. 2007;179(3):1979–1987. doi: 10.4049/jimmunol.179.3.1979. [DOI] [PubMed] [Google Scholar]
- 40.Said EA, Dupuy FP, Trautmann L, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nature Medicine. 2010;16(4):452–459. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trabattoni D, Saresella M, Biasin M, et al. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood. 2003;101(7):2514–2520. doi: 10.1182/blood-2002-10-3065. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Zhang Z, Zhang S, et al. B7-H1 up-regulation impairs myeloid DC and correlates with disease progression in chronic HIV-1 infection. European Journal of Immunology. 2008;38(11):3226–3236. doi: 10.1002/eji.200838285. [DOI] [PubMed] [Google Scholar]
- 43.Rodríguez-García M, Porichis F, de Jong OG, et al. Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. Journal of Leukocyte Biology. 2011;89(4):507–515. doi: 10.1189/jlb.0610327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchell AE, Colvin HM, Beasley RP. Institute of medicine recommendations for the prevention and control of hepatitis B and C. Hepatology. 2010;51(3):729–733. doi: 10.1002/hep.23561. [DOI] [PubMed] [Google Scholar]
- 45.Kantzanou M, Lucas M, Barnes E, et al. Viral escape and T cell exhaustion in hepatitis C virus infection analysed using Class I peptide tetramers. Immunology Letters. 2003;85(2):165–171. doi: 10.1016/s0165-2478(02)00224-9. [DOI] [PubMed] [Google Scholar]
- 46.Reignat S, Webster GJM, Brown D, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. The Journal of Experimental Medicine. 2002;195(9):1089–1101. doi: 10.1084/jem.20011723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boni C, Fisicaro P, Valdatta C, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. Journal of Virology. 2007;81(8):4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng G, Li S, Wu W, Tan X, Chen Y, Chen Z. PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Molecular Immunology. 2008;45(4):963–970. doi: 10.1016/j.molimm.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 49.Chen C, Qu QX, Huang JA, et al. Expression of programmed-death receptor ligands 1 and 2 may contribute to the poor stimulatory potential of murine immature dendritic cells. Immunobiology. 2007;212(3):159–165. doi: 10.1016/j.imbio.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 50.Urbani S, Amadei B, Tola D, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. Journal of Virology. 2006;80(22):11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. The Journal of Immunology. 2007;178(5):2714–2720. doi: 10.4049/jimmunol.178.5.2714. [DOI] [PubMed] [Google Scholar]
- 52.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. Journal of Virology. 2007;81(17):9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamoto N, Cho H, Shaked A, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathogens. 2009;5(2) doi: 10.1371/journal.ppat.1000313. Article ID e1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. Journal of Virology. 2009;83(5):2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allen SJ, Mott KR, Zandian M, Ghiasi H. Immunization with different viral antigens alters the pattern of T cell exhaustion and latency in herpes simplex virus type 1-infected mice. Journal of Virology. 2010;84(23):12315–12324. doi: 10.1128/JVI.01600-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen SJ, Hamrah P, Gate D, et al. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. Journal of Virology. 2011;85(9):4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frank GM, Lepisto AJ, Freeman ML, Sheridan BS, Cherpes TL, Hendricks RL. Early CD4+ T cell help prevents partial CD8+ T cell exhaustion and promotes maintenance of herpes simplex virus 1 latency. The Journal of Immunology. 2010;184(1):277–286. doi: 10.4049/jimmunol.0902373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takamura S, Tsuji-Kawahara S, Yagita H, et al. Premature terminal exhaustion of friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. The Journal of Immunology. 2010;184(9):4696–4707. doi: 10.4049/jimmunol.0903478. [DOI] [PubMed] [Google Scholar]
- 59.Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. The Journal of Immunology. 2006;176(5):3000–3009. doi: 10.4049/jimmunol.176.5.3000. [DOI] [PubMed] [Google Scholar]
- 60.Mitchell P, Germain C, Fiori PL, et al. Chronic exposure to Helicobacter pylori impairs dendritic cell function and inhibits Th1 development. Infection and Immunity. 2007;75(2):810–819. doi: 10.1128/IAI.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu Y-Y, Chen J-H, Kao J-T, et al. Expression of CD25high regulatory T cells and PD-1 in gastric infiltrating CD8+ T lymphocytes in patients with Helicobacter pylori infection. Clinical and Vaccine Immunology. 2011;18(7):1198–1201. doi: 10.1128/CVI.00422-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schreiber HA, Hulseberg PD, Lee J, et al. Dendritic cells in chronic mycobacterial granulomas restrict local anti-bacterial T cell response in a murine model. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011453. Article ID e11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stäger S. B7-H1 blockade increases survival of dysfunctional CD8+ T cells and confers protection against Leishmania donovaniinfections. PLoS Pathogens. 2009;5(5) doi: 10.1371/journal.ppat.1000431. Article ID e1000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhadra R, Gigley JP, Weiss LM, Khan IA. Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(22):9196–9201. doi: 10.1073/pnas.1015298108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith P, Walsh CM, Mangan NE, et al. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. The Journal of Immunology. 2004;173(2):1240–1248. doi: 10.4049/jimmunol.173.2.1240. [DOI] [PubMed] [Google Scholar]
- 66.Colley DG, Sasser LE, Reed AM. PD-L2+ dendritic cells and PD-1+ CD4+ T cells in schistosomiasis correlate with morbidity. Parasite Immunology. 2005;27(1-2):45–53. doi: 10.1111/j.1365-3024.2005.00742.x. [DOI] [PubMed] [Google Scholar]
- 67.Rollier CS, Paranhos-Baccala G, Verschoor EJ, et al. Vaccine-induced early control of hepatitis C virus infection in chimpanzees fails to impact on hepatic PD-1 and chronicity. Hepatology. 2007;45(3):602–613. doi: 10.1002/hep.21573. [DOI] [PubMed] [Google Scholar]
- 68.Finnefrock AC, Tang A, Li F, et al. PD-1 blockade in rhesus macaques: impact on chronic infection and prophylactic vaccination. The Journal of Immunology. 2009;182(2):980–987. doi: 10.4049/jimmunol.182.2.980. [DOI] [PubMed] [Google Scholar]
- 69.Song M-Y, Park S-H, Nam HJ, Choi D-H, Sung Y-C. Enhancement of vaccine-induced primary and memory CD8+ t-cell responses by soluble PD-1. The Journal of Immunotherapy. 2011;34(3):297–306. doi: 10.1097/CJI.0b013e318210ed0e. [DOI] [PubMed] [Google Scholar]
- 70.Ha SJ, Mueller SN, Wherry EJ, et al. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. The Journal of Experimental Medicine. 2008;205(3):543–555. doi: 10.1084/jem.20071949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mueller SN, Matloubian M, Clemens DM, et al. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(39):15430–15435. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Velu V, Titanji K, Zhu B, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berger R, Rotem-Yehudar R, Slama G, et al. Phase i safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clinical Cancer Research. 2008;14(10):3044–3051. doi: 10.1158/1078-0432.CCR-07-4079. [DOI] [PubMed] [Google Scholar]
- 74.Franceschini D, Paroli M, Francavilla V, et al. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. The Journal of Clinical Investigation. 2009;119(3):551–564. doi: 10.1172/JCI36604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MBA. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(51):20428–20433. doi: 10.1073/pnas.0811139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunology. 2009;10(1):29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Titanji K, Velu V, Chennareddi L, et al. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. The Journal of Clinical Investigation. 2010;120(11):3878–3890. doi: 10.1172/JCI43271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lázár-Molnár E, Chen B, Sweeney KA, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(30):13402–13407. doi: 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. The Journal of Immunology. 2011;186(3):1598–1607. doi: 10.4049/jimmunol.1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]