

Abstract
Abstract
Ischaemic stroke is a leading cause of death and disability worldwide. The complex cellular interactions leading from thromboembolic vessel occlusion to infarct development within the brain parenchyma in acute stroke are poorly understood, which translates into only one approved effective treatment, thrombolysis. Importantly, however, patients can develop progressive stroke despite reperfusion of previously occluded major intracranial arteries, a process referred to as ‘reperfusion injury’ which can be reproduced in the mouse model of transient middle cerebral artery occlusion (tMCAO). Although pathological platelet and coagulant activity have long been recognized to be involved in the initiation of ischaemic stroke, their contribution to infarct maturation remained elusive. Experimental evidence now suggests that early platelet adhesion/activation mechanisms involving the von Willebrand factor (vWF) receptor glycoprotein (GP) Ib, its ligand vWF, and the collagen receptor GPVI are critical pathogenic factors in infarct development following tMCAO, whereas platelet aggregation through GPIIb/IIIa is not. Further experimental work indicates that these pathways in conjunction with coagulation factor XII (FXII)-driven processes orchestrate a ‘thrombo-inflammatory’ cascade in acute stroke that results in infarct growth. This review summarizes these recent developments and briefly discusses their potential clinical impact.
Bernhard Nieswandt (left) is a cell biologist with expertise in platelet adhesion and activation mechanisms in thrombosis, haemostasis and inflammatory disease. He received his PhD from the University of Regensburg, Germany in 1997 having specialized in platelet surface receptor biology in genetic mouse models. In 1998 he became a research group leader at Witten-Herdecke University, Germany. In 2002 he moved his laboratory to the Rudolf Virchow Center, DFG-Research Center for Experimental Biomedicine at the University of Würzburg, Germany. Bernhard Nieswandt holds a chair of Vascular Medicine and is coordinator of the Collaborative Research Center 688 (‘Mechanisms and imaging of cardiovascular cell–cell interactions’). Guido Stoll (centre) and Christoph Kleinschnitz (right) are clinical scientists and active neurologists with expertise in pathophysiology of stroke, neuroimmunology and cellular neuroimaging. They chair the Research Group for Multiple Sclerosis and Neuroimmunology and the clinical stroke unit at the Department of Neurology of the University of Würzburg, Germany, and established an interdisciplinary stroke research and neuroimaging laboratory in Würzburg in 2002. G.S. is member of the board of directors of the German Heart Failure Center, Würzburg, and C.K. is scientific secretary of the Collaborative Research Center 688.
 |
Introduction
Cerebral ischaemia accounts for about 80% of all strokes, the other 20% are due to intracranial haemorrhage (Feigin et al. 2003). Atrial fibrillation and symptomatic extracranial artery stenoses represent the main sources of thromboembolism to the brain. Seconds after an embolus has occluded an intracranial vessel the lack of oxygen and glucose supply to the respective brain territory leads to acute neurological deficits such as hemiparesis or aphasia, a situation which is regarded a medical emergency. In the (rare) case of spontaneous thrombus resolution, symptoms are fully reversible and the event is termed transient ischaemic attack (TIA). Otherwise, a definite stroke evolves. In either circumstance patients require a complete cardiological and neurological examination due to the high risk of recurrent ischaemic events. Thus, one clinical aim is to identify the source of thromboembolism and to instigate either anticoagulation or anti-platelet treatment for secondary stroke prevention. Anticoagulation with warfarin, mainly prescribed in patients with atrial fibrillation, however, carries a considerable risk of bleeding complications which has led to the recent development of specific thrombin and factor Xa-inhibitors. Platelet inhibitors such as acetylsalicylic acid (ASA) or clopidogrel are more effective in patients with extracranial artery stenoses.
There is only one treatment option for acute stroke when persistent neurological deficit presents on arrival at the emergency room, namely thrombolysis with the ‘clot-buster’ agent tissue plasminogen activator (tPA; Saver et al. 2009). Unfortunately, tPA application is restricted to the first 4.5 h after the onset of stroke (Hacke et al. 2008). Beyond this ‘window of opportunity’, tPA-induced bleeding complications may outweigh the benefit of dissolving the vessel-occluding thrombus. Moreover, the efficacy of tPA is only moderate at best: for one patient to have a favourable outcome (a score of 0 or 1 on the modified Rankin scale), the number needed to treat (NNT) with tPA is 9 within the first 3 h post stroke. With the extended time window between 3 and 4.5 h, the NNT even raises to 14 (Hacke et al. 2008). Persistent vessel occlusion is a frequent cause of tPA treatment failure, but a considerable number of patients show secondary infarct growth despite successful vessel recanalization, a phenomenon referred to as ‘reperfusion injury’. The molecular mechanisms involved in secondary infarct growth are still poorly understood, but targeting ‘brain injury during reperfusion’ represents an important therapeutic goal (Stoll et al. 2008). In this review we summarize recent insights into the pathophysiological role of platelet receptors and related downstream signalling pathways as well as the intrinsic coagulation system in experimental cerebral ischaemia. By illustrating a link between thrombus formation and inflammatory pathways we introduce the tempting concept of stroke being a ‘thrombo-inflammatory’ disease. Based on these findings, novel pharmacological targets for both acute stroke treatment and secondary stroke prevention are highlighted.
All animal studies referred to in this review used the transient middle cerebral artery occlusion (tMCAO) model to induce ischaemic stroke in mice. In this model a small filament is advanced from the internal carotid artery to the origin of the middle cerebral artery (MCA) to occlude the vessel and induce tissue ischaemia. Although removal of the occluding filament, e.g. after 1 h, allows reperfusion, a complete MCA infarct, comprising the ipsilateral parietal neocortex as well as the basal ganglia, evolves within 24 h. Infarction can then be visualized by histological techniques or in vivo by magnetic resonance imaging (MRI) (Kleinschnitz et al. 2007; Pham et al. 2010).
Basic mechanisms of thrombus formation
At sites of vascular injury, the subendothelial extracellular matrix (ECM) is exposed to the flowing blood, which triggers adhesion and activation of blood platelets (primary haemostasis), followed by the activation of the coagulation system (secondary haemostasis). Primary and secondary haemostasis synergize to form a fibrin-rich thrombus that seals the wound and initiates wound healing (Ruggeri, 2002) (Fig. 1). The ECM contains multiple adhesive macromolecules, such as laminins, fibronectins and collagens. Platelets can bind these macromolecules via specific receptors that have distinct functions in the haemostatic cascade (Varga-Szabo et al. 2008b). The mechanisms of platelet adhesion at sites of injury are to a large extent determined by the prevailing rheological conditions. Shear forces generated between adjacent layers of the flowing blood, which are maximal at the wall, produce a drag that opposes platelet adhesion and aggregation. The initial tethering of platelets to the ECM of the injured vessel wall under conditions of high shear rates (>500 s−1), found for instance in small arteries and arterioles, is strictly dependent on the interaction between the platelet receptor glycoprotein (GP)Ib and von Willebrand factor (vWF) (Savage et al. 1998). The GPIb-vWF binding has a fast off-rate and does not produce stable platelet adhesion but rather reduces the velocity of the flowing platelets thereby allowing their interaction with highly thrombogenic collagens through the immunoglobulin superfamily receptor GPVI. GPVI is a powerful signal-transducing receptor exclusively found in platelets and their precursors, the megakaryocytes. GPVI ligation leads to full platelet activation, characterized by a rise in the cytosolic Ca2+ concentration ([Ca2+]i), cytoskeletal rearrangements resulting in a change from discoid to spheric shape, fusion of intracellular α- and dense granules with the plasma membrane and release of secondary platelet agonists, most notably adenosine diphosphate (ADP), adenosine triphosphate (ATP), serotonin and thromboxane A2. These agonists, together with locally produced thrombin, act on platelet-expressed G-protein-coupled receptors thereby enhancing cellular activation and recruitment of additional platelets from the bloodstream to the site of injury. This fine-tuned interplay of extra- and intracellular signalling events leads to the functional upregulation of integrin adhesion receptors, most notably integrin αIIbβ3 (also called GPIIb/IIIa), resulting in the establishment of stable platelet–ECM contacts and platelet aggregation, through the binding of plasma fibrinogen. These basic mechanisms of thrombus formation may in principle also apply to cerebral blood vessels during the course of ischaemic stroke (del Zoppo & Mabuchi, 2003), but it is not established that this is a major pathomechanism in stroke development.
Figure 1. Simplified model of thrombus formation.
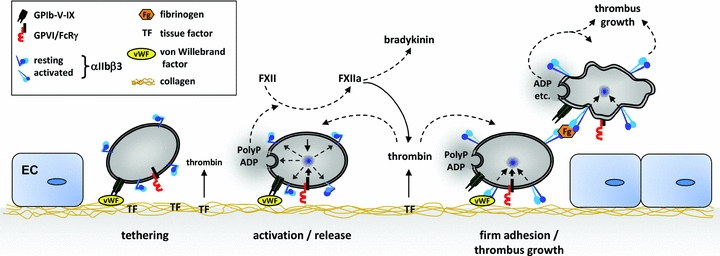
At sites of vascular injury the ECM becomes exposed to the flowing blood, allowing platelet adhesion and aggregation as well as coagulation. GPIb initiates haemostasis and thrombosis by recruiting platelets to the injured vessel wall, but it may also be involved in immune cell recruitment under inflammatory conditions. GPVI is the central activating platelet collagen receptor required for thrombus formation on the ECM but may also promote inflammation by triggering the release of PolyP. PolyP activate FXII leading to coagulation (via thrombin) and inflammation (via bradykinin). In parallel, thrombin generation is triggered by exposed tissue factor (TF, extrinsic pathway) also leading to coagulation and further platelet activation. Cellular activation and inside-out upregulation of GPIIb/IIIa (integrin αIIbβ3) affinity is essential for firm platelet adhesion and aggregation, the latter through binding of fibrinogen (Fg). EC, endothelial cell.
Anti-thrombotic treatment in clinical stroke
Excessive platelet activity and continuous platelet adherence to the brain microvasculature during reperfusion has long been recognized to be involved in experimental stroke (Choudhri et al. 1998; del Zoppo, 1998). The pathophysiological significance of these findings in human stroke patients, however, remained poorly understood as did the underlying mechanisms. Accordingly, it is still a matter of debate whether the modest clinical benefit of applying platelet inhibitors such as ASA or clopidogrel within the early phase of stroke (48 h) is due to prevention of recurrent thromboembolism originating from outside the brain (e.g. heart, atherosclerotic arteries) or local anti-thrombotic effects within the brain vasculature (Adams et al. 2007). An attempt to more effectively block platelet aggregation by antibodies against GPIIb/IIIa receptors failed: a clinical trial testing the monoclonal antibody abciximab had to be stopped prematurely due to excess bleeding-related mortality (Adams et al. 2008) and in an experimental study in which the JON/A F(ab)2 was applied intravenously 1 h before tCMAO more than 60% of mice died from intracranial haemorrhages (Kleinschnitz et al. 2007). Most importantly, surviving animals exhibited large cerebral infarcts similar to control mice, which can be taken as a first hint that final platelet aggregation via GPIIb/IIIa is not a mandatory step in stroke development in the reperfusion phase. A completely different picture emerged when early platelet adhesion and activation events rather than terminal platelet aggregation were targeted in the tMCAO model.
Platelet inhibition and experimental stroke: the GPIb–vWF–GPVI axis
Platelet tethering at sites of vascular injury must occur through a receptor that functions independently of cellular activation and allows rapid interactions that resist shear forces. This is achieved by GPIb-V-IX, a platelet-specific receptor complex encoded by four different genes, the α- and β-subunits of GPIb, GPIX and GPV (Bergmeier et al. 2000; Berndt et al. 2001). Lack or dysfunction of GPIb or GPIX gives rise to the Bernard Soulier syndrome, a congenital bleeding disorder characterized by mild thrombocytopenia and giant platelets (Berndt et al. 2001). The observed bleeding phenotype in GPIb-V-IX-deficient humans or mice is attributed to the loss of the extracellular domain of GPIbα which contains the binding sites for several ligands including vWF, thrombin, coagulation factor XI (FXI), FXII, P-selectin or Mac-1 (Berndt et al. 2001). The critical role of GPIb-IX-V in thrombus formation has been demonstrated in a model of permanent femoral arteriovenous shunt implantation in baboons by the use of F′(ab) fragments of antibodies directed against the vWF binding site on GPIbα (Cauwenberghs et al. 2000) and in mice lacking functional GPIbα (Bergmeier et al. 2006).
Recent studies have provided compelling evidence that GPIb might represent a novel target to efficiently prevent or treat acute ischaemic stroke. Both prophylactic (1 h before tMCAO) and therapeutic (1 h after tMCAO) blockade of the vWF binding site on GPIbα by intravenous injection of F′(ab) fragments of the anti-GPIbα antibody, p0p/B, dramatically protected mice from stroke progression following tMCAO (Fig. 2). This was associated with significantly better functional outcomes on day 1 (Kleinschnitz et al. 2007). Importantly, no increased incidence of intracranial haemorrhage was noted in these animals, although GPIbα inhibition resulted in increased tail bleeding times (Kleinschnitz et al. 2007) thereby confirming that there is no clear correlation between bleeding time and bleeding risk (Rodgers & Levin, 1990). Ultrahigh field MRI studies at 17.6 T employing perfusion-weighted imaging revealed that blockade of GPIb 1 h after tMCAO maintained blood flow during the reperfusion phase, while blood flow continuously decreased in control animals leading to large infarcts of the MCA territory after 24 h (Pham et al. 2011). In line with these findings, it was found that mice lacking phospholipase D1, which transduces activation signals downstream of vWF-occupied GPIb and thereby critically contributes to GPIb-dependent integrin αIIbβ3 activation and platelet adhesion under high shear, are markedly protected from stroke progression following tMCAO again without displaying increased bleeding (Elvers et al. 2010).
Figure 2. Blocking of early platelet adhesion and activation protects mice from acute ischaemic stroke.

Upper panel, representative 2,3,5-triphenyltetrazoliumchloride (TTC)-stained coronal brain sections on day 1 after tMCAO. Ischaemic infarctions (white areas) appear smaller in anti-GPIbα-treated mice, vWF−/− mice and anti-GPVI-treated mice compared with wild-type controls and this was confirmed by infarct volumetry (lower panel). In contrast, blocking of the final common pathway of platelet aggregation using anti-GPIIb/IIIa F′(ab)2 could not reduce stroke size indicating that mechanisms beyond platelet-derived thrombus formation are operative during infarct evolution. *P < 0.05; **P < 0.01 compared with wild-type mice (n = 8–10/group); one-way ANOVA, Bonferroni's multiple comparisons post test. (Adapted from Kleinschnitz et al. 2007, 2009.)
Given the disappointing results obtained with GPIIb/IIIa inhibitors (Kleinschnitz et al. 2007; Adams et al. 2008), the profound and apparently safe protection from infarct progression following tMACO achieved by inhibition of GPIbα or downstream signalling events was very unexpected and raised the question of whether the pathogenetic significance of GPIb is based on its central role in thrombus formation or other activities. Therefore, the role of the principal GPIb ligand vWF in stroke development was assessed. vWF is a multimeric glycoprotein that becomes immobilized on exposed collagens at sites of vessel injury and facilitates platelet recruitment under high shear by interacting with GPIbα. In addition, vWF contains a RGD (amino acid sequence arginine, glycine, aspartic acid) motif that serves as an adhesion site for platelet GPIIb/IIIa which contributes to firm platelet attachment and thrombus growth (Ruggeri, 2003). Lack of vWF impairs haemostasis in humans and mice and provides protection from experimental thrombosis in the latter (Denis et al. 1998; Ruggeri, 2003). Studies in vWF−/− mice revealed a central role of this adhesive protein in infarct progression following tMCAO (Kleinschnitz et al. 2009). Very similar to p0p/B-F′(ab)-treated mice, these animals were protected from stroke and showed significantly better neurological scores 24 h after ischaemia and this was, again, not associated with increased intracranial bleeding (Kleinschnitz et al. 2009). In a series of studies, De Meyer et al. could show that the observed pathogenic function of vWF requires its binding sites for GPIbα and collagen, but not the one for GPIIb/IIIa, thereby further supporting the hypothesis that thrombus growth may not be the only pathomechanism underlying infarct progression after tMCAO (De Meyer et al. 2010). In line with these experimental observations several prospective clinical studies identified high plasma levels of vWF as a strong and independent risk factor for stroke (Roldan et al. 2005; Wieberdink et al. 2010).
In further support of a decisive role of vWF–GPIb interactions, two additional groups reported independently that mice lacking a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) 13, which cleaves highly thrombogenic ultra-large vWF (>20 million kDa) to less active vWF, are more susceptible to infarct progression following tMCAO (Zhao et al. 2009; Fujioka et al. 2010; Nieswandt & Stoll, 2010). This was accompanied by increased accumulation of immune cells and thrombi in the ischaemic brain. Conversely, infusion of recombinant ADAMTS13 into wild-type mice was stroke-protective (Zhao et al. 2009). Autoantibodies against ADAMTS13 are frequently found in patients with thrombotic thrombocytopenic purpura which carry a high risk of ischaemic strokes (Sadler, 2008).
Although GPIb–vWF interaction triggers platelet activation, this stimulus is generally considered very weak (Jackson et al. 2003). Among the numerous constituents of the ECM, collagens are the most thrombogenic as they provide an adhesion substrate for platelets and directly activate the cells. This activation is mediated by GPVI, a type I transmembrane receptor belonging to the immunoglobulin superfamily that is exclusively expressed in platelets and megakaryocytes (Nieswandt & Watson, 2003). GPVI may be a promising anti-thrombotic target as the receptor can be depleted from the surface of circulating platelets through antibodies in humans (Boylan et al. 2003) and mice (Nieswandt et al. 2001), resulting in a ‘GPVI knockout-like’ phenotype and long-term anti-thrombotic protection in the latter (Massberg et al. 2003). Such GPVI-depleted mice displayed significantly reduced brain infarct volumes at day 1 after tMCAO (Kleinschnitz et al. 2007). Further studies revealed that mice lacking components of the machinery that mediates store-operated Ca2+ entry are likewise protected from stroke progression in the tMCAO model. Platelets isolated from these mice show a selective functional defect in response to GPVI agonists whereas other pathways are apparently not affected (Varga-Szabo et al. 2008a; Braun et al. 2009) thereby indirectly supporting the hypothesis that GPVI-mediated activation processes are important during stroke development. This is further confirmed by studies showing that elevated GPVI expression levels in platelets are associated with an increased risk of stroke occurrence in humans (Bigalke et al. 2009), and elevated levels of the soluble GPVI ectodomain have been detected in acute ischaemic stroke suggesting increased GPVI activation in this setting (Al-Tamimi et al. 2011).
Coagulation factor XII
Activated platelets facilitate the local activation of the coagulation cascade through two major pathways. Firstly, expression of procoagulant activity characterized by the exposure of negatively charged phosphatidylserine (PS). PS provides high-affinity binding sites for coagulation factors and, hence, facilitates the assembly of tenase and prothrombinase complexes, which generate factor Xa and thrombin, respectively (Heemskerk et al. 2002). Secondly, activated platelets release negatively charged inorganic polyphosphates (PolyP) that activate coagulation factor XII (FXII, Hageman factor) (Muller et al. 2009), which represents the starting point of the so-called intrinsic coagulation pathway. For more than 50 years FXII was believed to play no role in blood clotting in vivo, based on the fact that FXII-deficient humans do not show any bleeding abnormality. In line with this FXII-deficient mice display normal tail bleeding times and no signs of spontaneous bleeding. Surprisingly, however, FXII−/− mice are consistently unable to form occlusive thrombi in different thrombosis models, indicating that pathological thrombus formation and haemostasis may be driven by at least partially distinct pathways (Renne et al. 2005). In line with this novel concept, genetic disruption or pharmacological inhibition of FXII using the highly specific FXIIa inhibitor rHA-infestin-4 immediately before tMCAO profoundly reduced infarct size and this protection was not associated with an increased risk of intracranial bleeding (Kleinschnitz et al. 2006; Hagedorn et al. 2010). Perfusion-weighted neuroimaging revealed that stroke protection in FXII−/− mice was due to sustained patency of intracerebral vessels during the reperfusion phase (Pham et al. 2010). Accordingly, immunohistochemical analyses showed reduced fibrin formation in the infarct area in FXII-deficient mice compared to wild-type controls indicating that FXII-dependent thrombin generation occurs to a considerable extent during cerebral ischaemia (Kleinschnitz et al. 2006).
Thrombo-inflammation: linking the thrombotic cascade to innate immunity
As outlined above, cumulating evidence suggests that platelet attachment and activation through the GPIb–vWF–GPVI axis and subsequent FXII activation constitute a critical pathomechanism in acute cerebral ischaemia. However, although these events will eventually result in intravascular thrombus formation (Stoll et al. 2008; Varga-Szabo et al. 2008b), this appears not to be the key step in ischaemic lesion development during the reperfusion phase as strong platelet aggregation inhibitors, such as anti-GPIIb/IIIa antibodies, do not afford protection from stroke (Kleinschnitz et al. 2007; Adams et al. 2008). A number of recent observations rather support the idea that GPIb–vWF–GPVI–FXII-triggered pathways may promote detrimental inflammation in acute cerebral ischaemia.
It is well established that ischaemic stroke elicits a strong inflammatory response involving T-cells, monocytes/macrophages and neutrophils (Stoll et al. 1998; Dirnagl et al. 1999), which is time-restricted and occurs in ‘waves’. Early during cerebral ischaemia leukocytes are attracted by cell adhesion molecules expressed on the luminal endothelial surface and act within the endovascular space while infiltration into the parenchyma occurs at later stages.
There is recent evidence that platelet receptors GPIb and GPVI guide inflammation thereby providing a possible link between thrombotic activity and inflammation in ischaemic brain damage. GPIbα harbours a binding site for Mac-1 (an integrin expressed on neutrophils and monocytes (CD11b/CD18)) and mice deficient in Mac-1 are less susceptible to cerebral ischaemia/reperfusion injury (Soriano et al. 1999). An important role of GPIb (and vWF) in the recruitment of immune cells has recently been demonstrated in a model of acute peritonitis, but the underlying mechanisms have not been fully elucidated (Petri et al. 2010). Similarly, GPVI probably contributes to pro-inflammatory processes as well. Boilard et al. showed that GPVI-induced platelet microparticle formation promoted inflammation in experimental rheumatoid arthritis independently of thrombus formation (Boilard et al. 2010). Upon activation via GPVI, platelets are also a source of the proinflammatory cytokine interleukin-1α through which they can contribute to inflammation-mediated brain injury (Thornton et al. 2010). Moreover, platelets release PolyP as potent FXII activators (Muller et al. 2009). Activated FXII (FXIIa) in turn not only initiates coagulation, but also triggers inflammation through activation of the kallikrein–kinin system (KKS). The end product of the KKS is the potent proinflammatory mediator bradykinin (Muller & Renne, 2008). The hypothesis that FXII activation links thrombotic activity with inflammation (via the KKS) was further strengthened by the analysis of bradykinin receptor B1 (B1R) knockout mice (Fig. 3). These animals developed smaller infarctions and displayed markedly reduced brain oedema formation compared with wild-type controls after tMCAO (Austinat et al. 2009).
Figure 3. FXII plays a central role in stroke progression in mice.
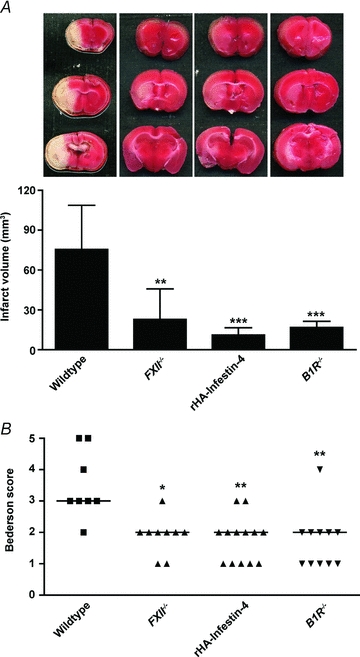
Infarct volumes as measured from TTC-stained coronal brain sections (A) and neurological Bederson score (B) on day 1 after tMCAO. Genetic ablation (FXII−/− mice) or pharmacological blockade (rHA-Infestin-4) of FXII immediately before stroke markedly protects mice from acute ischaemic brain damage (white areas) as does disruption of the bradykinin receptor B1 (B1R−/−). B1R belongs to the proinflammatory kallikrein–kinin system that is also activated by FXII. *P < 0.05; **P < 0.01; ***P < 0.0001 compared with wild-type mice (n = 8–10/group): one-way ANOVA, Bonferroni's multiple comparisons post test (A); Kruskal–Wallis test, Dunn's multiple comparison post test (B). Horizontal lines indicate median. (Adapted from Kleinschnitz et al. 2006; Austinat et al. 2009; Hagedorn et al. 2010.)
In conclusion, there is increasing evidence that thrombus formation and immune-mediated processes are strongly interrelated during cerebral ischaemia and significantly contribute to brain damage. The considerable impact of immune cells on stroke outcome is underlined by the fact that immunodeficient mice lacking T-cells are remarkably protected against focal brain ischaemia (Yilmaz et al. 2006; Kleinschnitz et al. 2010). We are currently only beginning to understand the cellular and molecular interactions linking thrombus formation with neuroinflammation. GPIb, GPVI and FXII are ‘hot’ candidates orchestrating ‘thrombo-inflammation’ and provide potential novel targets for stroke treatment and prevention.
Acknowledgments
Research in the authors' laboratories was supported by the Deutsche Forschungsgemeinschaft, Bonn (SFB 688 A1, A13 and B1).
Glossary
Abbreviations
- ASA
acetylsalicylic acid
- ECM
extracellular matrix
- FX
coagulation factor X
- FXI
coagulation factor XI
- FXII
coagulation factor XII
- GP
glycoprotein
- KKS
kallikrein–kinin system
- MCA
middle cerebral artery
- PolyP
inorganic polyphosphates
- tMCAO
transient middle cerebral artery occlusion
- tPA
tissue plasminogen activator
- vWF
von Willebrand factor
References
- Adams HP, Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL, Higashida RT, Jauch EC, Kidwell C, Lyden PD, Morgenstern LB, Qureshi AI, Rosenwasser RH, Scott PA, Wijdicks EF. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711. doi: 10.1161/STROKEAHA.107.181486. [DOI] [PubMed] [Google Scholar]
- Adams HP, Jr, Effron MB, Torner J, Davalos A, Frayne J, Teal P, Leclerc J, Oemar B, Padgett L, Barnathan ES, Hacke W. Emergency administration of abciximab for treatment of patients with acute ischemic stroke: Results of an international phase III trial. Abciximab in Emergency Treatment of Stroke Trial (AbESTT-II) Stroke. 2008;39:87–99. doi: 10.1161/STROKEAHA.106.476648. [DOI] [PubMed] [Google Scholar]
- Al-Tamimi M, Gardiner EE, Thom JY, Shen Y, Cooper MN, Hankey GJ, Berndt MC, Baker RI, Andrews RK. Soluble glycoprotein VI is raised in the plasma of patients with acute ischemic stroke. Stroke. 2011;42:498–500. doi: 10.1161/STROKEAHA.110.602532. [DOI] [PubMed] [Google Scholar]
- Austinat M, Braeuninger S, Pesquero JB, Brede M, Bader M, Stoll G, Renne T, Kleinschnitz C. Blockade of bradykinin receptor B1 but not bradykinin receptor B2 provides protection from cerebral infarction and brain edema. Stroke. 2009;40:285–293. doi: 10.1161/STROKEAHA.108.526673. [DOI] [PubMed] [Google Scholar]
- Bergmeier W, Piffath CL, Goerge T, Cifuni SM, Ruggeri ZM, Ware J, Wagner DD. The role of platelet adhesion receptor GPIbα far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci U S A. 2006;103:16900–16905. doi: 10.1073/pnas.0608207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeier W, Rackebrandt K, Schroder W, Zirngibl H, Nieswandt B. Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood. 2000;95:886–893. [PubMed] [Google Scholar]
- Berndt MC, Shen Y, Dopheide SM, Gardiner EE, Andrews RK. The vascular biology of the glycoprotein Ib-IX-V complex. Thromb Haemost. 2001;86:178–188. [PubMed] [Google Scholar]
- Bigalke B, Stellos K, Weig HJ, Geisler T, Seizer P, Kremmer E, Potz O, Joos T, May AE, Lindemann S, Gawaz M. Regulation of platelet glycoprotein VI (GPVI) surface expression and of soluble GPVI in patients with atrial fibrillation (AF) and acute coronary syndrome (ACS) Basic Res Cardiol. 2009;104:352–357. doi: 10.1007/s00395-009-0779-7. [DOI] [PubMed] [Google Scholar]
- Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O'Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan B, Rathore V, Paddock C, Friedman K, Curtis B, Stapleton M, Newman DK, Chen H, Kahn M, Newman PJ. JAQ-2/3-like ITP – a newly recognized clinical syndrome caused by autoantibody-mediated clearance of the GPVI/FcRγ-chain complex from the human platelet surface. Blood. 2003;102:12A. doi: 10.1182/blood-2004-03-0896. [DOI] [PubMed] [Google Scholar]
- Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, Bosl M, Stoll G, Nieswandt B. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113:2056–2063. doi: 10.1182/blood-2008-07-171611. [DOI] [PubMed] [Google Scholar]
- Cauwenberghs N, Meiring M, Vauterin S, van Wyk V, Lamprecht S, Roodt JP, Novák L, Harsfalvi J, Deckmyn H, Kotzé HF. Antithrombotic effect of platelet glycoprotein Ib-blocking monoclonal antibody Fab fragments in nonhuman primates. Arterioscler Thromb Vasc Biol. 2000;20:1347–1353. doi: 10.1161/01.atv.20.5.1347. [DOI] [PubMed] [Google Scholar]
- Choudhri TF, Hoh BL, Zerwes HG, Prestigiacomo CJ, Kim SC, Connolly ESJ, Kottirsch G, Pinsky DJ. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J Clin Invest. 1998;102:1301–1310. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ. The role of platelets in ischemic stroke. Neurology. 1998;51:S9–14. doi: 10.1212/wnl.51.3_suppl_3.s9. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- De Meyer SF, Schwarz T, Deckmyn H, Denis CV, Nieswandt B, Stoll G, Vanhoorelbeke K, Kleinschnitz C. Binding of von Willebrand factor to collagen and glycoprotein Ibα, but not to glycoprotein IIb/IIIa, contributes to ischemic stroke in mice – Brief report. Arterioscler Thromb Vasc Biol. 2010;30:1949–1951. doi: 10.1161/ATVBAHA.110.208918. [DOI] [PubMed] [Google Scholar]
- Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–9529. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers ME, Boesl M, Chen Q, Heemskerk JW, Stoll G, Frohman MA, Nieswandt B. Impaired αIIbβ3 integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci Signal. 2010;3:ra1. doi: 10.1126/scisignal.2000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2:43–53. doi: 10.1016/s1474-4422(03)00266-7. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, Nakano T, Muroi C, Fukushima H, Sugimoto M, Banno F, Kokame K, Miyata T, Fujiwara M, Okuchi K, Nishio K. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115:1650–1653. doi: 10.1182/blood-2009-06-230110. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Hagedorn I, Schmidbauer S, Pleines I, Kleinschnitz C, Kronthaler U, Stoll G, Dickneite G, Nieswandt B. Factor XIIa inhibitor recombinant human albumin Infestin-4 abolishes occlusive arterial thrombus formation without affecting bleeding. Circulation. 2010;121:1510–1517. doi: 10.1161/CIRCULATIONAHA.109.924761. [DOI] [PubMed] [Google Scholar]
- Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. 2002;88:186–193. [PubMed] [Google Scholar]
- Jackson SP, Nesbitt WS, Kulkarni S. Signaling events underlying thrombus formation. J Thromb Haemost. 2003;1:1602–1612. doi: 10.1046/j.1538-7836.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, De Meyer SF, Schwarz T, Austinat M, Vanhoorelbeke K, Nieswandt B, Deckmyn H, Stoll G. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. 2009;113:3600–3603. doi: 10.1182/blood-2008-09-180695. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–2330. doi: 10.1161/CIRCULATIONAHA.107.691279. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, Austinat M, Nieswandt B, Wiendl H, Stoll G. Early detrimental T cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, Renne C, Gailani D, Nieswandt B, Renne T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–518. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massberg S, Gawaz M, Gruner S, Schulte V, Konrad I, Zohlnhofer D, Heinzmann U, Nieswandt B. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–49. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renne T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, Renne T. Novel roles for factor XII-driven plasma contact activation system. Curr Opin Hematol. 2008;15:516–521. doi: 10.1097/MOH.0b013e328309ec85. [DOI] [PubMed] [Google Scholar]
- Nieswandt B, Schulte V, Bergmeier W, Mokhtari-Nejad R, Rackebrandt K, Cazenave JP, Ohlmann P, Gachet C, Zirngibl H. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193:459–470. doi: 10.1084/jem.193.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieswandt B, Stoll G. The smaller, the better: VWF in stroke. Blood. 2010;115:1477–1478. doi: 10.1182/blood-2009-12-255000. [DOI] [PubMed] [Google Scholar]
- Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102:449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, Goerge T, Schneider SW, Jones C, Nieswandt B, Wild MK, Vestweber D. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–4719. doi: 10.1182/blood-2010-03-276311. [DOI] [PubMed] [Google Scholar]
- Pham M, Helluy X, Kleinschnitz C, Kraft P, Bartsch AJ, Jakob P, Nieswandt B, Bendszus M, Stoll G. Sustained reperfusion after blockade of glycoprotein-receptor-Ib in focal cerebral ischemia: an MRI study at 17.6 Tesla. PLoS One. 2011;6:e18386. doi: 10.1371/journal.pone.0018386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham M, Kleinschnitz C, Helluy X, Bartsch AJ, Austinat M, Behr VC, Renne T, Nieswandt B, Stoll G, Bendszus M. Enhanced cortical reperfusion protects coagulation factor XII-deficient mice from ischemic stroke as revealed by high-field MRI. Neuroimage. 2010;49:2907–2914. doi: 10.1016/j.neuroimage.2009.11.061. [DOI] [PubMed] [Google Scholar]
- Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers RP, Levin J. A critical reappraisal of the bleeding time. Semin Thromb Hemost. 1990;16:1–20. doi: 10.1055/s-2007-1002658. [DOI] [PubMed] [Google Scholar]
- Roldan V, Marin F, Garcia-Herola A, Lip GY. Correlation of plasma von Willebrand factor levels, an index of endothelial damage/dysfunction, with two point-based stroke risk stratification scores in atrial fibrillation. Thromb Res. 2005;116:321–325. doi: 10.1016/j.thromres.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1:1335–1342. doi: 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 2008;112:11–18. doi: 10.1182/blood-2008-02-078170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- Saver JL, Gornbein J, Grotta J, Liebeskind D, Lutsep H, Schwamm L, Scott P, Starkman S. Number needed to treat to benefit and to harm for intravenous tissue plasminogen activator therapy in the 3- to 4.5-h window: joint outcome table analysis of the ECASS 3 trial. Stroke. 2009;40:2433–2437. doi: 10.1161/STROKEAHA.108.543561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. doi: 10.1161/01.str.30.1.134. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Stoll G, Kleinschnitz C, Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood. 2008;112:3555–3562. doi: 10.1182/blood-2008-04-144758. [DOI] [PubMed] [Google Scholar]
- Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1α drives cerebrovascular inflammation. Blood. 2010;115:3632–3639. doi: 10.1182/blood-2009-11-252643. [DOI] [PubMed] [Google Scholar]
- Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, Renne T, Stoll G, Nieswandt B. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008a;205:1583–1591. doi: 10.1084/jem.20080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008b;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- Wieberdink RG, van Schie MC, Koudstaal PJ, Hofman A, Witteman JC, de Maat MP, Leebeek FW, Breteler MM. High von Willebrand factor levels increase the risk of stroke: the Rotterdam study. Stroke. 2010;41:2151–2156. doi: 10.1161/STROKEAHA.110.586289. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, Scheiflinger F, Wagner DD. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114:3329–3334. doi: 10.1182/blood-2009-03-213264. [DOI] [PMC free article] [PubMed] [Google Scholar]