

Abstract
Abstract
Endogenous defence mechanisms by which the brain protects itself against noxious stimuli and recovers from ischaemic damage are a key target of stroke research. The loss of viable brain tissue in the ischaemic core region after stroke is associated with damage to the surrounding area known as the penumbra. Activation of the redox-sensitive transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) plays a pivotal role in the cellular defence against oxidative stress via transcriptional upregulation of phase II defence enzymes and antioxidant stress proteins. Although recent evidence implicates Nrf2 in neuroprotection, it is not known whether activation of this pathway within the neurovascular unit protects the brain against blood–brain barrier breakdown and cerebrovascular inflammation. Targeting the neurovascular unit should provide novel insights for effective treatment strategies and facilitate translation of experimental findings into clinical therapy. This review focuses on the cytoprotective role of Nrf2 in stroke and examines the evidence that the Nrf2–Keap1 defence pathway may serve as a therapeutic target for neurovascular protection.
Giovanni Mann (left) obtained his BSc in Zoology from George Washington University, USA and MSc and PhD in Physiology from University College London. As Professor of Vascular Physiology at King's College London, he has served as Chairman of the Executive Committee of The Physiological Society, President of the British Microcirculation Society and currently Secretary General of SFRRI, an Associate Editor for Free Radical Biology & Medicine, on Editorial Boards of The Journal of Physiology, Microcirculation, and Free Radical Research, and an Editorial Advisor for the Biochemical Journal. He chairs Heart Research UK Translational Sciences panel and serves on the Royal Society International Networks panel, and as an External Referee for BBSRC and MRC. He heads an active research group investigating the role of Nrf2 as a regulator of vascular antioxidant defences and redox signalling in diseases associated with oxidative stress such as gestational diabetes, pre-eclampsia and stroke. Alessio Alfieri (right) obtained his Pharmacy degree (2004) and his PhD in Drug Science (2007) from the University of Naples. After working at the University of Sheffield (2008–2010), he joined the Cardiovascular Division at King's College London. His research interests include vascular physiology and pharmacology and the molecular mechanisms underlying endothelial dysfunction and inflammation.
 |
Introduction
Stroke is the second most common cause of death and the leading cause of adult disability (Donnan et al. 2008; Endres et al. 2008; Balami et al. 2011). Despite advances in the understanding of the pathophysiology of cerebral ischaemia, therapeutic options remain limited, with only recombinant tissue-plasminogen activator (rt-PA) currently approved for the treatment of stroke (Lakhan et al. 2009), but its use is limited by a brief therapeutic window (3–4.5 h) and potential side effects (intracranial haemorrhage). In stroke, cerebral ischaemia triggers the pathological mechanisms, collectively known as the ischaemic cascade, causing rapid and irreversible neuronal injury within the ischaemic core. However, the surrounding hypoperfused brain tissue, known as the penumbra, can be salvaged if flow is restored and/or efficacious therapies are applied. Notably, reperfusion from recanalised cerebral vessels can cause tissue injury due to cerebral oedema, brain haemorrhage and neuronal death (Jung et al. 2010). Acute responses of brain tissue to cerebral ischaemia and its chronic pathogenic progression involve many pathways, with accumulating evidence implicating reactive oxygen species (ROS) and inflammation as pivotal mediators (Lakhan et al. 2009; Jung et al. 2010). Early events following ischaemic damage, such as excitotoxicity induced by glutamate, calcium overload and ROS-mediated oxidative stress rapidly result in cell death within the infarct core, whereas later events precipitated by pro-inflammatory and pro-apoptotic mediators (interleukin-1 (IL-1), cyclooxygenase-2 (COX-2), matrix metalloproteinases (MMPs), caspases) escalate the progression of damage to the ischaemic penumbra (Dirnagl et al. 2003; Candelario-Jalil, 2009). Protective mediators are also released in the early (GABA, adenosine) and delayed (interlleukin-10 (IL-10), B-cell lymphoma 2 (Bcl2), erythropoietin) phases of cerebral ischaemia, attenuating the damage to brain cells in the penumbra. Oxidative stress may function as a ‘switch mechanism’ tipping the balance between pro-death and pro-survival pathways in cerebral ischaemia (Crack & Taylor, 2005; Moskowitz et al. 2010).
Nrf2: a regulator of endogenous antioxidant defences
In addition to its high consumption of oxygen and glucose, the brain is enriched in peroxidisable fatty acids, iron and ascorbate (Zaleska & Floyd, 1985; Adibhatla & Hatcher, 2010). Moreover, several neurotransmitters are excitotoxic or auto-oxidizable (Halliwell, 2006). The brain has evolved endogenous defence mechanisms to counteract the damaging effects of ROS (Halliwell, 2001, 2011); however, antioxidant defences are much lower than other organs including liver and kidney (Marklund et al. 1982).
As summarised in Fig. 1, the redox-sensitive transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) plays a key role in the cellular defence against oxidative stress (Ishii et al. 2000, 2004; Kensler et al. 2007; Kaspar et al. 2009). Under quiescent conditions, Nrf2 is sequestered by its cytosolic repressor Keap1 (Kelch-like ECH-associated protein 1), a cytoskeletal protein that anchors and represses its transcriptional activity (Itoh et al. 1999; McMahon et al. 2003; Tong et al. 2007). Keap1 promotes rapid proteasomal degradation of Nrf2 via ubiquitination and also acts as a sensor to oxidative and electrophilic stress (Itoh et al. 1999). It has been suggested that alterations in the structure of Keap1 leads to dissociation of the Nrf2–Keap1 complex (Motohashi & Yamamoto, 2004), but site-specific modification of Keap1 may also cause an altered E3 ubiquitin ligase function and subsequent reduction in Nrf2 degradation (Tong et al. 2007).
Figure 1. Activation of the Nrf2–Keap1 defence pathway in oxidative stress.
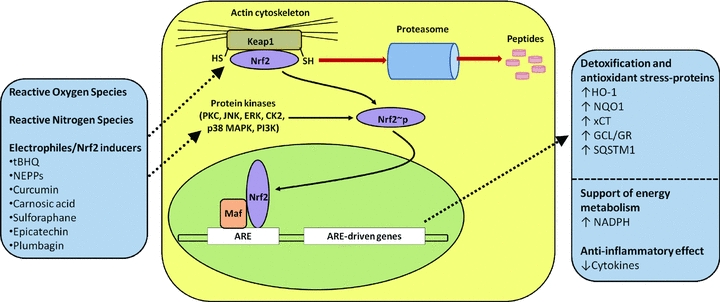
Nrf2 is normally retained in the cytosol bound to the actin-binding protein Keap1 and targeted for proteasomal degradation. Reactive oxygen species, reactive nitrogen species and endogenous and exogenous electrophiles/activators (see text for more details) can alter the Nrf2–Keap1 complex by modifying cysteine (-SH) residues on Keap1. Subsequent phosphorylation of Nrf2 by cytoplasmic kinases may increase its nuclear translocation, where it binds with small Maf proteins to the antioxidant response element (ARE). Induction of ARE-driven genes results in upregulation of haem oxygenase-1 (HO-1), NAD(P)H-quinone oxidoreductase-1 (NQO1), glutamate-cysteine ligase (GCL), glutathione reductase, sequestosome-1 (SQSTM1) and the cystine/glutamate anionic amino acid transporter (xCT). Nrf2/ARE-linked detoxification and antioxidant stress proteins restore the basal redox status in cells exposed to oxidative stress and inflammatory mediators. tBHQ, tert-butylhydroquinone; NEPPs, neurite outgrowth-promoting prostaglandins. Adapted from Itoh et al. (1999); Ishii et al. (2000); Kensler et al. (2007); Innamorato et al. (2008); Siow & Mann (2010).
Phosphorylation of serine/threonine residues in Nrf2 may be an alternative mechanism by which Nrf2 dissociates from Keap1 (Surh et al. 2008), and kinases, including protein kinase C (PKC), c-jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), casein kinase 2 (CK2), p38 mitogen-activated protein kinase (p38 MAPK) and phosphoinositide 3-kinase (PI3K), appear to modulate nuclear import and export of Nrf2 (Jain & Jaiswal, 2007; Surh et al. 2008). In summary, oxidation of redox-sensitive cysteines within Keap1 constitute the molecular basis for Nrf2 activation (Tong et al. 2007), but ROS-induced Nrf2 phosphorylation may provide an alternative mechanism (Burdette et al. 2010). Moreover, there are reports of crosstalk between Nrf2, cFOS, peroxisome proliferator-activated receptor γ (PPARγ), p53 and nuclear factor κB (NFκB) signalling pathways (Wakabayashi et al. 2010).
The induction of phase II defence enzymes and antioxidant stress proteins by Nrf2 is regulated via the antioxidant response element (ARE, or electrophile response element EpRE) in the promoter region of target genes (Fig. 1). Notably, many Nrf2 inducers exhibit hormetic properties with beneficial effects reported at nanomolar concentrations but toxic effects at higher concentrations (Mann et al. 2009; Siow & Mann, 2010).
Nrf2 is ubiquitously expressed (Moi et al. 1994) and, in the brain, may act as one of the most important defences against oxidative stress by modulating microglial dynamics (Rojo et al. 2010), protecting astrocytes (Vargas & Johnson, 2009) and neurons (Lee et al. 2003) from toxic insults, regulating the expression of inflammatory markers (Innamorato et al. 2008) and antioxidant enzymes (Shah et al. 2007; Yan et al. 2008). It has also been proposed that Nrf2 plays a protective role in neurodegenerative disorders, including Parkinson's (Cuadrado et al. 2009), Alzheimer's (Kanninen et al. 2008), and Huntington's (Stack et al. 2010) disease as well as traumatic brain injury (Yan et al. 2008).
This review focuses on the cytoprotective role of Nrf2 in stroke and examines the evidence that the Nrf2–Keap1 defence pathway may serve as a therapeutic target for neurovascular protection in cerebral ischaemia. In addition to reviewing the findings of previous studies in experimental stroke, we discuss possible therapeutic strategies aimed to protect the penumbra from cell death following cerebral ischaemia.
Stroke, the neurovascular unit and antioxidant defences
With its special anatomical characteristics, the brain circulation plays a critical role in the pathogenesis of cerebrovascular disorders (Abbott et al. 2010) (see Fig. 2). Microvascular alterations after ischaemia–reperfusion, including disruption of the blood–brain barrier, oedema and haemorrhage, can induce swelling of astrocytic foot processes and neurodegeneration. Despite its high energetic demands, the brain does not possess reserves of oxygen or nutrients such as ATP, glucose, glycogen, phosphocreatine, and thus relies on cerebral blood flow for normal function. Notably, the interaction of astrocytes and neurons with the vasculature is essential for local regulation of cerebral blood flow (Abbott et al. 2006; Attwell et al. 2010). The close relationship between neural activity and cerebral blood flow is described as neurovascular coupling, with the neurovascular unit comprised of neurons, glia (astrocytes, microglia, oligodendrocytes) and vascular cells (endothelial, smooth muscle, adventitial cells and pericytes) (Iadecola, 2004).
Figure 2. Communication between astrocytes, neurons and brain endothelial cells in the defence against oxidative stress.
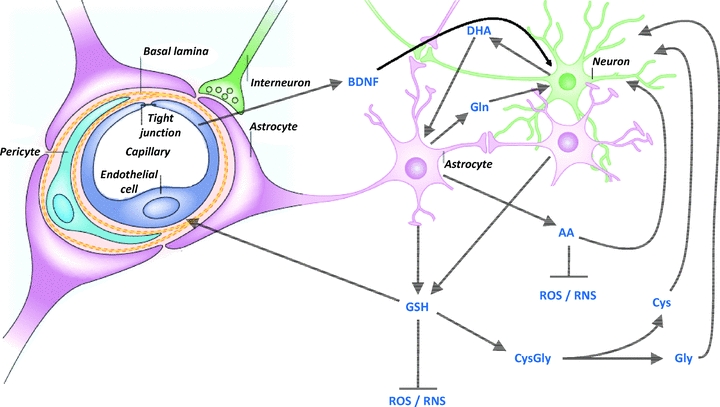
Glutathione (GSH) and its precursors, ascorbic acid (AA) and dehydroascorbate (DHA), and brain-derived neurotrophic factor (BDNF) provide protection of the neurovascular unit against free radical-mediated injury (see text for more details). Cys, cysteine; Gln, glutamine; Gly, glycine; RNS, reactive nitrogen species. Modified from Fig. 2 in Abbott et al. (2006) with permission from the Nature Publishing Group.
Despite knowledge of the cellular function of neurons, astrocytes and vascular cells, information on intercommunication between these cell types in response to ischaemic injury and the molecular mechanisms underlying damage and repair processes in the neurovascular unit following an ischaemic episode is limited. The fine match between neural energetic demands and vascular flow is disrupted in stroke. Oxidative stress is a major contributor to cerebrovascular dysfunction (Faraci, 2005). Interestingly, oxidative stress seems to be a common pathway within the neurovascular unit, affecting neurons (Niizuma et al. 2009), astrocytes (Simpson et al. 2010) and the endothelium (Rizzo & Leaver, 2010). Cellular defences in the brain involve a number of endogenous protective enzymes, including superoxide dismutase (SOD), glutathione peroxidase, catalase, thioredoxin, peroxiredoxins and haem oxygenases. In addition to these antioxidant enzymes, small non-protein compounds (glutathione, dietary vitamins C and E) essentially contribute to defend the brain against oxidative stress (Halliwell, 2001).
Ischaemia–reperfusion in the brain triggers oxidative and nitrative injury in the neurovascular unit (Gursoy-Ozdemir et al. 2004). Using fluorescent probes in mouse brains subjected to 2 h of ischaemia and 3 h of reperfusion, high levels of superoxide and peroxynitrate production have been observed in neurons, astrocytes and the endothelium. Oxidative and nitrative stress is also associated with markers of vascular injury, e.g. metalloproteinase-9, and blood–brain barrier breakdown, e.g. leakage of Evans blue, suggesting that oedema and haemorrhage may result from reactive oxygen species (ROS) and reactive nitrogen species (RNS) generated in the neurovascular unit during stroke. However, after longer time-points of reperfusion an infiltration of leukocytes may further exacerbate oxidative stress and inflammation.
Due to their more efficient synthesis of glutathione and ARE-linked gene expression, astrocytes are more protected than neurons against basal levels of oxidative stress (Vargas & Johnson, 2009). As astrocytes closely interact with neurons to provide protection from noxious stimuli, Nrf2 activation in astrocytes has been proposed as a therapeutic target for neuroprotection. As illustrated in Fig. 2, glutathione (GSH) released from astrocytes may protect neurons against oxidative stress. As neurons are not able to take up glutathione, additional mechanisms may underlie the neuroprotection afforded by astrocytes: (i) radical-scavenging action by GSH in the extracellular space; (ii) extracellular hydrolysis of GSH by γ-glutamyltranspeptidase present on the external surface of astrocytes, yielding cysteine and glycine, which in turn can be taken up by neurons and used for intracellular GSH synthesis; (iii) GSH may act as a neuromodulator/neurotransmitter by binding membrane receptors on neurons (Halliwell, 2001; Vargas & Johnson, 2009). Moreover, astrocytes also release glutamine, thus providing all the precursors for GSH synthesis in neurons (Vargas & Johnson, 2009). Another example of cross-talk in the antioxidant defences between neurons and astrocytes is ascorbate transport and recycling: neurons are able to take up ascorbate, whereas astrocytes take up the precursor dehydroascorbate (DHA) and then convert it to ascorbate intracellularly (Rice, 2000). This is considered as an important mechanism in brain homeostasis, as neurotransmission requires high ascorbate and low DHA concentrations in the extracellular fluid (Wilson, 2002). It has been suggested that neurons release DHA which is subsequently taken up and converted to ascorbate in astrocytes (Swanson et al. 2004). Moreover, astrocytes release ascorbate into the extracellular fluid, which can be taken up by neurons and/or elicit antioxidant actions extracellularly (Wilson, 2002).
Although brain endothelial cells are more resistant to oxidative stress and ischaemia compared to astrocytes and neurons (Lee et al. 2010), sub-lethal levels of free radicals affect brain endothelial cell function (Guo et al. 2008; Arai & Lo, 2009). Notably, the brain-derived neurotrophic factor released from brain endothelial cells protects neurons from oxidative stress and oxygen-glucose deprivation in vitro (a condition which resembles cerebral ischaemia) (Guo et al. 2008). Recent evidence shows that GSH released from astrocytes protects brain endothelial cells against hemin-induced apoptosis (Sukumari-Ramesh et al. 2010). However, it remains to be resolved whether GSH released from astrocytes acts as an extracellular free radical scavenger and/or exerts cytoprotective effects intracellularly after uptake by endothelial cells (Kannan et al. 2000). Notably, co-cultures of brain capillary endothelial cells and astrocytes show a decreased content of GSH compared to cells in monoculture, but this is accompanied by an increased antioxidant activity and reduced free radical-induced lipid peroxidation (Schroeter et al. 1999).
However, approaches exclusively targeted to protect neurons have proven to be of limited value in clinical trials (del Zoppo, 2010). Thus, future research and therapeutic interventions in stroke should target all components of the neurovascular unit, with an aim of trying to further resolve the time course of repair and injury processes, and whether neurodegeneration promotes vascular dysfunction or vice versa.
Nrf2-mediated neurovascular protection in stroke
Two research approaches have been employed in vivo to investigate the role of the Nrf2–Keap1 defence pathway in stroke: genetic modification of mice (Nrf2−/−) and use of activators of Nrf2-linked gene transcription. Several natural and synthetic compounds, including isothiocyanates, flavonols, heavy metals and hydroperoxides, are potent Nrf2-inducers (Rushmore & Kong, 2002; Shah et al. 2010) (Fig. 1). Liverman et al. (2004) first reported an upregulation of Nrf2 in the brain following oligaemia, a pathological condition characterized by blood flow reduction and oxidative stress in the absence of cell death, mimicking events in the ischaemic penumbra (Baron, 2001). In this study, Nrf2-positive neurons, while absent in brains from control mice, were detected following oligaemia in the Purkinje cells of the cerebellar cortex and pyramidal neurons of the cingulate cortex. Conflicting findings have been reported for Nrf2-deficient mice (see Table 1), with one study reporting that loss of Nrf2 exacerbates cortical infarction after 7 days, but not 24 h, after permanent middle cerebral artery occlusion (MCAO) (Shih et al. 2005). Notably, NAD(P)H-quinone oxidoreductase-1 (NQO1) and glutathione S-transferase activity is reduced in the brain of Nrf2-deficient mice. Another study, however, reports an increase in infarct volume (∼10%) 24 h after transient MCAO (Shah et al. 2007) (see Fig. 3). The discrepancy between these two studies may be explained by the difference in the mouse genetic background (C57B/SV129 vs. CD1) and/or by the type of occlusion used (permanent vs. transient). As a consequence of reperfusion, oxygen and other free radical species are rapidly generated (Allen & Bayraktutan, 2009) with oxidative stress providing a key stimulus for Nrf2-mediated neuroprotection. A delayed reperfusion and inflammatory response, particularly within the peri-infarct region and cortex, with secondary generation of oxidative stress and subsequent cell death, may explain the difference between the wild-type and Nrf2−/− genotypes after cerebral ischaemia–reperfusion injury (Nagayama et al. 2000; Carmichael, 2005).
Table 1.
Activation of the Nrf2–Keap1 defence pathway in experimental stroke
| Strain and species | Experimental model | Findings | Reference |
|---|---|---|---|
| Sprague–Dawley rats | Permanent MCAO–intraluminal filament method | Nrf2 and HO-1 expression was up-regulated between 3 and 72 h after cerebral ischaemia. Curcumin, 100 mg kg−1i.p., 15 min after the onset of stroke reduced cerebral infarct, neurological deficit and brain oedema at 24 h of ischaemia. | Yang et al. (2009) |
| Sprague–Dawley rats | MCAO–intraluminal filament method for 90 min | tBHQ 16.7 mg kg−1i.p. three times (−24, −16 and −8 h) before stroke reduced infarct size and sensorimotor deficit at 24 h and 1 month after ischaemia–reperfusion. | Shih et al. (2005) |
| Wistar rats | MCAO–intraluminal filament method for 90 min | ICV infusion of tBHQ 1 mm for 72 h reduced cerebral infarct size and sensorimotor deficit at 24 h after ischaemia–reperfusion. | Shih et al. (2005) |
| Long–Evans rats | MCAO and CCAO: vessel clip for 3 h | Sulforaphane 5 mg kg−1i.p. 15 min after the onset of ischaemia decreased the infarct volume at 3 days of reperfusion. | Zhao et al. (2006) |
| ICR mice | MCAO: intraluminal filament method for 60 min | Stroke induced Nrf2 expression in neurons of the peri-infarct region between 2 and 72 h, peaking at 8 h. Keap1 expression declined after stroke in neurons of both the peri-infarct and infarct region between 2 and 72 h. | Tanaka et al. (2011) |
| CD1 mice | MCAO: intraluminal filament method for 90 min | Loss of Nrf2 function in KO animals increased cerebral infarct and neurological deficit at 24 h of reperfusion. | Shah et al. (2007) |
| C57B/SV129 mice | MCAO: permanent vessel cauterization | Loss of Nrf2 function in KO animals increased cerebral infarct at 7 days but not 24 h after ischaemia. | Shih et al. (2005) |
| C57B/SV129 mice | Intracortical injection of endothelin-1 | Neuroprotection by dietary 1% tBHQ observed in wild-type was lost in Nrf2−/− animals. | Shih et al. (2005) |
| C57BL/6 mice | MCAO: intraluminal filament method for 2 h | NEPP11 1 mg kg−1i.p. 1 h before and 4 h after the onset of stroke reduced brain infarct at 24 h of reperfusion. | Satoh et al. (2006) |
| C57BL/6 mice | MCAO: intraluminal filament method for 2 h | Carnosic acid 1 mg kg−1i.p. 1 h before the onset of stroke reduced cerebral infarct at 24 h of reperfusion. | Satoh et al. (2008) |
| C57BL/6 mice | MCAO: intraluminal filament method for 1 h | Plumbagin 3 mg kg−1i.v. 6 and 24 h before (but not 1 h after) the onset of stroke reduced cerebral infarct and neurological deficit at 72 h of reperfusion. | Son et al. (2010) |
| C57BL/6 mice | MCAO: intraluminal filament method for 90 min | Epicatechin given orally by gavage 30 mg kg−1 90 min before the onset of stroke reduced cerebral infarct and neurological deficit at 24 h of reperfusion, and this effect was lost in Nrf2-deficient animals. Post-treatment with the same dose and by the same route 3.5 h but not 6 h after the onset of stroke reduced cerebral infarct and neurological deficit at 72 h of reperfusion. | Shah et al. (2010) |
| C57BL/6 mice | Intra-striatal injection of NMDA | Epicatechin given orally by gavage 30 mg kg−1 90 min before the onset of stroke reduced NMDA-induced excitotoxicity at 48 h. | Shah et al. (2010) |
Abbreviations: CCAO, common carotid artery occlusion; HO-1, haem oxygenase-1; ICV, intracerebroventricular; i.p., intraperitoneal; i.v., intravenous; KO, knock-out; MCAO, middle cerebral artery occlusion; NEPP11, neurite outgrowth-promoting prostaglandin; NMDA, N-methyl-d-aspartate; Nrf2, nuclear factor erythroid 2-related factor 2; tBHQ, tert-butylhydroquinone.
Figure 3. Cerebral infarct volumes in wild-type (WT) and Nrf2-deficient mice subjected to 90 min MCAO and 24 h reperfusion.
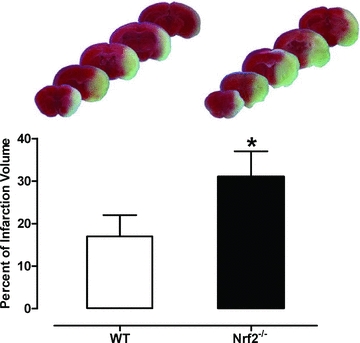
Upper panel: representative images of serial brain sections stained with TTC (2,3,5-triphenyltetrazolium chloride). The light areas denote the infarct region. Lower panel: quantification of infarct areas in WT and Nrf2−/− (mean ± SEM, P < 0.01; n = 8 per group). Adapted from Fig. 1 in Shah et al. (2007) with permission from Elsevier.
tert-Butylhydroquinone (tBHQ) was the first Nrf2 inducer shown to be neuroprotective against experimental stroke (Shih et al. 2005) (Table 1). This molecule is a metabolite of the dietary antioxidant butylated hydroxyanisole which possesses an oxidisable 1,4-diphenolic structure that activates Nrf2, and it can increase Nrf2 phosphorylation via PKC (Huang et al. 2000). As shown in Fig. 4, tBHQ pre-treatment reduced cortical infarct size following transient MCAO in rats either after local intracerebroventricular delivery or multiple systemic intraperitoneal injections (Shih et al. 2005). Furthermore, dietary administration of tBHQ also protects mice from endothelin-1-induced ischaemia/reperfusion injury, and this effect is lost in Nrf2-deficient animals. Thus, despite being a potent inducer of phase-2 defence enzymes via Nrf2 in astrocytes in vitro, protection afforded by tBHQ in vivo requires high doses and longer-term administration to reduce the damaging effects of stroke.
Figure 4. Reduction in cerebral infarct area in stroke following pre-treatment with the Nrf2-inducer tert-butylhydroquinone.
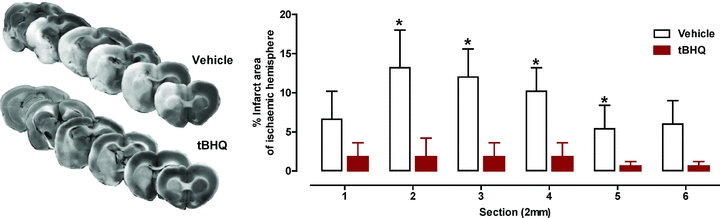
Left panel: representative images of serial brain sections stained with TTC. Right panel: effect of intracerebral ventricular delivery of tert-butylhydroquinone (tBHQ, 1 mm for 72 h from pump implant) before 90 min MCAO and 24 h of reperfusion. Infarct areas are shown between section 1 (anterior) and section 2 (posterior). Data are expressed as mean ± SEM, *P < 0.05; n = 7–9. Adapted from Fig. 2 in Shih et al. (2005) with permission from The Journal of Neuroscience.
Several studies of stroke highlight the ability of drugs or natural plant-derived compounds to protect against stroke via activation of the Nrf2–Keap1 defence pathway (see Fig. 1). Neurite outgrowth-promoting prostaglandins (NEPPs) were synthesised based on the chemical structure of anti-cancer cyclopentenone prostaglandin derivatives and characterized by their neurotrophic effects. NEPP11 was shown to protect neurons in vitro against oxidative stress (Satoh et al. 2003) and to activate the Nrf2–Keap1 pathway in the HT22 neuronal cell line (Satoh et al. 2006). Moreover, NEPP11 reduces cerebral infarction by 30–50% after transient MCAO (Satoh et al. 2006). As summarised in Table 1, curcumin, a low molecular weight polyphenol found in turmeric, elicits antioxidant and anti-inflammatory actions; curcumin administration reduces infarct area, brain oedema and neurological deficits after 24 h of ischaemia (Yang et al. 2009). At this same time-point after ischaemia, Nrf2 protein levels are increased in neurons and astrocytes in the infarcted brain cortex. Carnosic acid, another polyphenolic compound with anti-inflammatory and free radical scavenging properties, reduced the infarct volume in mice subjected to transient MCAO and 24 h of reperfusion (Satoh et al. 2008). Notably, oral administration of the flavonol (−)-epicatechin prior to transient MCAO reduces infarct volume and neurological deficits after 24 h of reperfusion, but this protective effect is lost in Nrf2 and haem oxygenase-1 (HO-1)-deficient mice (Shah et al. 2010). It is interesting that the toxic plant repellent plumbagin protects mice from cerebral ischaemia and associated neurological deficits, but is not effective when administered 1 h after the onset of cerebral ischaemia (Son et al. 2010). Plumbagin-induced activation of Nrf2 was shown in vitro to involve PI3K and MAPK signalling pathways. Sulforaphane, abundantly present in cruciferous vegetables and readily bioavailable in rodents and humans, crosses the blood–brain barrier and is a well-known activator of Nrf2 (McWalter et al. 2004). Administration of sulforaphane after the onset of transient MCAO in mice reduces infarct size measured after 72 h of reperfusion (Zhao et al. 2006).
Therefore, a number of compounds known to activate the Nrf2–Keap1 defence pathway provide neuroprotection in experimental models of stroke. Notably, most of the Nrf2 inducers tested, including curcumin, NEPP11, plumbagin and sulforaphane, increased the levels of HO-1 in the brain. However, all of these compounds may also have additional biological activities and in most of the cases (e.g. NEPP11, carnosic acid, plumbagin and sulforaphane), it remains to be established whether protection in cerebral ischaemia is mediated entirely via Nrf2.
Analysis of Nrf2 mRNA and protein expression in rat brains after permanent MCAO reveals a time-dependent increase, starting at 3 h, peaking at 24 h and declining after 48 and 72 h (Yang et al. 2009). A recent study (Tanaka et al. 2011) investigated temporal changes in expression of Nrf2, Keap1 and the downstream antioxidant proteins thioredoxin (Trx) and HO-1 in murine brains after 60 min of transient MCAO and reported a difference in expression between the peri-infarct and the infarct region. Keap1 was found in the cytoplasm of cells and its expression declined in a time-dependent manner after transient MCAO. In both the peri-infarct and infarct regions, expression levels decreased from 2 to 24 h after reperfusion and remained at low levels after 72 h (see Fig. 5A). Although negligible levels of Nrf2 expression were detected in sham brain sections, Nrf2 was detected in both the cytoplasm and nucleus of the peri-infarct region 2 h after MCAO, peaking at 8 h and then declining at 24 and 72 h (Tanaka et al. 2011). Both Nrf2 and Keap1 seemed to be mainly expressed in neurons (Fig. 5B and C) and not in astroglial or microglial cells (data not shown). Moreover, GSH levels and Trx and HO-1 expression was low or absent in sham brain sections but increased in the peri-infarct region at 24 and 72 h after reperfusion. Notably, expression of Nrf2, Trx and HO-1 was very low in the infarct compared to peri-infarct region, possibly due to oxidative stress in these areas (Tanaka et al. 2011). As the infarct region is characterized by tissue necrosis and cell death (Yuan, 2009), loss of cellular activity may underlie the lack of free radical production and antioxidant enzyme expression. Moreover, as the area of lesion and relevant time-dependent progression of damage was not reported in this study, it remains unclear whether Nrf2 activation and subsequent antioxidant protein expression influence the recruitment of the penumbra into the infarct core following experimental stroke. Indeed, activation of the Nrf2–Keap1 pathway in the penumbra seems to be a major protective mechanism against oxidative stress-induced cell death.
Figure 5. Keap1 and Nrf2-positive cells in the peri-infarct and infarct region of mouse brains after MCAO.
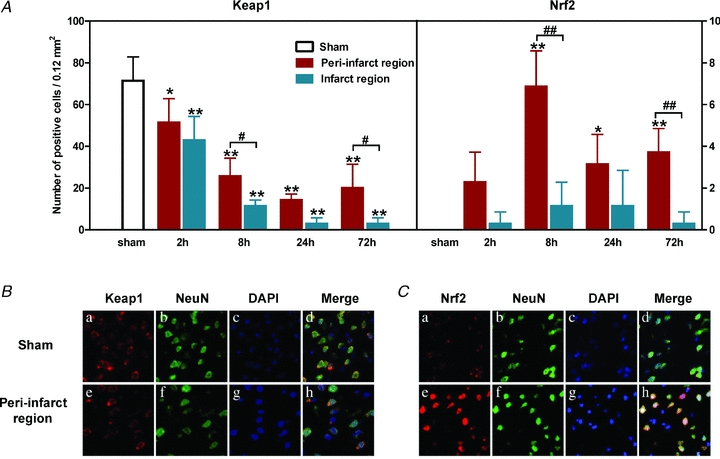
A, brain sections were immunostained at different times of reperfusion after 60 min of MCAO (mean ± SEM. *P < 0.05 and **P < 0.01 vs. sham; #P < 0.05 and ##P < 0.01 vs. infarct region. n = 5 per group). Double immunofluorescent staining for the neural marker NeuN and Keap1 (B) and NeuN and Nrf2 (C) in brain sections from control (sham) mice and in the peri-infarct region of mice subjected to 60 min MCAO and 8 h of reperfusion. DAPI (4',6-diamidino-2-phenylindole) was used for nuclear staining. Adapted from Figs 1 and 2 in Tanaka et al. (2011) with permission from Elsevier.
Future research initiatives need to define the relative distribution of Nrf2 in different cell types of the neurovascular unit. Notably, traumatic brain injury induces Nrf2 activation in microvessels (Zhao et al. 2007) and larger arteries of the cerebral vasculature (Zhao et al. 2010), protecting the brain from blood–brain barrier breakdown. Thus, Nrf2 may improve cerebral vascular function in larger vessels as well as the blood–brain barrier. It has recently been reported that nerve blood flow is improved by Nrf2 activation (Negi et al. 2011). As anti-inflammatory and athero-protective roles for Nrf2 have already been described in the peripheral circulation (Siow & Mann, 2010), it is possible that the Nrf2–Keap1 pathway elicits neurovascular protection via an improvement of cerebral blood flow.
Conclusions and future perspectives
The Nrf2–Keap1 defence pathway serves as a master regulator of endogenous antioxidant defences, and hence has been investigated as a potential therapeutic target for protection of the neurovascular unit in stroke. We have reviewed the evidence that Nrf2 is neuroprotective in experimental stroke, and that its activation may prevent the ischaemic penumbra from cell death. However, further research is required to establish the ‘therapeutic window’ during which activation of the redox-sensitive transcription factor Nrf2 is able to afford protection against cerebral ischaemia. Nrf2 inducers have been shown to be protective when administered before or after the onset of experimental stroke, but it seems likely that Nrf2-mediated increases in the activity of cytoprotective proteins require time. Based on the current understanding of the onset and progression of repair and injury processes in stroke and the need for alternative clinical treatment (Endres et al. 2008), studies are warranted to examine whether preconditioning of the Nrf2–Keap1 defence pathway in vivo offers significant protection against vascular dementia, where people experience small strokes over years. In our current studies, we have established that Nrf2 is expressed in different cell types of the neurovascular unit, and thus Nrf2 may have the potential not only to maintain cerebral blood flow, but also the survival of astrocytes and neurons following cerebral ischaemia–reperfusion injury in stroke.
As ageing-related changes in the brain are associated with an increased incidence of stroke (Chen et al. 2010), it is worth noting that both expression and activity of Nrf2 are diminished in ageing mice (Suh et al. 2004; Collins et al. 2009; Duan et al. 2009) and patients (Cheng et al. 2011; Demirovic & Rattan, 2011). Understanding the molecular mechanisms regulating Nrf2-mediated redox signalling in both young and aged rodent models of stroke should provide valuable insights for potential therapies targeted to protect the neurovascular unit.
Acknowledgments
The authors wish to acknowledge The Henry Smith Charity (RG 20092511) and the British Heart Foundation (FS/09/056) for funding our current research projects focused on the role of Nrf2 in neurovascular protection in stroke.
Glossary
Abbreviations
- ARE
antioxidant response element
- DHA
dehydroascorbate
- GSH
glutathione
- HO-1
haem oxygenase-1
- Keap1
Kelch-like ECH-associated protein 1
- MCAO
middle cerebral artery occlusion
- Nrf2
nuclear factor erythroid 2-related factor 2
- MAPK
mitogen-activated protein kinase
- PKC
protein kinase C
- ROS
reactive oxygen species
- tBHQ
tert-butylhydroquinone
- Trx
thioredoxin
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12:125–169. doi: 10.1089/ars.2009.2668. [DOI] [PubMed] [Google Scholar]
- Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–4355. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balami JS, Chen RL, Grunwald IQ, Buchan AM. Neurological complications of acute ischaemic stroke. Lancet Neurol. 2011;10:357–371. doi: 10.1016/S1474-4422(10)70313-6. [DOI] [PubMed] [Google Scholar]
- Baron JC. Perfusion thresholds in human cerebral ischemia: historical perspective and therapeutic implications. Cerebrovasc Dis. 2001;11(Suppl. 1):2–8. doi: 10.1159/000049119. [DOI] [PubMed] [Google Scholar]
- Burdette D, Olivarez M, Waris G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J Gen Virol. 2010;91:681–690. doi: 10.1099/vir.0.014340-0. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs. 2009;10:644–654. [PubMed] [Google Scholar]
- Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RL, Balami JS, Esiri MM, Chen LK, Buchan AM. Ischemic stroke in the elderly: an overview of evidence. Nat Rev Neurol. 2010;6:256–265. doi: 10.1038/nrneurol.2010.36. [DOI] [PubMed] [Google Scholar]
- Cheng X, Siow RC, Mann GE. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Pratico D, Harrison DG, Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–e54. doi: 10.1161/CIRCRESAHA.108.188771. [DOI] [PubMed] [Google Scholar]
- Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med. 2005;38:1433–1444. doi: 10.1016/j.freeradbiomed.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Moreno-Murciano P, Pedraza-Chaverri J. The transcription factor Nrf2 as a new therapeutic target in Parkinson's disease. Expert Opin Ther Targets. 2009;13:319–329. doi: 10.1517/13543780802716501. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267:156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirovic D, Rattan SI. Curcumin induces stress response and hormetically modulates wound healing ability of human skin fibroblasts undergoing ageing in vitro. Biogerontology. 2011 doi: 10.1007/s10522-011-9326-7. DOI: 10.1007/s10522-011-9326-7. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhang R, Guo Y, Jiang Y, Huang Y, Jiang H, Li C. Nrf2 activity is lost in the spinal cord and its astrocytes of aged mice. In Vitro Cell Dev Biol Anim. 2009;45:388–397. doi: 10.1007/s11626-009-9194-5. [DOI] [PubMed] [Google Scholar]
- Endres M, Engelhardt B, Koistinaho J, Lindvall O, Meairs S, Mohr JP, Planas A, Rothwell N, Schwaninger M, Schwab ME, Vivien D, Wieloch T, Dirnagl U. Improving outcome after stroke: overcoming the translational roadblock. Cerebrovasc Dis. 2008;25:268–278. doi: 10.1159/000118039. [DOI] [PubMed] [Google Scholar]
- Faraci FM. Oxidative stress: the curse that underlies cerebral vascular dysfunction? Stroke. 2005;36:186–188. doi: 10.1161/01.STR.0000153067.27288.8b. [DOI] [PubMed] [Google Scholar]
- Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35:1449–1453. doi: 10.1161/01.STR.0000126044.83777.f4. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Free radicals and antioxidants -quo vadis? Trends Pharmacol Sci. 2011;32:125–130. doi: 10.1016/j.tips.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, De Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain AK, Jaiswal AK. GSK-3β acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, Niizuma K, Katsu M, Okami N, Yoshioka H, Sakata H, Goeders CE, Chan PH. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41:172–179. doi: 10.1007/s12035-010-8102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Chakrabarti R, Tang D, Kim KJ, Kaplowitz N. GSH transport in human cerebrovascular endothelial cells and human astrocytes: evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000;852:374–382. doi: 10.1016/s0006-8993(99)02184-8. [DOI] [PubMed] [Google Scholar]
- Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Yla-Herttuala S, Levonen AL, Koistinaho J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci. 2008;39:302–313. doi: 10.1016/j.mcn.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BJ, Egi Y, van Leyen K, Lo EH, Arai K. Edaravone, a free radical scavenger, protects components of the neurovascular unit against oxidative stress in vitro. Brain Res. 2010;1307:22–27. doi: 10.1016/j.brainres.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- Liverman CS, Cui L, Yong C, Choudhuri R, Klein RM, Welch KM, Berman NE. Response of the brain to oligemia: gene expression, c-Fos, and Nrf2 localization. Brain Res Mol Brain Res. 2004;126:57–66. doi: 10.1016/j.molbrainres.2004.02.028. [DOI] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J Nutr. 2004;134:3499S–3506S. doi: 10.1093/jn/134.12.3499S. [DOI] [PubMed] [Google Scholar]
- Mann GE, Bonacasa B, Ishii T, Siow RC. Targeting the redox sensitive Nrf2-Keap1 defense pathway in cardiovascular disease: protection afforded by dietary isoflavones. Curr Opin Pharmacol. 2009;9:139–145. doi: 10.1016/j.coph.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Marklund SL, Westman NG, Lundgren E, Roos G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982;42:1955–1961. [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Lan J, Henshall DC, Chen D, O'Horo C, Simon RP, Chen J. Induction of oxidative DNA damage in the peri-infarct region after permanent focal cerebral ischemia. J Neurochem. 2000;75:1716–1728. doi: 10.1046/j.1471-4159.2000.0751716.x. [DOI] [PubMed] [Google Scholar]
- Negi G, Kumar A, Sharma SS. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-κB and Nrf2 cascades. J Pineal Res. 2011;50:124–131. doi: 10.1111/j.1600-079X.2010.00821.x. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem. 2009;109(Suppl. 1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- Rizzo MT, Leaver HA. Brain endothelial cell death: modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol Neurobiol. 2010;42:52–63. doi: 10.1007/s12035-010-8132-6. [DOI] [PubMed] [Google Scholar]
- Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- Satoh T, Baba M, Nakatsuka D, Ishikawa Y, Aburatani H, Furuta K, Ishikawa T, Hatanaka H, Suzuki M, Watanabe Y. Role of heme oxygenase-1 protein in the neuroprotective effects of cyclopentenone prostaglandin derivatives under oxidative stress. Eur J Neurosci. 2003;17:2249–2255. doi: 10.1046/j.1460-9568.2003.02688.x. [DOI] [PubMed] [Google Scholar]
- Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2008;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc Natl Acad Sci U S A. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter ML, Mertsch K, Giese H, Muller S, Sporbert A, Hickel B, Blasig IE. Astrocytes enhance radical defence in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett. 1999;449:241–244. doi: 10.1016/s0014-5793(99)00451-2. [DOI] [PubMed] [Google Scholar]
- Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Dore S. The flavanol (–)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab. 2010;30:1951–1961. doi: 10.1038/jcbfm.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Haynes LJ, Theaker R, Gelsthorpe C, Baxter L, Forster G, Lace GL, Shaw PJ, Matthews FE, Savva GM, Brayne C, Wharton SB. Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol Appl Neurobiol. 2010;36:25–40. doi: 10.1111/j.1365-2990.2009.01030.x. [DOI] [PubMed] [Google Scholar]
- Siow RC, Mann GE. Dietary isoflavones and vascular protection: activation of cellular antioxidant defenses by SERMs or hormesis? Mol Aspects Med. 2010;31:468–477. doi: 10.1016/j.mam.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA, Johnson JA, Greig NH, Mattson MP. Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem. 2010;112:1316–1326. doi: 10.1111/j.1471-4159.2009.06552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M, Beal MF. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington's disease. Free Radic Biol Med. 2010;49:147–158. doi: 10.1016/j.freeradbiomed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumari-Ramesh S, Laird MD, Singh N, Vender JR, Alleyne CH, Jr, Dhandapani KM. Astrocyte-derived glutathione attenuates hemin-induced apoptosis in cerebral microvascular cells. Glia. 2010;58:1858–1870. doi: 10.1002/glia.21055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008;74:1526–1539. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian F, Shang J, Matsuura T, Abe K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011;1370:246–253. doi: 10.1016/j.brainres.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27:7511–7521. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17. doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who's listening? Antioxid Redox Signal. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JX. The physiological role of dehydroascorbic acid. FEBS Lett. 2002;527:5–9. doi: 10.1016/s0014-5793(02)03167-8. [DOI] [PubMed] [Google Scholar]
- Yan W, Wang HD, Hu ZG, Wang QF, Yin HX. Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci Lett. 2008;431:150–154. doi: 10.1016/j.neulet.2007.11.060. [DOI] [PubMed] [Google Scholar]
- Yang C, Zhang X, Fan H, Liu Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009;1282:133–141. doi: 10.1016/j.brainres.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–477. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaleska MM, Floyd RA. Regional lipid peroxidation in rat brain in vitro: possible role of endogenous iron. Neurochem Res. 1985;10:397–410. doi: 10.1007/BF00964608. [DOI] [PubMed] [Google Scholar]
- Zhao J, Kobori N, Aronowski J, Dash PK. Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci Lett. 2006;393:108–112. doi: 10.1016/j.neulet.2005.09.065. [DOI] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XD, Zhou YT, Zhang X, Wang XL, Qi W, Zhuang Z, Su XF, Shi JX. Expression of NF-E2-related factor 2 (Nrf2) in the basilar artery after experimental subarachnoid hemorrhage in rabbits: a preliminary study. Brain Res. 2010;1358:221–227. doi: 10.1016/j.brainres.2010.08.035. [DOI] [PubMed] [Google Scholar]