

Abstract
Abstract
The best neuroprotectant for acute ischaemic stroke would always be the rapid return of oxygen and glucose to physiological levels. This is currently provided by thrombolysis which restores blood flow to the ischaemic region. The attempt to confer neuroprotection by targeting the brain parenchyma has shown promise in experimental stroke models, but has unequivocally failed to translate to the clinic. Neuroprotective therapy primarily targets the biochemical cascade that produces cell death following cerebral ischaemia. However, these agents may also alter signal transduction that controls cerebral blood flow, for example glutamate, which may affect the outcome after ischaemia. In these cases, neuroprotection may potentially be due to the improved access to oxygen and glucose rather than biochemical prevention of cell death. Improvement in cerebral blood flow is an important but often overlooked effect of neuroprotective therapy, analogous to the protective effects of drug-induced hypothermia. This short review will discuss cerebral blood flow alteration and protection of the brain in the context of ischaemic preconditioning, oxygen sensing and thrombolysis. Future neuroprotection studies in cerebral ischaemia require stringent monitoring of cerebral blood flow, plus other physiological parameters. This will increase the chances that any protection observed may be able to translate to human therapy.
Alastair Buchan (left) studied at Cambridge, Oxford and Harvard Universities and graduated from Medical School in 1980. After training positions at University of Oxford, Imperial College London and University Hospital University of Western Ontario, he specialized as a neurologist before undertaking a postdoctoral fellowship at Cornell University Medical Centre, New York City. This was followed by appointments as Assistant Professor at the University of Western Ontario, Associate Professor at the University of Ottawa and then Professor of Stroke Research at the University of Calgary. Back at Oxford since 2005, he is now Dean of the Medical School and Professor of Stroke Medicine in the Nuffield Department of Medicine. Brad Sutherland (right) obtained his BSc(Hons) in Pharmacology (2004) and his PhD in Pharmacology (2009) from the Department of Pharmacology & Toxicology, University of Otago, New Zealand. Currently a Postdoctoral Research Fellow in the Acute Stroke Programme in the Nuffield Department of Clinical Medicine, University of Oxford, his work is funded by the Fondation Leducq and forms part of a Transatlantic Network of Excellence investigating the control of cerebral blood flow and how this is disrupted following a stroke. Research interests include identifying neuroprotective strategies that can reduce brain injury following stroke, and the pharmacological mechanisms behind these effects.
 |
Introduction
During acute ischaemic stroke, the early restoration of oxygen and glucose to the ischaemic region is the best ‘neuroprotective therapy’. This is currently provided clinically by thrombolysis which reinstates blood flow by breaking down the clot or embolus. Targeting the biochemical cascade that occurs following ischaemia to restrict injury to the brain parenchyma has been successfully trialled in many pre-clinical studies (O'Collins et al. 2006). However, no neuroprotectant that directly targets the brain parenchyma has achieved success in the clinic, with many reasons being cited for these translational failures (Hoyte et al. 2004). Many pre-clinical neuroprotective studies were of low methodological quality (O'Collins et al. 2006), with physiological parameters not being measured. Studies have shown that some neuroprotectants alter physiological parameters, such as temperature, blood pressure and glucose, which may contribute to their neuroprotective effects. One example is the NMDA receptor antagonist MK-801 which induced hypothermia to produce neuroprotection (Buchan & Pulsinelli, 1990). Reducing temperature is a well-established protective mechanism and if it is not adequately controlled in pre-clinical studies, it can mask genuine neuroprotection of specific compounds. In addition, MK-801 also significantly increased cerebral blood flow (CBF) which contributed to its neuroprotective effects (Buchan et al. 1992). Therefore, the question is raised, do neuroprotective agents produce neuroprotection by actively inhibiting the ischaemic cascade thereby preventing cell death following an injurious insult (pharmacological neuroprotection), or do these agents alter physiological parameters such as CBF and temperature (physiological neuroprotection)? In this review, we will focus on the hypothesis that neuroprotective agents may be reducing injury through alteration of CBF during or following ischaemia in animal models, an effect not necessarily translated to humans. Other physiological parameters will not be further discussed here. A selection of compounds that produce neuroprotection while augmenting CBF in pre-clinical studies are listed in Table 1.
Table 1.
A selection of neuroprotective strategies that reduce neuronal injury and improve CBF following focal cerebral ischaemia in animals
| Strategy | Action | Model | Infarct volume | CBF change | Time of administration | Route of administration | Reference |
|---|---|---|---|---|---|---|---|
| Phenylephrine | α-1 agonist, hypertensive | Mouse transient MCAO | Reduced | Increased | 10 or 60 min post MCAO onset | i.v. infusion | Shin et al. (2008) |
| MK-801 | NMDA receptor antagonist | Rat permanent & transient MCAO | Reduced | Increased | 30 min prior to MCAO | i.p. | Buchan et al. (1992) |
| CGS-19755 | NMDA receptor antagonist | Rat permanent MCAO | Reduced | Increased | At MCAO onset | i.v. infusion | Takizawa et al. (1991) |
| G-CSF & GM-CSF | Cytokine & haematopoietic | Mouse permanent MCAO | Reduced | Increased | 7 days prior to MCAO | s.c. | Sugiyama et al. (2011) |
| Exercise | Many mechanisms | Rat transient MCAO | Reduced | Increased | 3 weeks prior to MCAO | Treadmill | Zwagerman et al. (2010) |
| IPC | Many mechanisms | Mouse transient MCAO | Reduced | Increased | 3 days prior to MCAO | Short MCAO | Hoyte et al. (2006) |
| IPC | Many mechanisms | Rat permanent MCAO | Reduced | Increased | 24 h prior to MCAO | Short MCAO | Zhao & Nowak (2006) |
| DMOG | PHD inhibitor | Rat permanent & transient MCAO | Reduced | Increased | 20 h prior to permanent MCAO or at reperfusion following transient MCAO | i.p. (permanent MCAO), i.v. (transient MCAO) | Nagel et al. (2011) |
| VEGF | Angiogenic | Rat transient MCAO | Reduced | Increased angiogenesis | 24 h post MCAO onset | i.c.v. infusion | Sun et al. (2003) |
| EPO | Haematopoietic and erythropoietic | Mouse permanent MCAO | Reduced | Increased | 30 min prior to MCAO | i.p. | Li et al. (2007) |
DMOG, dimethyloxalylglycine; EPO, erythropoietin; G-CSF, granulocyte colony-stimulating-factor; GM-CSF, granulocyte-macrophage colony-stimulating-factor; i.c.v. intracerebroventricular; IPC, ischaemic preconditioning; MCAO, middle cerebral artery occlusion; PHD, prolyl hydroxylase; VEGF, vascular endothelial growth factor.
CBF changes following ischaemia
Ischaemia has profound effects on CBF levels. Antegrade blood flow ceases during arterial occlusion, but collateral vessels may sustain cerebral perfusion in the arterial bed (Liebeskind, 2007). Ischaemic damage occurs when collaterals fail to provide adequate perfusion leading to symptom onset (Liebeskind, 2007). The largest flow reduction is observed in the ischaemic core territory, while the reduction is less marked in the surrounding penumbra (Girouard & Iadecola, 2006). Autoregulation of CBF is lost during ischaemia meaning arterial blood pressure changes have a large impact on CBF, with recent evidence showing that pharmacologically induced hypertension improves CBF and subsequently reduces injury following middle cerebral artery occlusion (MCAO) (Shin et al. 2008). The neurovascular control of CBF is also disrupted during ischaemia, with the loss of coupling between neural activity and haemodynamic effects (Bundo et al. 2002).
Blood flow remains attenuated until the occluding clot or embolus has been removed or dissolved. Upon reperfusion, such as following treatment with the thrombolytic recombinant tissue plasminogen activator (rtPA), a significant hyperaemia within the ischaemic region immediately occurs (Fig. 1). However, this is followed by a post-ischaemic hypoperfusion (Fig. 1), which can last for hours. This is described as the ‘no-reflow phenomenon’ and has been attributed to the narrowing of capillaries (Hauck et al. 2004) and loss of both arteriolar dilating mechanisms and cerebrovascular reactivity (Leffler et al. 1989). Pericytes are susceptible to ischaemic injury resulting in contraction of capillaries causing attenuated CBF, even after reperfusion (Yemisci et al. 2009). In addition, restoration of blood flow following ischaemia can exacerbate damage to neurons and the vasculature, which is termed ‘reperfusion injury’. Reperfusion injury is due to the oxygen ‘surge’ and free radical overproduction (Aronowski et al. 1997) leading to blood–brain barrier (BBB) breakdown and oedema formation (Heo et al. 2005). These effects produce further dysregulation of CBF due to neuronal, glial and pericyte injury (Attwell et al. 2010) and can antagonize any beneficial effects produced by recanalization of the vessel (Aronowski et al. 1997).
Figure 1. CBF during ischaemia and reperfusion and subsequent injury in the rat model of MCAO.
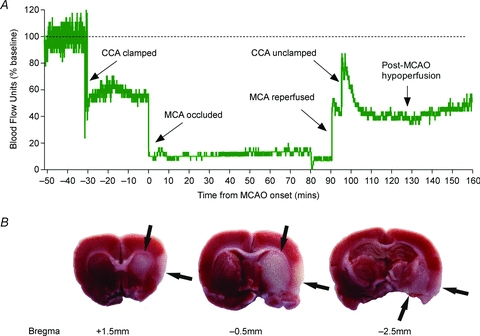
A, methodology of the MCAO technique and ethical regulations associated with these experiments have been previously described (Nagel et al. 2011). Laser Doppler flowmetry was used to measure relative CBF over the right somatosensory cortex of a male Wistar rat. Baseline CBF was normalized to 100% blood flow units (BFU). Upon temporary common carotid artery (CCA) ligation, CBF was reduced to 60% BFU. A silicon-coated 4-0 monofilament was then inserted through the external carotid artery and advanced up the internal carotid artery to occlude the right middle cerebral artery (MCA). MCAO was confirmed by a sharp decrease in CBF to < 20% BFU, and this was maintained for 90 min. Reperfusion of the MCA was achieved through retraction of the monofilament, when a sharp increase in CBF and a small hyperaemia was observed. After 5 min of MCA reperfusion (to allow removal of the monofilament), the CCA was unclamped to allow full reperfusion to the ischaemic area, which produced a further increase in CBF and hyperaemia lasting approximately 10 min. This was followed by a post-ischaemic hypoperfusion at 50% BFU during the next 60 min. B, cerebral injury was observed 24 h post-ischaemia onset in the striatum and the cortex (arrows) from the same animal as A using triphenyl-tetrazolium staining.
Link between CBF and neuroprotection
A number of pathways are involved in the control of CBF (Attwell et al. 2010), with some also involved in the biochemical cascade leading to cell death following ischaemia. One example is glutamate, a neurotransmitter that produces excitotoxicity but also activates pathways that regulate CBF (Fig. 2). In neurons, glutamate activates NMDA receptors, which increases intracellular Ca2+ and vasodilates arterioles through nitric oxide (NO) and prostaglandins (PGs). In astrocytes, glutamate activates metabotropic glutamate receptors (mGluRs), which releases arachidonic acid (AA) derivatives resulting in either dilatation or constriction of vessels depending on the metabolite. Many NMDA receptor antagonists were neuroprotective pre-clinically but failed to translate to clinical neuroprotection. Both competitive (Takizawa et al. 1991) and non-competitive (Buchan et al. 1992) NMDA receptor antagonists produced significant increases in CBF while attenuating injury. This suggests that NMDA receptor antagonists may confer neuroprotection through augmenting CBF in affected brain areas. Other neuroprotective strategies also have similar effects on blood flow (see Table 1). Granulocyte colony-stimulating-factor (G-CSF) and granulocyte-macrophage colony-stimulating-factor (GM-CSF) were neuroprotective following focal ischaemia, which was coupled with an improvement in cerebral perfusion and arteriogenesis (Sugiyama et al. 2011). Animals that underwent exercise prior to ischaemia had improved CBF upon reperfusion, which was associated with a reduction in injury (Zwagerman et al. 2010). This links the improvement of CBF with reduction in injury. There is a possibility that many neuroprotective agents modulate CBF following ischaemia and protection may in part be due to the greater access to oxygen and glucose rather than targeting the ischaemic cascade to prevent cell death. It is imperative that all pre-clinical cerebral ischaemia studies measure CBF to assess what effects these agents are having on cerebral perfusion. Many techniques can be used including laser Doppler/Speckle flowmetry, magnetic resonance imaging, perfusion-weighted imaging and autoradiography.
Figure 2. The control of CBF by glutamate through astrocytes and neurons.
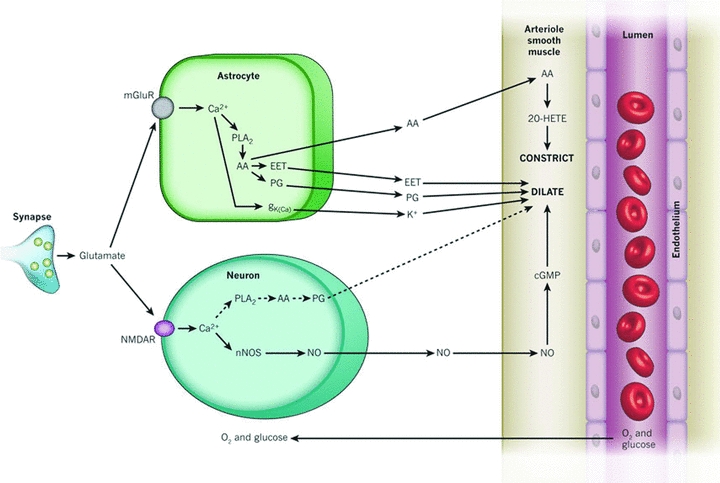
Glutamate released from synapses can modulate vascular tone and the subsequent supply of oxygen and glucose through neurons and astrocytes. In neurons, glutamate activates NMDA receptors increasing [Ca2+]i and either producing NO through neuronal nitric oxide synthase (nNOS) or prostaglandins (PG) through phospholipase A2 (PLA2) and cyclooxygenase-2 to dilate vessels. In astrocytes, glutamate acts on metabotropic glutamate receptors (mGluR) which increases [Ca2+]i and generates arachidonic acid (AA). Three metabolites are derived from AA: PG and EETs in astrocytes which dilate vessels, and 20-HETE in smooth muscle which constricts vessels. Ca2+-gated K+ channels (gK(Ca)) on astrocytic endfeet are also activated which releases K+ to dilate vessels. This figure has been reproduced with permission from Attwell et al. (2010) and Nature Publishing Group.
Changes in CBF with neuroprotective therapy may contribute to some protective effects seen in animal models of cerebral ischaemia, and these effects need to be closely scrutinized before proceeding to clinical trials with a putative neuroprotective compound. There are some drugs that produced neuroprotection but had no effects on CBF such as the antioxidant NXY-059 (Zhao et al. 2001), suggesting that CBF augmentation only forms part of the neuroprotective response. In addition, many of these CBF-enhancing neuroprotective drugs have also failed to translate into clinical success. This limited efficacy in humans is probably not due to the biological actions of the drug as significant effects were seen in animals, but due to the differences in methodology between clinical and pre-clinical trials such as time window, and the heterogeneity of human ischaemic stroke. Reasons for these translational failures have been reported previously (Hoyte et al. 2004; Endres et al. 2008). Acute stroke clinical trials assessing neuroprotective drugs only measured clinical outcome without assessing pharmacological effects. It is important to establish clinically if the drugs are producing the desired biochemical/physiological changes (such as CBF alteration) as well as any clinical benefit. Biomarkers that are directly involved in the ischaemic cascade could be used as a surrogate of ischaemia and potentially titrate any neuroprotective effect. Unfortunately, such biomarkers have currently not yet been identified. Measuring additional outcomes will provide clues to reasons for translational failure of neuroprotectants and how translation can be improved to make neuroprotection a clinical reality.
Ischaemic preconditioning changes in CBF
In addition to neuroprotective agents, ischaemic preconditioning (IPC) is an established experimental protocol that confers resistance to ischaemia, partially by altering CBF (Dirnagl et al. 2009). IPC is induced by a short period of ischaemia that improves the tolerance of the brain to subsequent injurious ischaemia (Chen et al. 1996). Protection is observed in two time windows: an early one, occurring within minutes of the IPC stimulus and a late one at 24–72 h post IPC. The contribution of CBF to the increased resistance of the IPC brain has only been correlated with the late window of protection. Rodent studies have demonstrated that IPC animals had an attenuated drop in regional CBF (rCBF) in the ipsilateral hemisphere during MCAO compared to naive animals, which resulted in smaller infarct volumes (Hoyte et al. 2006; Zhao & Nowak, 2006).
The ability of preconditioned animals to maintain rCBF appears to be mediated by the induction of genes and pathways involved in the preservation of the vasculature's integrity. Genes participating in vasculogenesis and vasoregulation, such as endothelial nitric oxide synthase (eNOS), vascular endothelial growth factor (VEGF) and erythropoietin (EPO) are activated by IPC (Gustavsson et al. 2007b). VEGF stimulates vasculogenesis, increasing the vascular density (Gustavsson et al. 2007a), while both eNOS (Atochin et al. 2003) and EPO (Li et al. 2007) improve rCBF. VEGF and EPO also activate the phosphoinositide 3-kinase (PI3k)-Akt pathway, which can regulate eNOS activity (Hashiguchi et al. 2004). NO produced by eNOS causes vasodilatation and decreases leukocyte–endothelial interactions (Atochin et al. 2003). eNOS is required for ischaemic tolerance by IPC since IPC did not produce protection against ischaemia in eNOS knockout mice (Atochin et al. 2003).
Overall, alterations of CBF contribute to the neuroprotective capacity of IPC. However, this physiological effect is a manifestation of changes in gene expression and protein activity, which maintain the integrity and increase the tolerance of the neurovascular unit to severe ischaemia.
CBF changes through oxygen sensors and downstream pathways
Hypoxia-inducible factor (HIF), the master regulator of oxygen homeostasis, is an attractive pharmacological target that could be used to enhance cerebral perfusion (Semenza, 1999). HIF is composed of two subunits, α and β. Under normoxia, iron- and 2-oxoglutarate-dependent oxygenases utilize oxygen to catalyse the hydroxylation of HIF-α, which then targets HIF-α for proteasomal degradation; during ischaemia, the lack of oxygen prevents the hydroxylation of HIF-α, and allows HIF to induce the transcription of a number of target genes including VEGF and EPO (Harten et al. 2010). Iron chelators or 2-oxoglutarate-dependent oxygenase inhibitors can stabilize and activate HIF, which have been shown to produce neuroprotection and are associated with the improvement of CBF after reperfusion (Harten et al. 2010). Administration of dimethyloxalylglycine (DMOG), an inhibitor of 2-oxoglutarate-dependent oxygenases, reduced infarct volume following both permanent (Fig. 3) and transient MCAO (Nagel et al. 2011). Pretreatment with DMOG produced less severe ischaemia during permanent MCAO that was inversely correlated with injury (Fig. 3), while DMOG post-treatment following 60 min MCAO upregulated CBF at 24 h (Nagel et al. 2011). In humans, infusion of desferroxamine (DFO), an iron chelator, led to cerebral vasodilatation and improved CBF, and was temporally associated with HIF-1 activation (Sorond et al. 2009). DFO also increased forearm blood flow and improved vasoreactivity in patients with endothelial dysfunction (Duffy et al. 2001).
Figure 3. The effects of dimethyloxalylglycine (DMOG) on cerebral injury and CBF following permanent MCAO.
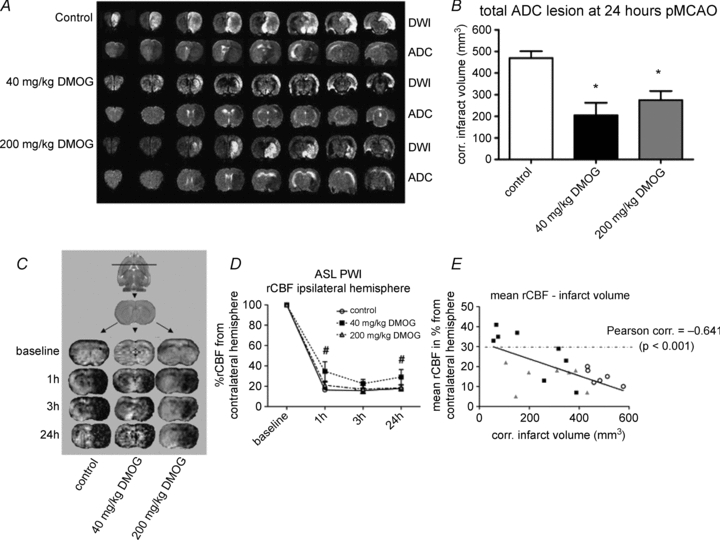
A, one representative animal per group is presented by diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) maps. In DWI, infarcts are detected as areas of hyperintensity; in ADC maps, infarcts are detected as areas of hypointensity. B, quantification of ADC infarct volumes 24 h post-MCAO onset show that both 40 mg kg-1 DMOG and 200 mg kg-1 DMOG reduced infarct volume compared to control. *P < 0.05 versus control. C, arterial spin labelling: perfusion-weighted imaging was used to create rCBF maps for each group 1 h, 3 h and 24 h post-MCAO. Areas with reduced blood flow are detected as areas of hypointensity. D, rCBF values decreased significantly for all groups following occlusion. During occlusion, rCBF values were not significantly different between groups, but at 1 and 24 h there was a tendency towards higher rCBF values in the 40 mg kg-1 DMOG group compared with the other groups. #P < 0.1 versus control. E, mean rCBF over time was inversely correlated (P < 0.001) with final infarct volume, with the 40 mg kg-1 DMOG group producing higher rCBF (50% of animals were above the critical threshold of 30% rCBF of baseline) associated with lower infarcts. This figure has been adapted and reproduced with permission from Nagel et al. (2011) and Nature Publishing Group.
Many genes (eNOS, VEGF and EPO) that are activated by IPC are HIF target genes (Harten et al. 2010), and can have an effect on outcome following ischaemia through perfusion changes and angiogenesis. Mice with vasodilatory eNOS knocked out had larger infarcts than wild-type mice, had more severe ischaemia (Huang et al. 1996), and had diminished ischaemia-induced angiogenesis and pericyte recruitment (Yu et al. 2005). Moreover, administration of NO donors increased CBF which was associated with a reduction in infarct volume following permanent ischaemia (Willmot et al. 2005). VEGF is a pivotal regulator in angiogenesis, and first appears in CNS pericytes within 24 h of low oxygen exposure, and in pericapillary astrocytes by 4 days (Dore-Duffy & LaManna, 2007). VEGF administration intracerebroventricularly reduced infarct volume and neurological deficit following MCAO while increasing neurogenesis and angiogenesis (Sun et al. 2003). Conversely, VEGF can activate matrix metalloprotease-9 leading to vascular permeability and BBB breakdown (Valable et al. 2005). EPO levels increase substantially within hours of hypoxic stimulation (Jelkmann, 1992) and act upon vascular cells to foster angiogenesis (Li et al. 2004). EPO administration to mice that underwent MCAO reduced infarct volume, which was associated with an increase in angiogenesis and rCBF (Li et al. 2007). However, a recent multi-centre EPO stroke trial failed to provide neuroprotection (Ehrenreich et al. 2009). This reinforces the notion that neuroprotection of EPO seen in pre-clinical studies might be due to confounding factors such as an increase in CBF.
HIF activation following ischaemia promotes angiogenesis, which is capable of overcoming the detrimental effects of arterial occlusion by augmenting rCBF which is determined by the quantity of microvessels (Fraisl et al. 2009). Capillary restructuring requires at least a week, and thus does not play a part in transient disturbances to the balance between oxygen delivery and energy demand (LaManna et al. 2004). Nevertheless, in subacute and chronic phases, the increase in capillary density, and thus the increase in glucose transporter-1 protein, results in an increase in the transport of glucose across the BBB, compensating for the increased glucose consumption in hypoxia (LaManna et al. 2004).
CBF alteration with thrombolysis
The only FDA-approved thrombolytic for acute ischaemic stroke is rtPA which restores CBF, but its application is limited to within 4.5 h from ischaemic onset (Hacke et al. 2008). rtPA produces a decrease in eNOS expression (Kilic et al. 2005) which may contribute to the post-ischaemic hypoperfusion (Fig. 1) (Kilic et al. 2001), and also leads to dysfunctional vascular reactivity to vasoactive mediators (Cipolla et al. 2000). In addition, rtPA can directly affect the integrity of the BBB via the low density lipoprotein receptor-related protein (Yepes et al. 2003) leading to the dissociation of astrocytic endfeet from cerebral vessels (Yamashita et al. 2009) and vasogenic oedema (Goto et al. 2007). rtPA also potentiates NMDA receptor activation (Nicole et al. 2001) leading to neurotoxicity (Harston et al. 2010).
On the other hand, rtPA is involved in neurovascular regulation by enhancing the coupling between NMDA receptor activity and the synthesis of neuronal NO (Park et al. 2008). rtPA is formulated in a high concentration of l-arginine, the substrate for NO production. l-Arginine has a number of beneficial effects on CBF, mainly related to endothelial NO activation. l-Arginine increases CBF following i.v. infusion in healthy volunteers (Pretnar-Oblak et al. 2006), and in animal models of cerebral ischaemia (Willmot et al. 2005), which can reduce injury. Ischaemic vessels also preferentially dilate with l-arginine compared to vessels in non-ischaemic tissue (He et al. 1995). It is currently unknown whether administering rtPA with l-arginine is beneficial, but l-arginine may be fuelling rtPA neurotoxicity through the NO pathway (Harston et al. 2010).
Administering rtPA with a neuroprotective agent could improve post-ischaemic CBF, reduce cerebral injury, restrict adverse effects and extend the therapeutic time window. Numerous agents have additive effects with rtPA in animal models of cerebral ischaemia, including free radical scavengers, matrix metalloprotease inhibitors, anti-excitotoxic and anti-inflammatory agents (Kaur et al. 2004). Unfortunately, none of these approaches have reached clinical practice. Identifying combination strategies that can counteract rtPA-induced post-ischaemic hypoperfusion and neurotoxicity as well as providing a neuroprotective effect may lead to a reduction in injury and improved functional outcome following stroke.
Conclusion
Many neuroprotective treatments for ischaemic stroke are successful pre-clinically in animal models but have failed to translate into clinical benefit. A number of these neuroprotective agents appear to be modulating CBF, which may improve access to oxygen and glucose in the brain leading to their observed protective effect, rather than a biochemically mediated prevention of cell death. However, even neuroprotective drugs that augment CBF in animal studies have failed to translate into clinical neuroprotection, which may be due to methodological differences rather than drug action. This suggests that most likely CBF improvement may be important, but not necessary, for neuroprotection. Neuroprotective strategies that have multiple mechanisms of action including augmenting CBF will present the most attractive option for neuroprotective acute stroke treatment. Any future neuroprotective therapy will be used in conjunction with thrombolysis, which restores blood flow to the affected area. Future pre-clinical studies must use careful monitoring of CBF, and other physiological parameters, to identify if neuroprotective agents have effects on these parameters in addition to their specific mechanism of action. Also, clinical studies should use additional measures to assess the pharmacological effects of any neuroprotective drug as well as clinical outcome. By ascertaining these effects, genuine or ‘true’ neuroprotection can be identified, which should aid in translation to human studies and ultimately clinical benefit.
Acknowledgments
Funding for this work was received from the Fondation Leducq, Medical Research Council UK, the Dunhill Medical Trust, and the National Institute for Health Research Biomedical Research Centre. All authors drafted, revised and finalized this manuscript. The authors have no conflicts of interest to disclose.
Glossary
Abbreviations
- BBB
blood–brain barrier
- CBF
cerebral blood flow
- CCA
common carotid artery
- DMOG
dimethyloxalylglycine
- eNOS
endothelial nitric oxide synthase
- EPO
erythropoietin
- G-CSF
granulocyte colony-stimulating-factor
- GM-CSF
granulocyte-macrophage colony-stimulating-factor
- HIF
hypoxia inducible factor
- IPC
ischaemic preconditioning
- MCAO
middle cerebral artery occlusion
- NO
nitric oxide
- rCBF
regional cerebral blood flow
- rtPA
recombinant tissue plasminogen activator
- VEGF
vascular endothelial growth factor
References
- Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke. 2003;34:1299–1303. doi: 10.1161/01.STR.0000066870.70976.57. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan A, Pulsinelli WA. Hypothermia but not the N-methyl-d-aspartate antagonist, MK-801, attenuates neuronal damage in gerbils subjected to transient global ischemia. J Neurosci. 1990;10:311–316. doi: 10.1523/JNEUROSCI.10-01-00311.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan AM, Slivka A, Xue D. The effect of the NMDA receptor antagonist MK-801 on cerebral blood flow and infarct volume in experimental focal stroke. Brain Res. 1992;574:171–177. doi: 10.1016/0006-8993(92)90814-p. [DOI] [PubMed] [Google Scholar]
- Bundo M, Inao S, Nakamura A, Kato T, Ito K, Tadokoro M, Kabeya R, Sugimoto T, Kajita Y, Yoshida J. Changes of neural activity correlate with the severity of cortical ischemia in patients with unilateral major cerebral artery occlusion. Stroke. 2002;33:61–66. doi: 10.1161/hs0102.101816. [DOI] [PubMed] [Google Scholar]
- Chen J, Graham SH, Zhu RL, Simon RP. Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:566–577. doi: 10.1097/00004647-199607000-00006. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Lessov N, Clark WM, Haley EC., Jr Postischemic attenuation of cerebral artery reactivity is increased in the presence of tissue plasminogen activator. Stroke. 2000;31:940–945. doi: 10.1161/01.str.31.4.940. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore-Duffy P, LaManna JC. Physiologic angiodynamics in the brain. Antioxid Redox Signal. 2007;9:1363–1371. doi: 10.1089/ars.2007.1713. [DOI] [PubMed] [Google Scholar]
- Duffy SJ, Biegelsen ES, Holbrook M, Russell JD, Gokce N, Keaney JF, Jr, Vita JA. Iron chelation improves endothelial function in patients with coronary artery disease. Circulation. 2001;103:2799–2804. doi: 10.1161/01.cir.103.23.2799. [DOI] [PubMed] [Google Scholar]
- Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, Schellinger PD, Bohn M, Becker H, Wegrzyn M, Jahnig P, Herrmann M, Knauth M, Bahr M, Heide W, Wagner A, Schwab S, Reichmann H, Schwendemann G, Dengler R, Kastrup A, Bartels C. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–e656. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- Endres M, Engelhardt B, Koistinaho J, Lindvall O, Meairs S, Mohr JP, Planas A, Rothwell N, Schwaninger M, Schwab ME, Vivien D, Wieloch T, Dirnagl U. Improving outcome after stroke: overcoming the translational roadblock. Cerebrovasc Dis. 2008;25:268–278. doi: 10.1159/000118039. [DOI] [PubMed] [Google Scholar]
- Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell. 2009;16:167–179. doi: 10.1016/j.devcel.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Goto H, Fujisawa H, Oka F, Nomura S, Kajiwara K, Kato S, Fujii M, Maekawa T, Suzuki M. Neurotoxic effects of exogenous recombinant tissue-type plasminogen activator on the normal rat brain. J Neurotrauma. 2007;24:745–752. doi: 10.1089/neu.2006.0183. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Mallard C, Vannucci SJ, Wilson MA, Johnston MV, Hagberg H. Vascular response to hypoxic preconditioning in the immature brain. J Cereb Blood Flow Metab. 2007a;27:928–938. doi: 10.1038/sj.jcbfm.9600408. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Wilson MA, Mallard C, Rousset C, Johnston MV, Hagberg H. Global gene expression in the developing rat brain after hypoxic preconditioning: involvement of apoptotic mechanisms? Pediatr Res. 2007b;61:444–450. doi: 10.1203/pdr.0b013e3180332be4. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Harston GW, Sutherland BA, Kennedy J, Buchan AM. The contribution of L-arginine to the neurotoxicity of recombinant tissue plasminogen activator following cerebral ischemia: a review of rtPA neurotoxicity. J Cereb Blood Flow Metab. 2010;30:1804–1816. doi: 10.1038/jcbfm.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12:459–480. doi: 10.1089/ars.2009.2870. [DOI] [PubMed] [Google Scholar]
- Hashiguchi A, Yano S, Morioka M, Hamada J, Ushio Y, Takeuchi Y, Fukunaga K. Up-regulation of endothelial nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2004;24:271–279. doi: 10.1097/01.WCB.0000110539.96047.FC. [DOI] [PubMed] [Google Scholar]
- Hauck EF, Apostel S, Hoffmann JF, Heimann A, Kempski O. Capillary flow and diameter changes during reperfusion after global cerebral ischemia studied by intravital video microscopy. J Cereb Blood Flow Metab. 2004;24:383–391. doi: 10.1097/00004647-200404000-00003. [DOI] [PubMed] [Google Scholar]
- He Z, Ibayashi S, Nagao T, Fujii K, Sadoshima S, Fujishima M. l-Arginine ameliorates cerebral blood flow and metabolism and decreases infarct volume in rats with cerebral ischemia. Brain Res. 1995;699:208–213. doi: 10.1016/0006-8993(95)00907-8. [DOI] [PubMed] [Google Scholar]
- Heo JH, Han SW, Lee SK. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. 2005;39:51–70. doi: 10.1016/j.freeradbiomed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Hoyte L, Kaur J, Buchan AM. Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol. 2004;188:200–204. doi: 10.1016/j.expneurol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Hoyte LC, Papadakis M, Barber PA, Buchan AM. Improved regional cerebral blood flow is important for the protection seen in a mouse model of late phase ischemic preconditioning. Brain Res. 2006;1121:231–237. doi: 10.1016/j.brainres.2006.08.107. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- Jelkmann W. Erythropoietin: structure, control of production, and function. Physiol Rev. 1992;72:449–489. doi: 10.1152/physrev.1992.72.2.449. [DOI] [PubMed] [Google Scholar]
- Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab. 2004;24:945–963. doi: 10.1097/01.WCB.0000137868.50767.E8. [DOI] [PubMed] [Google Scholar]
- Kilic E, Bahr M, Hermann DM. Effects of recombinant tissue plasminogen activator after intraluminal thread occlusion in mice: role of hemodynamic alterations. Stroke. 2001;32:2641–2647. doi: 10.1161/hs1101.097381. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Bahr M, Hermann DM. Tissue plasminogen activator-induced ischemic injury is reversed by NMDA antagonist MK-801 in vivo. Neurodegener Dis. 2005;2:49–55. doi: 10.1159/000089283. [DOI] [PubMed] [Google Scholar]
- LaManna JC, Chavez JC, Pichiule P. Structural and functional adaptation to hypoxia in the rat brain. J Exp Biol. 2004;207:3163–3169. doi: 10.1242/jeb.00976. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Busija DW, Mirro R, Armstead WM, Beasley DG. Effects of ischemia on brain blood flow and oxygen consumption of newborn pigs. Am J Physiol Heart Circ Physiol. 1989;257:H1917–H1926. doi: 10.1152/ajpheart.1989.257.6.H1917. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Erythropoietin on a tightrope: balancing neuronal and vascular protection between intrinsic and extrinsic pathways. Neurosignals. 2004;13:265–289. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu Z, Keogh CL, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043–1054. doi: 10.1038/sj.jcbfm.9600417. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS. Understanding blood flow: the other side of an acute arterial occlusion. Int J Stroke. 2007;2:118–120. doi: 10.1111/j.1747-4949.2007.00117.x. [DOI] [PubMed] [Google Scholar]
- Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D, Sibson NR, Pugh C, Buchan AM. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132–143. doi: 10.1038/jcbfm.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Park L, Gallo EF, Anrather J, Wang G, Norris EH, Paul J, Strickland S, Iadecola C. Key role of tissue plasminogen activator in neurovascular coupling. Proc Natl Acad Sci U S A. 2008;105:1073–1078. doi: 10.1073/pnas.0708823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretnar-Oblak J, Zaletel M, Zvan B, Sabovic M, Pogacnik T. Cerebrovascular reactivity to L-arginine in patients with lacunar infarctions. Cerebrovasc Dis. 2006;21:180–186. doi: 10.1159/000090530. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- Shin HK, Nishimura M, Jones PB, Ay H, Boas DA, Moskowitz MA, Ayata C. Mild induced hypertension improves blood flow and oxygen metabolism in transient focal cerebral ischemia. Stroke. 2008;39:1548–1555. doi: 10.1161/STROKEAHA.107.499483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorond FA, Shaffer ML, Kung AL, Lipsitz LA. Desferroxamine infusion increases cerebral blood flow: a potential association with hypoxia-inducible factor-1. Clin Sci (Lond) 2009;116:771–779. doi: 10.1042/CS20080320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama Y, Yagita Y, Oyama N, Terasaki Y, Omura-Matsuoka E, Sasaki T, Kitagawa K. Granulocyte colony-stimulating factor enhances arteriogenesis and ameliorates cerebral damage in a mouse model of ischemic stroke. Stroke. 2011;42:770–775. doi: 10.1161/STROKEAHA.110.597799. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa S, Hogan M, Hakim AM. The effects of a competitive NMDA receptor antagonist (CGS-19755) on cerebral blood flow and pH in focal ischemia. J Cereb Blood Flow Metab. 1991;11:786–793. doi: 10.1038/jcbfm.1991.136. [DOI] [PubMed] [Google Scholar]
- Valable S, Montaner J, Bellail A, Berezowski V, Brillault J, Cecchelli R, Divoux D, Mackenzie ET, Bernaudin M, Roussel S, Petit E. VEGF-induced BBB permeability is associated with an MMP-9 activity increase in cerebral ischemia: both effects decreased by Ang-1. J Cereb Blood Flow Metab. 2005;25:1491–1504. doi: 10.1038/sj.jcbfm.9600148. [DOI] [PubMed] [Google Scholar]
- Willmot M, Gray L, Gibson C, Murphy S, Bath PM. A systematic review of nitric oxide donors and L-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide. 2005;12:141–149. doi: 10.1016/j.niox.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Kamiya T, Deguchi K, Inaba T, Zhang H, Shang J, Miyazaki K, Ohtsuka A, Katayama Y, Abe K. Dissociation and protection of the neurovascular unit after thrombolysis and reperfusion in ischemic rat brain. J Cereb Blood Flow Metab. 2009;29:715–725. doi: 10.1038/jcbfm.2008.164. [DOI] [PubMed] [Google Scholar]
- Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Demuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Nowak TS., Jr CBF changes associated with focal ischemic preconditioning in the spontaneously hypertensive rat. J Cereb Blood Flow Metab. 2006;26:1128–1140. doi: 10.1038/sj.jcbfm.9600269. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Cheng M, Maples KR, Ma JY, Buchan AM. NXY-059, a novel free radical trapping compound, reduces cortical infarction after permanent focal cerebral ischemia in the rat. Brain Res. 2001;909:46–50. doi: 10.1016/s0006-8993(01)02618-x. [DOI] [PubMed] [Google Scholar]
- Zwagerman N, Sprague S, Davis MD, Daniels B, Goel G, Ding Y. Pre-ischemic exercise preserves cerebral blood flow during reperfusion in stroke. Neurol Res. 2010;32:523–529. doi: 10.1179/016164109X12581096796431. [DOI] [PubMed] [Google Scholar]