

Abstract
Early diagnosis and appropriate treatment are key elements of malaria control programs in endemic areas. A major step forward in recent years has been the production and use of rapid diagnostic tests (RDTs) in settings where microscopy is impracticable. Many current RDTs target the Plasmodium falciparum histidine-rich protein 2 (PfHRP2) released in the plasma of infected individuals. These RDTs have had an indisputably positive effect on malaria management, but still present several limitations, including the poor characterization of the commercial monoclonal antibodies (mAbs) used for PfHRP2 detection, variable sensitivity and specificity and high costs. RDT use is further limited by impaired stability caused by temperature fluctuations during transport and uncontrolled storage in field-based facilities. To circumvent such drawbacks, an alternative could be the development of well-characterized, stabilized recombinant antibodies, with high binding affinity and specificity. Here, we report the characterization of the cDNA sequences encoding the Fab fragment of F1110 and F1546, two novel anti-PfHRP2 mAbs. FabF1546 was produced in the Escherichia coli periplasm. Its properties of binding to the parasite and to a recombinant PfHRP-2 antigen were similar to those of the parental mAb. As the affinity and stability of recombinant antibodies can be improved by protein engineering, our results open a novel approach for the development of an improved RDT for malaria diagnosis.
Key words: Plasmodium falciparum, malaria, histidine-rich protein, monoclonal antibodies, recombinant Fab, rapid diagnostic test
Introduction
Plasmodium falciparum malaria remains one of the leading causes of morbidity and mortality in tropical areas.1 Early diagnosis is a key element of malaria control programs, as it allows prompt and appropriate treatment of clinical malaria, reducing the risk of progression to severe disease. In addition, the artemisin-based combination therapies are expensive and their high cost increases the need for simple and accurate parasite-based diagnosis for malaria.2–5 Microscopic diagnosis of blood specimens is sensitive and specific, but difficult to apply in the field because of the need for specific equipment and experienced technical staff that are rarely available at the community level and time-consuming slide inspection for accurate quantification and species determination.6 Alternative immunodiagnostic approaches that are suitable for use in field conditions have been developed. A major advance in the recent years has been the deployment of rapid diagnostic tests (RDT) in settings in which microscopy is not possible.7–9
Most of the currently available RDTs for malaria are based on detection of the P. falciparum histidine-rich protein 2 (PfHRP2) by monoclonal antibodies (mAbs).10–14 The PfHRP2 protein contains central repeats, rich in alanine and histidine residues, the number of which varies between parasite clones. This abundant protein is soluble and heat-stable, produced specifically by P. falciparum, and absent from other malaria parasites infecting humans. It is an interesting and sensitive target antigen for detecting this species in biological fluids.15–17 Several companies manufacture PfHRP2-based RDTs, the performances of which have recently been assessed and compared.18,19 Comparative data from a large panel of malaria RDTs against P. falciparum samples adjusted at low and high parasite densities showed that only about one third (13/33) of commercial tests have a good sensitivity at low parasite density (200 parasites/µL of blood).18 As a general rule, current malaria RDTs have an acceptable sensitivity and specificity when parasite density exceeds 100 parasites/µL and are much less sensitive in conditions of lower parasitemia.7,8
PfHRP2-based RDTs have had an indisputably positive effect on malaria management in terms of patient care, but they remain subject to several limitations.7,18–20 Their cost, which reaches that of an artemisinin-based combination therapy, is unaffordable for most populations at risk.4,5,21 The RDTs produced by different manufacturers differ in terms of sensitivity and specificity in field conditions and show some batch to batch variations that necessitate quality controls by reference laboratories. The performance of RDTs is adversely affected by temperature fluctuation during transport and uncontrolled storage in field facilities. Sensitivity may be lost before the expiry date due to the instability of mAb components that denature at elevated temperature or detach from the solid nitrocellulose support at excessive humidity. Most manufacturers guarantee a shelf life of one to two years at temperatures below 40°C, but these conditions are not met in all endemic areas, particularly in Sub-Saharan Africa, where temperatures regularly rise above 45°C.18–20,22,23
Recombinant technologies can be used to produce cheap, well-defined, stabilized proteins, including antibodies with high binding affinity and specificity. The use of recombinant antibodies could potentially overcome the limitations of current RDTs.24,25 Surprisingly, little attention has been paid to this approach so far. Here, we describe two new mouse mAbs directed against and specific for PfHRP2. We report the cloning REPORTof the cDNAs encoding their Fab fragments, their nucleotide sequence and the expression and characterization of the recombinant Fabs. The ability of a recombinant Fab fragment to bind native and recombinant PfHRP2 compared well with the parental mAb. Our results provide the first molecular characterization of antibodies with specificity for PfHRP2 and a solid basis for improving their performance and stability for applications in malaria diagnosis.
Results
Characterization of novel anti-PfHRP2 monoclonal antibodies.
Hybridoma cell lines were constructed from mice that had been immunized with P. falciparum asexual blood stages. We screened the secreted antibodies for their reactivity towards PfHRP2 by western blotting and indirect immunofluorescence experiments. A representative western blot is shown in Figure 1 (left) for two of the selected mAbs, mAbF1110 and mAb1546, which belonged to the IgGκ isotype. As reported previously for mAbs reacting with PfHRP2, a multiple banding pattern, ranging from 50–35 kDa, was observed with mAbF1110 and mAbF1546. These antibodies recognized predominantly 50 and 37 kDa protein species, corresponding to the PfHRP2 and PfHRP3 polypeptides, respectively. They also reacted with a 33 kDa species, whose intensity varied depending on the particular antigen extract. We assumed that it corresponded to a proteolysis product of larger polypeptides, possibly PfHRP2 or PfHRP3. However, we cannot rule out the possibility that this species resulted from cross-reaction with another as yet unidentified parasite histidine-rich polypeptide.
Figure 1.

Reactivity of mAbF1546 and mAbF1110 with P. falciparum as assayed by western blots and indirect immunofluorescence. The western blot assayed the reactivity of mAbF1546 (lane 1) and mAbF1110 (lane 2) with P. falciparum crude antigenic extracts, subjected to electrophoresis under denaturing conditions. Apparent MW (kDa) are shown on the left side of the immunoblot. The indirect immunofluorescence staining patterns for mAbF1546 (part 1) and mAbF1110 (part 2) were performed on air-dried blood stages of P. falciparum. Antigenic preparations were obtained from 3D7 parasites.
Air-dried erythrocytes infected with P. falciparum late stages showed a typical coarse fragmented or dotted pattern of fluorescence in indirect immunofluorescence microscopy, whereas the ring forms of the parasite showed a weaker and more diffuse fluorescence (Fig. 1, right). These patterns are consistent with antibody reactivity to PfHRP2.15,17,26,27 The FITC conjugate alone or supernatants from non-fused melanoma cells did not produce any significant positive signal on air-dried parasites at similar dilutions. This absence of signal indicated that the tested antibodies were specific for infected red blood cells (IRBC). Air dried fixation was preferred over acetone treatment as the former is known to better preserve the integrity of parasite proteins while keeping them mostly in a native form. The specificity of mAbF1110 and mAbF1546 for PfHRP2 was confirmed by using a recombinant PfHRP2 protein, fused with the maltose binding protein (see below).
To verify the sensitivity and linearity of HRP-2 detection by the selected mAbs, antigenic extracts containing known amounts of in vitro-cultured P. falciparum-parasitized erythrocytes were prepared and examined by sandwich ELISA using mAbF1110 or mAbF1546 mAbs as capture antibodies, biotinylated mAbF1546 as the primary antibody, and an avidin-peroxydase conjugate as a detection reagent. The mean reactivity for duplicate measures of parasite densities equivalent to 500–0.25 parasite per µl are shown in Figure 2. Typical positive dose response curves were observed whether the homologous mAbF1546 (Fig. 2A) or heterologous mAbF1110 (Fig. 2B) was used for immunocapture. In all cases, the assay was found suitable for the detection of PfHRP2, freed from IRBC by hypotonic shock or mild denaturation with 0.05% SDS. Measures were slightly higher after treatment with 0.05% SDS. This observation indicated that SDS at low concentration might help PfHRP2 recovery without affecting the recognition by mAbs. The detection limit of this assay was comprised between one to four parasites/µl with a linear range of 4–125 parasites/µl. The fact that mAb (F1546) can be used for both capture and detection in a single assay, indicated that the targeted epitope was likely repeated and easily accessible. MAbF1546 could therefore be used either in combination with F1110 or alone for antigen immunocapture and detection without significant decrease in sensitivity. This may have interesting application for diagnosis purpose.
Figure 2.

Reactivity of mAbF1546 and sandwich ELISA with mAbF1546 and mAbF1110. Microtiter plates were coated with either mAbF1546 (A) or mAbF1110 (B) as capture antibody at a 10 µg.mL−1 in PBS, then reacted with decreasing concentrations of soluble PfHRP2 containing fractions equivalent to 500–0.25 IRBC µL−1 prepared by hypotonic lysis (SCHIZ-H20) or mild denaturation with 0.05% SDS in PBS (SCHIZ-0.05% SDS) of IRBC. Bound antigen was finally revealed using biotin labeled mAbF1546 and avidin peroxydase as indicated in the Materials and Methods section. The negative control (C neg) consists of equivalent preparations of uninfected human erythrocytes treated by 0.05% SDS and assayed in similar conditions.
Sequence analysis.
The VL-CL and VH-CH1 antibody gene fragments, derived from mouse monoclonal hybridoma cell lines F1110 and F1546, were generated by amplification of the corresponding cDNAs by PCR, using a set of degenerate primers. The CKDNA-3′ and LC7-5′ primers amplified a 678 bp PCR product from both F1546 and F1110 cDNAs, corresponding to their VL-CL domain. The VH-CH1 fragments were produced at high yield with a single 3′-primer, CIgG1, and two different 5′-primers, VHI (for F1546) and VHIIB (for F1110), producing 657 and 654 bp fragments from the F1546 and F1110 cDNAs, respectively. The various VL-CL and VH-CH1 amplification fragments were inserted into the pGEM-T vector. Three to five independent clones were sequenced and aligned for each molecular species making it possible to generate a consensus sequence for each chain. Pair-wise alignment of the F1546 and F1110 VL-CL and VH-CH1 consensus protein sequences showed that the monoclonal hybridoma cell lines F1546 and F1110 used the same CL-VL, but that their CH1-VH1 sequences differed, particularly in the variable regions, which displayed only 38% identity at protein level (Fig. 3). Thus, despite their similar pattern of reactivity on immunoblotting and immunofluorescence and their identical CL-VL germline, the monoclonal hybridoma cell lines F1110 and F1546 are different.
Figure 3.

Alignment of the deduced consensus amino-acid sequences of the light chain (VL-CL) and heavy chain (VH-CH1) from anti-PfHRP2 hybridomas secreting mAbF1110 and mAbF1546. Differences in amino-acid residues are boxed in black and residues of the same group are shaded in gray. Dashes indicate gaps introduced by ClustalW for optimization of the alignment.
Computational analysis of the VH and VL sequences.
The VH and VL domain sequences were analyzed with IMGT V-Quest software which identifies the immunoglobulin germline V, D and J genes from which a specific immunoglobulin chain is derived.28 Sequence analysis of the VL fragment shared by hybridoma cell lines F1546 and F1110 confirmed that it belonged to the mouse K chain subgroup. Maximum sequence identities of 96.2% (280/291 nt) and 97.3% (36/37 nt) were observed with the V gene IGKV3-5*01 [K02161] and IGKJ1*1 [V00777] alleles, respectively (Fig. 4A). Fourteen base pairs differed from the germline sequence, including five silent mutations. These mutations led to the following amino acid substitutions: D1E, I2L and L4M in FR1; S28N and S31R in CDR1; P46A in FR2; Y103F in FR3; and Q106R and D110V in CDR3. The F1110-VH sequence was most closely related to the IGHV1S22*01 [X02063] allele, with an identity of 96.1% (268/279 nt). Fourteen base pair differences, with respect to the closest germline sequence IGHV1S22*01, were observed in the F1110-VH domain, resulting in eight amino-acid substitutions: Q5E, S9A and K20M in FR1; T65A in CDR2; G76A, M89L and D98G in FR3; and R106N in CDR3 (Fig. 4B). The F1546-VH sequence was the closest to the IGHV3-8*02 [AJ972403] allele, with which it displayed 97.5% (269/276 nt) identity. Eleven base pair differences were detected in F1546-VH fragment compared to the IGHV3-8*02 germline sequence, leading to seven amino-acid substitutions: Q5E in FR1; Y52F and Y55S in FR2; S74G, K84N and Q86H in FR3; and R106 N in CDR3 (Fig. 4C). Amino-acid substitutions at positions Q5E and R106N were observed in both VH sequences, suggesting that a glutamine residue in position 5 and an asparagines residue in position 106 in CDR3 may be essential for antibody stability or antigen recognition. The CDRs of antibodies, and CDR3 in particular, are generally responsible for high-affinity binding. The J and D regions of both mAbs were the most similar to the IGHJ2*01 [V00770] and IGHD4-1*01 alleles, respectively.
Figure 4.


Nucleotide and deduced amino-acid sequences for the genes encoding the variable domains of mAbF1546 and mAbF1110. Differences between these sequences and the closest germline nucleotide sequences and corresponding amino acid sequences are shown. In (A), the VL domains from mAbF1546 and mAbF1110 are compared with the VL germline sequence IGKV3-5*01. The J gene region is also indicated. In (B), the VH domain of mAbF1110 is compared with the germline sequence IGHV1-S22*01, with detailed D and J gene regions. In (C), the VH domain of mAbF1546 is aligned with the germline sequence IGHV3-8*02, with the D and J genes.
Fab expression.
The pF1546 and pF1110 plasmids harboring the assembled VL-CL and VH1-CH1 fragments cloned into the pPE1 vector were used to transform the E. coli HB2151 strain.29,30 FabF1546-H6 and FabF1110-H6 antibody fragment have a C-terminal hexahistidine tag. After mild IPTG induction, a soluble recombinant Fab fragment was harvested from periplasmic extracts and purified. The purified fractions were analyzed by SDS-PAGE under reducing or non reducing conditions followed by immunoblotting. The crude periplasmic extracts gave a complex pattern of bands in the lower part of the gel, with two major bands in the 48 and 23 kDa regions (Fig. 5, lanes 1 and 2). Chromatography-purified Fab fragments migrated as a single band in the 48 kDa region of the gel under non reducing conditions, corresponding to intact recombinant Fab, and as a 26 kDa band after reduction, corresponding to the VL-CL and VH-CH1 fragments (lanes 3 and 4, and lanes 5 and 6, respectively). Somewhat larger yields were obtained for FabF1546-H6 fragment. We therefore selected the FabF1546-H6 fragment for further studies of binding properties.
Figure 5.

FabF1546-H6 and FabF1110-H6 productions in E. coli. Samples were fractionated by SDS-PAGE on 7.5–15% polyacrylamide gels (Biorad) and stained with Coomassie Blue. Molecular markers (kDa) are shown in lane M. Crude periplasmic extracts from recombinant HB2151 strains, producing recombinant Fab-H6 fragments, were subjected to electrophoresis under non-reducing conditions (lanes 1 and 2). The affinity-purified recombinant Fab-H6 were also subjected to electrophoresis under non-reducing conditions (lanes 3 and 4) or reducing conditions (lanes 5 and 6). Lanes 1, 3 and 5, FabF1546-H6; lanes 2, 4 and 6, FabF1110-H6.
Recombinant MalE-PfHRP2.
The MalE-PfHRP2 hybrid protein was produced in a soluble state and at high yield by the induced recombinant E. coli strain HB2151 (pER1). The MalE-PfHRP2 was recognized by human sera from endemic areas and by all commercial RDTs targeting PfHRP2 that we tested (data not shown). An example of such a reactivity is shown in Figure 6 for the reagents provided in the CareStart Malaria Combo test (Access Bio-Inc.,). The performance of the CareStart device, which is based on both HRP2 (Pf specific) and pLDH (pan specific) detections, has been evaluated recently in comparison with other commercial malaria RDT and ranked amongst the best.18 The soluble extract from induced HB2151(pER1) cells reacted strongly with P. falciparum test zone 1 of the device, based on PfHRP2 recognition, and with the control zone C, whereas the panspecific test zone 2, based on pLDH, did not react, as expected (lane 2). This pattern is similar to the pattern of P. falciparum-infected blood samples, demonstrating that the RDT reactivity of the recombinant MalE-PfHRP2 protein is similar to the native PfHRP2 produced by wild P. falciparum parasites. A control supernatant from non-transformed HB2151 cells, grown and induced with IPTG in similar conditions, did not react with the test zone 1 whereas a band was detected in the control zone C (lane 1).
Figure 6.
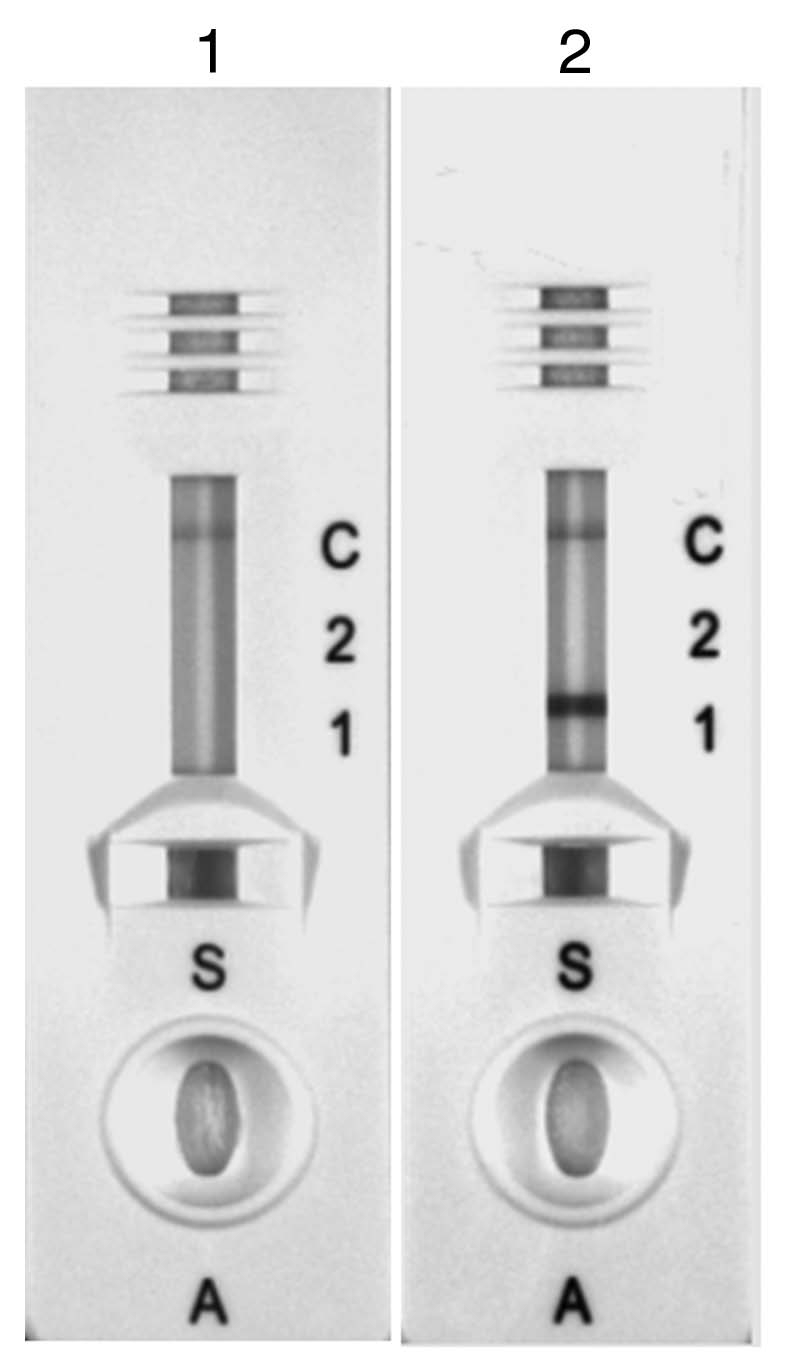
Reactivity of the recombinant MalE-PfHRP2 protein in the commercial CareStart Malaria Combo test. Crude soluble fraction prepared from non-transfected HB2151 control cells (lane 1) and crude soluble fraction from HB2151 (pER1) cells expressing MalE-PfHRP2 grown with IPTG (lane 2).
Binding of the recombinant Fab to PfHRP2.
The binding properties of the recombinant FabF1546-H6 fragment were assessed by western blotting and ELISA. Western blots of proteins separated under non reducing conditions showed that FabF1546-H6 recognized a protein that had an apparent molecular mass (MM) of 50 kDa in P. falciparum antigenic extracts, corresponding to the theoretical mass of PfHRP2 (Fig. 7, lane 1), and that co-migrated with the protein recognized by the full mAbF1546 (lane 2). Likewise, FabF1546-H6 recognized a protein with a MM of 76.5 kDa, corresponding to the theoretical mass of MalE-PfHRP2 (42 + 34.5 kDa) (lane 3) in a crude extract from strain HB2151(pER1). A similar pattern was observed on strips incubated with the parental mAbF1546 (lane 4). The reactivity of FabF1546-H6 was further examined by ELISA (Fig. 8). As we did not attempt to quantify precisely the antigen-binding properties of FabF1546-H6 but rather to check its functionality, periplasmic fluids were reacted directly with precoated wells. In brief, a crude soluble extract of P. falciparum, immobilized on a 96-well plate, was incubated with a series of two-fold dilutions, from 1/10 to 1/10,240, of a periplasmic extract from HB2151(pF1546) cells, producing FabF1546-H6. Dilutions of the periplasmic extract, up to 1/160, reacted with the parasite extract and gave a signal above background (Fig. 8A). The binding specificity of FabF1546-H6 was similarly analyzed by direct ELISA, using purified recombinant MalE-PfHRP2 at concentrations ranging from 20 µg.mL−1 to 1.5 ng.mL−1. A concentration-dependent response was again clearly observed, with reactivity above the technical threshold at MalE-PfHRP2 concentrations down to 20 ng.mL−1 (Fig. 8B). This sensitivity was in the same range as that previously reported for an HRP2-specific ELISA assay based on a recombinant HRP2 and two mAbs, 1E1 and 2G12.15,31 We observed that the recombinant antibody fragment had a lower reactivity with native and recombinant PfHRP2 than the parental mAb. This difference in reactivity comes probably from the fact that IgGs are bivalent and Fabs monovalent. Moreover, we used alkaline phosphatase conjugates, which have lower catalytic turnovers that horseradish peroxidase conjugates. Overall, our results indicated that the pattern of PfHR2 recognition by FabF1546-H6 and the parental mAb were essentially identical.
Figure 7.

Reactivity of parental mAbF1546 and recombinant FabF1546 to a crude extract of P. falciparum antigenic extract in western blots. A crude P. falciparum antigenic extract (lanes 1 and 2) and the periplasmic fluid of induced HB2151 (pER1) expressing MalE-PfHRP2 (lanes 3 and 4) were subjected to SDS-PAGE on a 7.5–15% polyacrylamide gel run under non reducing conditions. They were probed with the parental mAbF1546 (lanes 2 and 4, ascitic fluid at a 1/1,000 dilution) and the recombinant FabF1546-H6 (lanes 1 and 3, periplasmic fraction at a 1/10 dilution).
Figure 8.

Binding specificities of the recombinant FabF1546-H6 as determined by ELISA on parasite and recombinant PfHRP2. In part A, microplate wells were coated with a crude P. falciparum soluble extract (protein concentration adjusted to 20 µg.mL−1 with PBS) and reacted with two-fold dilutions of a periplasmic extract of HB2151(pF1546) from 1/10 to 1/10,240. In part B, wells were coated with various concentrations of affinity-purified MalE-PfHRP2 protein, from 1.5 ng.mL−1 to 20 µg.mL−1, and reacted with a periplasmic extract of HB2151 (p1546) at a 1/10 dilution. Bound antibodies or fragments were detected with an alkaline phosphatase-conjugated Fab-specific anti mouse IgG. Doted lines correspond to the technical background level (mean blank values + 2SD).
Discussion
Several millions of malaria RDTs, mostly specific for P. falciparum, are purchased every year for malaria control programs in endemic countries, but the world consumption of commercial RDTs will continue to increase in the coming years because of the recently published WHO guidelines recommend a parasitological confirmation of diagnosis in all patients suspected of having malaria before treatment.32 Implementation of this recommendation will require mobilization of important financial and human resources.33,34 The advent of RDTs has made such testing possible when microscopy would be difficult or impossible. Many current RDTs are based on the detection of P. falciparum HPR2, a soluble parasite antigen specific to P. falciparum that is considered the primary immunological target for P. falciparum malaria testing. A large body of information from field trials that assessed the impact on RDT specificity and sensitivity of parameters such as manufacturer, parasite polymorphism and stability to heat, or comparing the performance of RDTs with conventional methods such as microscopy has recently accumulated.7–9,13,18–20 Paradoxically, little information about the PfHRP2 antibodies used in these commercial tests has been published.35 For example, the isotype (IgG or IgM), subclass, epitope targeted (unique or repeated), molecular structure and laboratory origin of these antibodies remain essentially unknown. All the PfHRP2 antibodies in current use were developed at the end of the 1980s and most commercial RDTs are probably manufactured using reagents purchased from few suppliers producing the mAbs on a large scale. Given the increasing demand for RDTs, a better characterization of antibodies used for malaria diagnosis and distribution of this information to RDT users are required.18,19
In this study, we produced and characterized two novel mAbs against PfHRP2. In indirect immunofluorescence studies, these antibodies reacted over the entire infected cell, producing a granular pattern of staining typical of antibodies reacting with PfHRP2. In immunoblot experiments, both mAbs identified a multiplet of proteins, with two major bands at 37 kDa and 50 kDa. An additional band was also detected at about 35 kDa. The identification of multiple bands is consistent with previous reports on mAbs reacting with PfHRP2. In ELISA, the F1110 and F1546 antibodies could be used either alone or together, for antigen immunocapture and detection with no significant loss of sensitivity. The targeted epitopes are therefore repeated and possibly different. They are likely linear because recognition by these antibodies was not affected by SDS or treatment with reducing agents. A study evaluating series of five peptides, derived from the central part of the PfHRP2 molecule, showed that only antibodies raised against the CGDHHAADAHHATDAHHGC peptide cross-reacted with PfHRP3.36 Additional studies showed that most anti-HRP2 mAbs recognized DAHHAHHA as the major epitope, with possible substitutions replacing the first and last amino acids by Y or V.27 We anticipate that the F1110 and F1546 antibodies react with closely related epitopes, but careful mapping is required to confirm this hypothesis.
Commercially available tests are sensitive and specific, but there is still a need to establish simple and reliable procedures for ensuring that malaria RDTs meet high criteria of quality before their distribution in the field. Surprisingly, only the enzymatic reaction is currently controlled on nitrocellulose strips (capture of labeled antibodies by bound antibody). There is no control of mAb reactivity with the targeted antigen.7,19,37 Quality control procedures carried out by WHO at reference laboratories in areas of endemic malaria, are based on the use of calibrated parasite samples obtained from patients. However, patient parasites are polymorphic and therefore the sequence of PfHRP2 and possibly its expression levels may vary between parasite clones. These variations may bias the correlation between parasite number and HRP2 amount.27,35 The production of a recombinant protein mimicking the target parasite epitope would circumvent this problem. The recombinant MalE-PfHRP2 hybrid protein described here was recognized by all the RDTs tested targeting PfHRP2, and by the mAbs produced and characterized in this study. It could be used as a positive control for assessing the reactivity of these antibodies. This recombinant PfHRP2 protein mimicking the entire molecule thus provides interesting perspectives for the design of new protocols for RDT quality control and quality assurance.
Questions are regularly raised about the reproducibility, reliability, cost and stability of the currently available commercial RDTs. In particular, exposure to high temperature is likely to alter the performance and shelf-life of malaria RDTs.7,18,22,23 Manufacturers generally recommend storage at temperatures lower than 30°C to maximize stability. Unfortunately, these conditions are difficult to meet in areas of endemic malaria, in which RDTs are frequently exposed to temperatures exceeding 40°C and even more during storage and transportation. A recent comparative study showed that a significant proportion of commercially available RDTs continued to react positively with malaria parasites after six months of incubation at 45°C, at least when assayed using samples with high parasite densities.18 The extent to which exposure to a temperature of 45°C would reduce sensitivity and the shelf-life of RDTs has to be better documented as a point of care RDT should also be able to detect parasite carriers harboring low background infections.18 Actually, it appears that temperature fluctuations may have a greater effect on the viability of RDTs than storage at high temperatures. If RDTs are to function correctly in the conditions prevailing in areas of endemic malaria, they must be stable at temperatures of about 45–50°C for one to two years.
One way of overcoming these limitations is to produce recombinant antibody fragments. Indeed, the use of smaller fragments rather than complete antibodies has several advantages in diagnostic assays. It may decrease unwanted background signals on strips. In addition, microbiological systems can be used to produce a homogeneous protein both rapidly and cheaply. Large quantities of functional antibody fragments, sufficient for use in diagnostic applications, can therefore be obtained with careful optimization of the production conditions. It is also possible to refine the design of the fragment without altering the affinity for its antigen. The constant or variable domains can be engineered to improve the labeling or affinity properties of the antibody fragment for sensitive immunoassay applications, especially in detecting asymptomatic carriers who represent an important reservoir from which parasite infections may spread and interfere with the global malaria eradication program.22,23,29,32,38 Finally, the stability of recombinant antibody fragments can be improved by designing or selecting mutations conferring resistance to denaturation. Improvements in stability should make RDT devices more robust to heat and significantly increase their shelf-life. Fab fragments are generally considered to be more stable than scFv, due to the presence of two disulfide bonds. Several approaches have been described for the prediction of stabilizing mutations in recombinant antibody fragments. These methods are based on the observation that the effects of the mutations are additive, making it possible to adjust their combinations and achieve optimal stability.39
We described here the first steps on the path towards the engineering of antibodies reacting with PfHRP2 by recombinant techniques. We identified cDNAs encoding the variable domains of two mAbs, F1546 and F1110, directed against PfHRP2. These Fab fragments have the same VL but different heavy-chain VH-CDR structures, suggesting that they react with different epitopes. Both Fab fragments were produced in good yields in the periplasm of E. coli. Yields were particularly high for FabF1546-H6, which was further characterized here. The recombinant FabF1546-H6 displayed a binding specificity for the parasite and recombinant HRP2 proteins similar to that of the parental mAb. The proteins were recognized in a concentration-dependent manner by ELISA and produced similar banding patterns on western blots, probed with either the parental mAb or with the recombinant Fab fragment.
In conclusion, the mAbs and Fab fragments directed against PfHRP2 described here offer attractive perspectives for the development of improved rapid diagnostic tests for malaria. Inclusion of the recombinant PfHRP2 antigen would provide the added value of a positive antigen/antibody reaction control and even open the possibility of designing a quantitative assay of the amount of antigen present in the sample rather than a presence/absence testing. Although further studies are required to assess the potential of our results for use in diagnostic applications, they constitute an important step towards the development of a novel generation of diagnostic tests for malaria.
Materials and Methods
Parasites.
The 3D7 and the Palo Alto Marburg (PAM) strains of P. falciparum were used in this work.40 Parasites were maintained in asynchronous cultures in human blood group O+ red blood cells in RPMI 1640 supplemented with 10% AB human serum, 2 g.L−1 glucose, 20 mg.L−1 hypoxanthine, 9.1 g.L−1 hepes 1 M, 2 g.L−1 NaHCO3 and 2.5 mg.L−1 gentamicin, essentially as described by Trager and Jensen.41 These strains regularly produce high parasitemias in vitro and express the PfHRP2 and PfHRP3 antigens.
Monoclonal antibodies.
MAbs used in this study were produced in BALB/c mice immunized with schizonts of P. falciparum as described elsewhere.42 The hybridomas were initially screened for reactivity with P. falciparum on air-dried parasites by indirect immunofluorescence. The F1110 and F1546 mAbs were further selected on the basis of their reactivity for PfHRP2 using recombinant antigen. Culture supernatants or ascitic fluids from BALb/c mice bearing the hybridomas were used as sources of mAbs as indicated in the legend of the figures. Isotyping of mouse immunoglobulins was performed using culture supernatant on Isostrips™ mouse mAb isotyping Kit (Santa Cruz Biotechnology) according to manufacturer recommendations.
Immunofluorescence assay.
The immunofluorescence assay was performed using air-dried 3D7 parasites as follows. Smears of infected red blood cells (IRBC) adjusted to 1% parasitemia with PBS were made on multispot microscope slides and stored at −80°C until use. Air-dried spots where thawed and reacted with a 1:10 dilution of culture supernatant of mAbs F1546 and F1110 then incubated for 30 min. at 37°C. Slides were washed 3 times with PBS and stained for additional 30 min. at 37°C with a 1:50 dilution of a fluorescein-conjugate goat anti-mouse IgG (F9006 Sigma). The slides were mounted in PBS containing 10% glycerol and examined at X600 with a fluorescent microscope.
Sandwich ELISA assay for the reactivity of F1110 and F1546 mAbs.
Microtiter plates (Maxisorp, Nunc) were coated overnight at 4°C with 100 µl/well of the capture antibody F1110 or F1546 adjusted at 10 µg.mL−1 in PBS. The plates were washed and saturated with 200 µl/well of 1% BSA in PBS-0.05% Tween20 (PBS-T) for 1 h and 100 µl of decreasing concentrations of a soluble antigenic fraction were added. These fractions were prepared from a schizont-rich culture of P. falciparum (PAM strain at 78% of mature forms) synchronized by several rounds of sorbitol treatment and Plasmagel flotation. The culture was harvested at 9% parasitemia and IRBC were extensively washed with RPMI to remove human serum and secreted soluble proteins. Cells were recounted after washings then serially diluted in RPMI at concentrations equivalent to 5,00,000 to 250 IRBC mLµ1. After centrifugation of the diluted samples, the cell pellets were resuspended in an equal volume of buffered water or PBS containing 0.05% SDS. Complete lysis was finally achieved by resuspending the IRBC vigorously, and the samples were centrifuged at 10,000 g for 10 min. at 4°C to eliminate cellular debris. An equivalent preparation of uninfected human erythrocytes was prepared and used as a negative control. For the assay, duplicate samples of 100 µl of the hemolysates were added to the wells and incubated for 2 h. After additionnal washings, bound antigens were detected by incubation with 100 µl/well of a 1:5,000 dilution of biotinyled F1546 antibodies (1 mg.mL−1). Detection was achieved by addition of avidin-peroxidase (A3141, Sigma) and TMB microwell Peroxydase substrate (KPL) according to manufacturer recommendations. The reaction was stopped by addition of HCL 2N and O.D. values were read spectrophotometrically at 450 nm. Otherwise indicated all incubations steps were performed at room temperature and three washings were systematically performed with PBS-T.
Electrophoresis and immunoblots.
The samples were resuspended in Laemmli sample buffer (Biorad) with or without dithiothreitol, and boiled for 5 min. Samples were electrophoretically separated using the Criterion Precast Gel System on XT Bis-Tris 10% or 4–12% resolving gels (Biorad). The extracts were stained with Commassie brilliant blue or transferred onto nitrocellulose membranes by electroelution O/N at 30V. Blots were processed according to standard procedures and blocked with 50 mM Tris, pH 8, containing 0.15 M NaCl, 0.05% Tween 20, 5% non-fat milk. They were further incubated with mAbs or recombinant antibody fragments. Bound antibodies were detected by using an alkaline phosphatase conjugate of anti-mouse IgG whole molecule (Goat Anti-Mouse IgG, S3721, Promega, France) or an anti-mouse IgG (Fab specific)-Alkaline Phosphatase antibody produced in goat (A1293, Sigma, France) adjusted at 1/1,00,00 dilution. Binding was monitored using a BCIP/NBT mixture substrate (S3771, Promega) according to manufacturer recommendations.
Nucleic acids extraction and cDNA synthesis.
Total nucleic acids were extracted from P. falciparum infected erythrocytes using the High Pure PCR template preparation kit (Roche Applied Science) following manufacturer's protocol. For each sample, 200 µL of cell suspension were processed yielding a 200 µL final volume of nucleic acids in elution buffer. The sample was then treated by Dnase I Rnase free (Roche Applied Science) to eliminate genomic DNA. After an incubation step of 10 min at 37°C the Dnase was inactivated by boiling for 15 min. Hybridoma cells were grown in DMEM medium supplemented with 20% fetal calf serum. Cells were recovered by centrifugation and the pellet containing approximately 107 hybridoma cells was resuspended in 1 ml TRIzol reagent (Invitrogen) and conserved at −80°C. Total RNA was extracted according to manufacturer instructions. Briefly, after thawing the cells, 200 µL of chloroform were added to the sample. After vigorous shaking the tubes were centrifuged at 10,000 g for 15 min at 4°C, RNA was precipitated with isopropyl alcohol and recovered by centrifugation at 10,000 g for 10 min. The pellet was washed with 75% ethanol and resuspended in 100 µl of RNAse free water. The purity of the preparation was estimated from the A260/280 ratio. Two to 4 µL aliquots of these preparations were finally reverse transcribed using random hexamer priming and MMLV reverse transcriptase using the Improm II™ Reverse Transcription System Kit (Promega) and kept frozen at −20°C.
Amplification, cloning and expression of the MalE-PfHRP2 gene.
The full-length PfHRP2 sequence was PCR amplified from 3D7 cDNA using HRP2 forward and reverse primers:
HRP2 forward: 5′-TAG AAT TCA TGG TTT CCT TCT CAA AAA ATA AAG TA-3′.
HRP2 reverse: 5′-TAG GAT CCT ATA TTA TAA ATT TAA TGG CGT AG-3′.
Amplification was performed in 50-µL reaction volume containing 2 µL cDNA, 1 µM each primer, 250 µM of each dNTP, 2 mM of MgCl2 and 2.5 U of Taq polymerase. Samples were subjected to an initial denaturing step at 94°C for 120 s followed by 35 rounds of denaturing at 94°C for 30 s, annealing at 55°C for 60 s and synthesis at 72°C for 90 s and a final elongation step at 72°C for 10 min. The 920 bp PCR product was size-fractionated by agarose by gel electrophoresis, purified using a Qiaquick PCR purification Kit (Qiagen) and cloned into a pGEM-T vector (Promega) according to manufacturer recommendations. Positive clones were screened by PCR and the insert from three independent clones was sequenced. All had the same sequence encoding a protein identical to the published 3D7 sequence (embl accession number AL844506.2). The 3D7 PfHRP2 insert was then subcloned into a pMAL-c2X vector (New England BioLabs) between the EcoRI and BamHI sites and used to transform XL1-Blue competent cells. Positive clones were identified by PCR and the construct was verified by restriction analysis and DNA sequencing. The resulting plasmid named pER1, encoded a fusion protein between the MalE protein from E. coli and PfHRP2, MalE-PfHRP2. pER1 was introduced by transformation into the E. coli strain HB2151.29 The production of MalE-PfHRP2 was induced by 0.3 mM IPTG for 2 h according to standard protocol (pMAL™ protein fusion and purification system instruction manual, New England Biolabs).
Amplification and cloning of the cDNA fragments encoding VL-CL and VH-CH1 of hybridomas F1546 and F1110.
VL-CL and VH1-CH1 sequences were PCR amplified from reversed transcribed hybridoma cDNA by using a panel of degenerate primers containing appropriate restriction sites enabling the various PCR products to be inserted into the expression vector pPE1.30 The sequences encoding the F1546 and F1110 light chains (VL-CL) were amplified in good yields using the CK and LC7 primers containing XbaI and SacI restriction sites, respectively:
CK: 5′-GCG CCG TCT AGA ATT AAC ACT CAT TCC TGT TGA A-3′
LC7: 5′-CCA GTT CCG AGC TCG TGA TGA CAC AGT CTC CA-3′.
Amplification of the heavy chains was achieved by combining a single primer, CIgG1 located in the CH1 hinge region and containing a SpeI restriction site, with two different primers, VHI and VHIIB located in a partially conserved region, containing a XhoI restriction site and designed to amplify the gene region encoding VH-CH1 of F1110 and F1546, respectively:
CIgG1: 5′-AGG CTT ACT AGT ACA ATC CCT CAC AAT-3′
VHI: 5′-CAG GTC CAA CTC GAG CAG CCT GGG GC-3′
VHIIB 5′GAG GTG CAG CTC GAG GAG TCA GGA CC-3′.
The amplifications were performed in 50 µL reaction buffer containing 2 µL cDNA, 1 µM each primer, 250 µM of each dNTP, 2 mM of MgCl2 and 2.5 U of Taq polymerase (Promega). Samples were subjected to the following program: 94°C for 2 min, 58°C or 60°C for 1 min and 72°C for 1 min, then 39 rounds of denaturation at 94°C for 30 s, annealing at 58°C or 60°C for 1 min and extension at 72°C for 1 min, and an additional extension step at 72°C for 10 min. using a Mastercycler Gradient (Eppendorf). Annealing temperature at 58°C was used for heavy chain amplification whereas hybridization at 60°C was used for light chains amplification. PCR products were gel purified and ligated into pGEMT vector. XL1-Blue E. coli competent cells were transformed and selected by PCR and restriction digestion. The plasmid DNAs from five independent clones were sequenced to establish a consensus sequence for each PCR fragment. As the VH-CH1 from F1546 cDNA was found to contain an additional internal XhoI site, the fragment was then reamplified for subcloning, replacing the XhoI site in VHIIB primer by a SalI restriction site compatible with the XhoI cloning site in the pPEI vector.30 This change altered neither the ORF nor translated sequence.
Subcloning and expression of recombinant Fab fragments.
Clones that corresponded to the consensus sequence were digested with restriction enzymes SacI/XbaI and the XhoI/SpeI (F1110) or SalI/SpeI (F1546) restriction sites, for VL-CL and VH1-CH1, respectively and analyzed by agarose gel electrophoresis. The inserts corresponding to VL-CL and VH-CH1 gene sequences were excised and purified using the QIA quick Gel Extraction Kit (Qiagen) then assembled in the pPE1 vector. pPE1 is designed to express Fab antibody fragments and a hexahistidine (H6) in a format VL-CL::VH-CH-H6 where - and :: represent a covalent bond and a non covalent association, respectively, as previously described.30 The Fab expression is under control of promoter lac. Each step of the assembling was verified by restriction analysis and the final constructs were sequenced. In each case, the sequence of the inserted DNA fragments matched the consensus sequences and ORF was as predicted. These constructs were used to transform XL1-Blue competent cells and positive clones were kept at −80°C in glycerol for long term storage. The FabF1546-H6 and FabF1110-H6 were produced in strain HB2151 as described.29,30,39 Briefly, the bacteria were grown overnight at 30°C in glucose rich SBG10 medium to minimize toxicity then resuspended in SBG1 medium and further incubated at 22°C until the culture reached OD600 nm = 0.5. The bacteria were then induced with 0.2 mM IPTG for two hours, harvested by centrifugation and treated for 30 min with 1 mg.mL−1 polymyxin B to collect the perisplamic fluid. FabF1546-H6 and FabF1110-H6 were purified from the periplasmic fluids by affinity chromatography on Ni-NTA Spin Columns (Qiagen) according to the manufacturer recommendations.
Direct ELISA assay of the binding properties of FabF1546-H6.
Maxisorp™ high-protein binding capacity ELISA plates (Nunc, France) were coated overnight at +4°C with 100 mL of either a crude parasite antigenic extract at a concentration of 20 mg.mL−1 in PBS, or with decreasing quantities of affinity purified MalE-PfHRP2 recombinant protein in PBS. The parasite extract was prepared from the PAM strain. Briefly, the culture was harvested at the schizont stage and washed once in culture medium. Infected red blood cells were re-suspended in Plasmagel to enrich the culture in mature parasite stages. After 30 min at 37°C, supernatants were washed twice in RPMI and the cell pellet was resuspended in buffered water containing a cocktail of protease inhibitors. After two cycles of freezing and thawing, the lysate was finally centrifuged for 10 min at 10,000 g. The supernatant containing water soluble parasite antigens was quantified for protein content by using the Biorad protein assay. The Fab fragments were prepared as periplasmic fluids that resulted from the treatment of the E. coli producing cells with polymyxin B and dialysis against PBS. Plates were saturated for 2 h at RT with 200 µL PBS, pH 7.2, containing 5% bovine serum albumin (BSA). The plates were washed twice with PBS-T. Then, 100 µL of periplasmic fluid, diluted in PBS-T containing 1% BSA (PBS-T-BSA), were dispensed into the wells. The plates were incubated for 1 h at room temperature and washed three times with PBS-T. Binding of Fabs to antigenic preparations was detected by addition of 100 µL of an Anti-Mouse IgG (Fab specific)-Alkaline Phosphatase antibody produced in goat (A1293, Sigma) at a 1:10,000 dilution in PBS-T. The plates were washed three times and 100 µL of 5′-bromo-4-chloro-3-indolyl-phosphate substrate (BluePhosR substrate system, KPL) was added. The plates were incubated for 10 min after which the enzymatic reaction was stopped. The absorbance of solutions was determined at 650 nm.
Acknowledgements
This work was funded by Sanofi-Aventis and the French Ministry of Research and New Technologies “Fonds Dédié pour Combattre les Maladies Parasitaires” and by the Institut Pasteur International Network. E. Ravaoarisoa was supported by a special grant from the Institut Pasteur International Network. We would like to acknowledge Drs. P.H. David (Institut Pasteur), A. Talarmin and D. Menard (Institut Pasteur de Madagascar) for their continuous support and helpful discussions. We are grateful to Dr. M. Randrianarivelojosia for valuable comments on the manuscript. We also thank M. Guillotte and I. Vigan for their expertise in large scale in vitro production of P. falciparum parasites.
Abbreviations
- scFv
single-chain variable fragment
- FR
framework region
- CDR
complementarity determining regions
- Pf
Plasmodium falciparum
- HRP
histidine-rich protein
- VH
variable heavy
- CH
constant heavy
- VL
variable light
- CL
constant light
- MMLV
moloney murine leukemia virus
- IRBC
infected red blood cell
- PAM
palo alto marburg
- Fab
fragment antibody
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/12438
References
- 1.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Payne D. Use and limitations of light microscopy for diagnosing malaria at the primary health care level. Bull World Health Organ. 1988;66:621–626. [PMC free article] [PubMed] [Google Scholar]
- 3.Mwangi TW, Ross A, Snow RW, Marsh K. Case definitions of clinical malaria under different transmission conditions in Kilifi District, Kenya. J Infect Dis. 2005;191:1932–1939. doi: 10.1086/430006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olivar M, Develoux M, Chegou Abari A, Loutan L. Presumptive diagnosis of malaria results in a significant risk of mistreatment of children in urban Sahel. Trans R Soc Trop Med Hyg. 1991;85:729–730. doi: 10.1016/0035-9203(91)90432-x. [DOI] [PubMed] [Google Scholar]
- 5.Amexo M, Tolhurst R, Barnish G, Bates I. Malaria misdiagnosis: effects on the poor and vulnerable. Lancet. 2004;364:1896–1898. doi: 10.1016/S0140-6736(04)17446-1. [DOI] [PubMed] [Google Scholar]
- 6.Zikusooka CM, McIntyre D, Barnes KI. Should countries implementing an artemisinin-based combination malaria treatment policy also introduce rapid diagnostic tests? Malar J. 2008;7:176. doi: 10.1186/1475-2875-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.WHO, author. New perspectives, malaria diagnosis: report of a joint WHO/USAID informal consultation. Oct 25–27, 1999. Geneva: WHO; 2000. 2000. WHO/CDS/RBM/2000.14, WHO/MAL/2000.1091. [Google Scholar]
- 8.Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev. 2002;15:66–78. doi: 10.1128/CMR.15.1.66-78.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wongsrichanalai C. Rapid diagnostic techniques for malaria control. Trends Parasitol. 2001;17:307–309. doi: 10.1016/s1471-4922(01)01925-0. [DOI] [PubMed] [Google Scholar]
- 10.Beadle C, Long GW, Weiss WR, McElroy PD, Maret SM, Oloo AJ, et al. Diagnosis of malaria by detection of Plasmodium falciparum HRP-2 antigen with a rapid dipstick antigen-capture assay. Lancet. 1994;343:564–568. doi: 10.1016/s0140-6736(94)91520-2. [DOI] [PubMed] [Google Scholar]
- 11.Taylor DW, Voller A. The development and validation of a simple antigen detection ELISA for Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg. 1993;87:29–31. doi: 10.1016/0035-9203(93)90409-j. [DOI] [PubMed] [Google Scholar]
- 12.Shiff CJ, Minjas J, Premji Z. The ParaSight-F test: a simple rapid manual dipstick test to detect Plasmodium falciparum infection. Parasitol Today. 1994;10:494–495. doi: 10.1016/0169-4758(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 13.Murray CK, Bell D, Gasser RA, Wongsrichanalai C. Rapid diagnostic testing for malaria. Trop Med Int Health. 2003;8:876–883. doi: 10.1046/j.1365-3156.2003.01115.x. [DOI] [PubMed] [Google Scholar]
- 14.Forney JR, Magill AJ, Wongsrichanalai C, Sirichaisinthop J, Bautista CT, Heppner DG, et al. Malaria rapid diagnostic devices: performance characteristics of the ParaSight F device determined in a multisite field study. J Clin Microbiol. 2001;39:2884–2890. doi: 10.1128/JCM.39.8.2884-2890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parra ME, Evans CB, Taylor DW. Identification of Plasmodium falciparum histidine-rich protein 2 in the plasma of humans with malaria. J Clin Microbiol. 1991;29:1629–1634. doi: 10.1128/jcm.29.8.1629-1634.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desakorn V, Dondorp AM, Silamut K, Pongtavornpinyo W, Sahassananda D, Chotivanich K, et al. Stage-dependent production and release of histidine-rich protein 2 by Plasmodium falciparum. Trans R Soc Trop Med Hyg. 2005;99:517–524. doi: 10.1016/j.trstmh.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Howard RJ, Uni S, Aikawa M, Aley SB, Leech JH, Lew AM, et al. Secretion of a malarial histidine-rich protein (Pf HRP II) from Plasmodium falciparum-infected erythrocytes. J Cell Biol. 1986;103:1269–1277. doi: 10.1083/jcb.103.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.WHO, author. Malaria Rapid diagnostic test performance. Results of WHO product testing of malaria RDTs: Round1. 2008. www.wpro.who.int/NR/rdonlyres/ED81BDE9-B812-4B80-8408-3A129A6365C4/0/OMSFINDRapportMalaria200900514v25.pdf.
- 19.WHO, author. Towards quality testing of malaria rapid diagnostic tests: evidence and methods. Western Pacific Region, Manila, Philippines: WHO; 2006. [Google Scholar]
- 20.Bell D, Wongsrichanalai C, Barnwell JW. Ensuring quality and access for malaria diagnosis: how can it be achieved? Nat Rev Microbiol. 2006;4:7–20. doi: 10.1038/nrmicro1525. [DOI] [PubMed] [Google Scholar]
- 21.WHO, author. Sources and Prices of Selected Products for the Prevention, Diagnosis and treatment of malaria. A joint WHO RBM, UNICEF, UNAIDS PSI, MSH project. http://apps.who.int/medicinedocs/en/d/Js6174e/
- 22.Chiodini PL, Bowers K, Jorgensen P, Barnwell JW, Grady KK, Luchavez J, et al. The heat stability of Plasmodium lactate dehydrogenase-based and histidine-rich protein 2-based malaria rapid diagnostic tests. Trans R Soc Trop Med Hyg. 2007;101:331–337. doi: 10.1016/j.trstmh.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Jorgensen P, Chanthap L, Rebueno A, Tsuyuoka R, Bell D. Malaria rapid diagnostic tests in tropical climates: the need for a cool chain. Am J Trop Med Hyg. 2006;74:750–754. [PubMed] [Google Scholar]
- 24.Loo L, Robinson MK, Adams GP. Antibody engineering principles and applications. Cancer J. 2008;14:149–153. doi: 10.1097/PPO.0b013e318173a5d5. [DOI] [PubMed] [Google Scholar]
- 25.Rapley R. The biotechnology and applications of antibody engineering. Mol Biotechnol. 1995;3:139–154. doi: 10.1007/BF02789110. [DOI] [PubMed] [Google Scholar]
- 26.Rock EP, Marsh K, Saul AJ, Wellems TE, Taylor DW, Maloy WL, et al. Comparative analysis of the Plasmodium falciparum histidine-rich proteins HRP-I, HRP-II and HRP-III in malaria parasites of diverse origin. Parasitology. 1987;95:209–227. doi: 10.1017/s0031182000057681. [DOI] [PubMed] [Google Scholar]
- 27.Lee N, Baker J, Andrews KT, Gatton ML, Bell D, Cheng Q, et al. Effect of sequence variation in Plasmodium falciparum histidine-rich protein 2 on binding of specific monoclonal antibodies: Implications for rapid diagnostic tests for malaria. J Clin Microbiol. 2006;44:2773–2778. doi: 10.1128/JCM.02557-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brochet X, Lefranc MP, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36:503–508. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carter P, Bedouelle H, Winter G. Improved oligonucleotide site-directed mutagenesis using M13 vectors. Nucleic Acids Res. 1985;13:4431–4443. doi: 10.1093/nar/13.12.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bedouelle H, Belkadi L, England P, Guijarro JI, Lisova O, Urvoas A, et al. Diversity and junction residues as hotspots of binding energy in an antibody neutralizing the dengue virus. Febs J. 2006;273:34–46. doi: 10.1111/j.1742-4658.2005.05045.x. [DOI] [PubMed] [Google Scholar]
- 31.Leke RF, Djokam RR, Mbu R, Leke RJ, Fogako J, Megnekou R, et al. Detection of the Plasmodium falciparum antigen histidine-rich protein 2 in blood of pregnant women: implications for diagnosing placental malaria. J Clin Microbiol. 1999;37:2992–2996. doi: 10.1128/jcm.37.9.2992-2996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.WHO, author. Guidelines for the treatment of malaria: second edition. 2010. www.who.int/malaria/publications/atoz/9789241547925/en/index.html.
- 33.Perkins MD, Bell DR. Working without a blindfold: the critical role of diagnostics in malaria control. Malar J. 2008;7:5. doi: 10.1186/1475-2875-7-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.WHO, author. Diagnosis and management of severe malaria. Jun 2002; Trial edition. 2002. www.malaria.org.zw/Case/cm26.pdf.
- 35.Baker J, McCarthy J, Gatton M, Kyle DE, Belizario V, Luchavez J, et al. Genetic diversity of Plasmodium falciparum histidine-rich protein 2 (PfHRP2) and its effect on the performance of PfHRP2-based rapid diagnostic tests. J Infect Dis. 2005;192:870–877. doi: 10.1086/432010. [DOI] [PubMed] [Google Scholar]
- 36.Zerpa NC, Wide A, Noda J, Bermudez H, Pabon R, Noya OO. Immunogenicity of synthetic peptides derived from Plasmodium falciparum proteins. Exp Parasitol. 2006;113:227–234. doi: 10.1016/j.exppara.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 37.Versteeg I, Mens PF. Development of a stable positive control to be used for quality assurance of rapid diagnostic tests for malaria. Diagn Microbiol Infect Dis. 2009;64:256–260. doi: 10.1016/j.diagmicrobio.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 38.Smith KA, Nelson PN, Warren P, Astley SJ, Murray PG, Greenman J. Demystified recombinant antibodies. J Clin Pathol. 2004;57:912–917. doi: 10.1136/jcp.2003.014407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monsellier E, Bedouelle H. Improving the stability of an antibody variable fragment by a combination of knowledge-based approaches: validation and mechanisms. J Mol Biol. 2006;362:580–593. doi: 10.1016/j.jmb.2006.07.044. [DOI] [PubMed] [Google Scholar]
- 40.Fandeur T, Bonnefoy S, Mercereau-Puijalon O. In vivo and in vitro derived Palo Alto lines of Plasmodium falciparum are genetically unrelated. Mol Biochem Parasitol. 1991;47:167–178. doi: 10.1016/0166-6851(91)90176-7. [DOI] [PubMed] [Google Scholar]
- 41.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 42.Doury JC, Goasdoue JL, Tolou H, Martelloni M, Bonnefoy S, Mercereau-Puijalon O. Characterisation of the binding sites of monoclonal antibodies reacting with the Plasmodium falciparum rhoptry protein RhopH3. Mol Biochem Parasitol. 1997;85:149–159. doi: 10.1016/s0166-6851(96)02819-8. [DOI] [PubMed] [Google Scholar]