

Abstract
Abstract
The brain responds to noxious stimulation with protective signalling. Over the last decades, a number of experimental strategies have been established to study endogenous brain protection. Pre-, per-, post- and remote ‘conditioning’ are now widely used to unravel the underlying mechanisms of endogenous neuroprotection. Some of these strategies are currently being tested in clinical trials to protect the human brain against anticipated damage or to boost protective responses during or after injury. Here we summarize the principles of ‘conditioning’ research and current efforts to translate this knowledge into effective treatment of patients. Conditioning to induce protected brain states provides an experimental window into endogenous brain protection and can lead to the discovery of drugs mimicking the effects of conditioning. Mechanisms of endogenous brain tolerance can be activated through a wide variety of stimuli that signal ‘danger’ to the brain. These danger signals lead to the induction of regulator and effector mechanisms, which suppress death and induce survival pathways, decrease metabolism, as well as increase substrate delivery. We conclude that preclinical research on endogenous brain protection has greatly benefited from conditioning strategies, but that clinical applications are challenging, and that we should not prematurely rush into ill-designed and underpowered clinical trials.
Philipp Mergenthaler is a research fellow at the Department of Experimental Neurology, Charité University Medicine Berlin. His main research interests are the interdependence of the regulation of glucose metabolism and cell death and the molecular mechanisms of endogenous neuroprotection. Ulrich Dirnagl focuses on stroke, cerebral blood flow regulation and brain imaging. In preclinical models as well as clinical trials he explores mechanisms by which brain ischaemia leads to cell death, and develops novel methods to intercept mechanisms of damage in acute brain damage, as well as to foster regeneration and repair of the lesions. At the Charité University Medicine Berlin he serves as Director of the Department of Experimental Neurology, and Deputy Executive Director of the Center for Stroke Research.
 |
‘That which does not kill us makes us stronger’ Friedrich Nietzsche, Ecce Homo – Warum ich so weise bin 2 (1908)
The multiple flavours of ‘conditioning’
Cells, tissues, organs, as well as whole organisms respond to sublethal stress by activating protective signalling cascades (Dirnagl et al. 2003). In aerobic organisms, strong evolutionary pressure for the development of endogenous mechanisms of protection is generated by hypoxia, as well as by infection and inflammation. Protective responses may be generated either in anticipation of a stressor, during stress against its immediate harmful consequences, or as a response to delayed secondary mechanisms after stress. Various medical disciplines, in particular cardiology, neurology/neurosurgery, anaesthesiology, as well as transplantation medicine, are studying the pathways underlying this signalling of endogenous protective responses. It is the ultimate goal of this research to develop therapeutic organ protection based upon nature's own strategies (Dirnagl et al. 2009; Keep et al. 2010; Gidday, 2010). Early milestones in this quest were the discoveries that pre-exposure to hypoxia can prolong anoxic survival by preserving brain metabolism (Dahl et al. 1964); that brain can adapt to anoxia by hypoxic pre-exposure (Schurr et al. 1986); and the description of ischaemic preconditioning in ischaemic myocardium (Murry et al. 1986) and brain (Kitagawa et al. 1990). Since then, various types of organ ‘conditioning’ have been described (Fig. 1): preconditioning, in which the conditioning stimulus (e.g. ischaemia, hypoxia, metabolic inhibition or inflammation below the threshold of damage) is given several days (‘delayed preconditioning’) or minutes (‘classic preconditioning’) before a noxious stimulus presents (e.g. ischaemia); perconditioning, in which the conditioning stimulus is given while the noxious stimulus is still present; postconditioning, in which the conditioning stimulus is given shortly after the noxious stimulus (e.g. after reperfusion), and remote conditioning, in which not the organ which is affected by the noxious stimulus is conditioned, but another, remote organ or bodily system (e.g. limb ischaemia to induce protection of heart or brain). Robust experimental protocols have been developed for each of these types of conditioning, and a number of the underpinning signalling pathways have been established (Kirino, 2002; Gidday, 2006; Dirnagl & Meisel, 2008; Obrenovitch, 2008; Zhao, 2009; and see below). Some of these conditioning strategies are either directly applicable to patients (e.g. remote conditioning by limb ischaemia), or can be pharmacologically mimicked, such as prolyl-hydroxylase (PHD) inhibitors activating hypoxia inducible factor (HIF)-related pathways, or growth and survival factors such as erythropoeitin (EPO) or granulocyte colony stimulating factor (G-CSF). A number of clinical trials have been concluded in cardiology, and several are underway in patients with brain disease (see below and Table 1). At least in cardiology, where clinical development of conditioning-related strategies is most advanced, their translation into effective therapies has so far been hugely disappointing (Ludman et al. 2010). It is therefore timely to ask what we know about the mechanisms underlying brain conditioning, and what the chances are that brain conditioning will become a clinical reality in the near future.
Figure 1. ‘Conditioning’ paradigms to protect the brain.

Typically, preconditioning uses a sublethal stimulus given minutes or days before the insult against which it aims to protect. Stuttering reperfusion is the prototypical per- or postconditioning strategy, by which one aims to prevent ‘reperfusion damage’ by repetitively opening and blocking brain perfusion before permanent reperfusion is allowed. Remote ischaemia is another per- or postconditioning strategy which typically produces repetitive, short phases of ischaemia of a peripheral limb to induce humoral and neural mechanisms of protection of a remote organ, such as the brain. Pharmacological mimics are drugs that either boost endogenous protective signalling cascades (such as the HIF pathway), or exogenously provide the effectors of endogenous protection, such as EPO.
Table 1.
Examples of currently recruiting clinical trials with neurological endpoints as listed at ClinicalTrials.gov (as of 3/2011): preconditioning, remote conditioning and agents that mimic endogenous neuroprotection
| Trial name | Condition | Intervention | NCT registration |
|---|---|---|---|
| Preconditioning for aneurismal subarachnoid haemorrhage | Subarachnoid haemorrhage | Remote limb preconditioning | NCT01110239 |
| Remote ischaemic preconditioning in subarachnoid haemorrhage (RIPC-SAH) | Subarachnoid haemorrhage | Remote ischaemic preconditioning | NCT01158508 |
| Aneurysmal subarachnoid haemorrhage | |||
| Cerebral vasospasm | |||
| Intracranial aneurysm | |||
| The neuroprotection of sevoflurane preconditioning on intracranial aneurysm surgery | Brain ischaemia | Sevoflurane continuous inhalation | NCT01204268 |
| Effect of remote ischaemic preconditioning on clinical outcomes in CABG surgery (ERICCA) | Coronary heart disease | Remote ischaemic preconditioning | NCT01247545 |
| Effect of remote ischaemic preconditioning on cognitive function after cardiac surgery | Cardiac surgery | Remote ischaemic preconditioning | NCT00877305 |
| New acute treatment for stroke – the effect of remote PERconditioning | Acute stroke | Remote preconditioning | NCT00975962 |
| Neuroprotective study of electroacupuncture pretreatment in patients undergoing cardiac surgery | Stroke Brain injuries | Electroacupuncture pretreatment | NCT01020266 |
| Thrombolysis and deferoxamine in middle cerebral artery occlusion (TANDEM-1) | Acute ischaemic stroke | Deferoxamine | NCT00777140 |
| AX200 for the treatment of ischaemic stroke (AXIS 2) | Acute ischaemic stroke | Filgrastim (G-CSF) | NCT00927836 |
A window into endogenous brain protection
Pre-, per-, post-, remote as well as pharmacological (‘mimics’) conditioning serve as highly valuable tools to understand the mechanisms of endogenous brain protection. It appears that these mechanisms are independent of the conditioning strategy, or have at least a vast overlap. This reflects the fact that these mechanisms have evolved as unspecific responses to a number of challenges to the organism (hypoxia, injury, infection). In the following we discuss them briefly and without reference to specific conditioning strategies (Fig. 2).
Figure 2. General principles of action of ‘conditioning strategies’ to protect the brain.
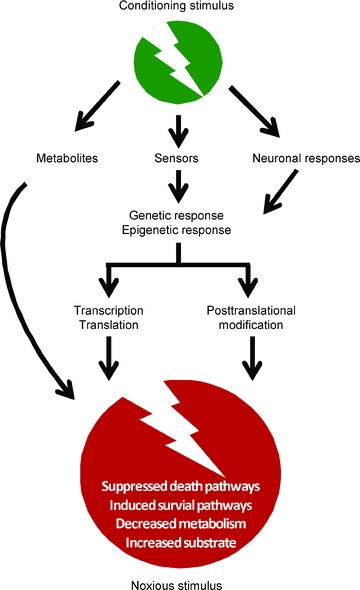
A pre-, per-, post-, remote-conditioning stimulus may either: directly protect the brain via release of locally or remotely acting metabolites (e.g. adenosine); after activation of sensors (e.g. HIF-1) lead to a complex signalling cascade which may include genetic as well as epigenetic responses; or activate genetic and epigenetic responses via neuronal pathways (e.g. activating the sympathetic nervous system or the hypothalamic–pituitary axis). The signalling pathways of the various conditioning strategies may converge in similar or even identical effector mechanisms, such as suppressed death pathways, induced survival pathways, decreased metabolism (‘hibernation’), and increased substrate delivery.
Sensors of danger
Mechanisms of endogenous brain tolerance can be activated through a wide variety of stimuli that signal ‘danger’ to the brain. Both hypoxia and infection endanger the entire organism and are fundamental challenges for most organisms and hence for organs and their cells. Therefore, multiple redundant cascades to adapt to these conditions have evolved. Many aspects of (anti-)inflammatory and hypoxic signalling overlap, and cascades mediating endogenous tolerance are very similar in different organs. For comprehensive overviews the reader is referred to Dirnagl et al. (2003); Gidday (2006); Dirnagl & Meisel (2008); Obrenovitch (2008), and the references therein.
Hypoxia-inducible factor-1 (HIF-1) is the key regulator of cellular oxygen homeostasis. Under hypoxic conditions HIF-1 activates highly conserved transcriptional profiles ultimately geared to adapt cellular homeostasis to reduced oxygen availability. Among others, these adaptations include changes in cellular energy metabolism, regulation of Bcl2-family proteins, cell proliferation, cell cycle control and vasomotor control or angiogenesis (Sharp & Bernaudin, 2004; Semenza, 2009). Likewise, toll-like receptors (TLRs), which are present on many if not all mammalian cells, are responsible for inducing cellular responses to counteract infection.
HIF-1 induces adaptation to decreased oxygen on two levels. First, cellular homeostasis, including mitochondrial respiration, is adapted to hypoxic conditions rather quickly (within hours, Semenza, 2010). Second, chronic hypoxia induces angiogenesis to increase blood supply to hypoxic tissue through a variety of HIF-1-dependent factors such as vascular endothelial growth factor (VEGF; Semenza, 2009). In general, under normoxic conditions the HIF-1α subunit is targeted to rapid proteasomal degradation through post-translation modification by prolyl-hydroxylation, whereas under hypoxic conditions HIF-1α is stabilized and HIF-1-dependent transcription is initialized (Sharp & Bernaudin, 2004; Semenza, 2009). Furthermore, transcription of HIF-1α is increased upon growth-factor signalling, in particular upon activation of the PI3K–Akt–mTOR (mTOR, mammalian target of rapamycin) pathway (DeBerardinis et al. 2008). While the brain has developed some unique sensors of systemic hypoxia, such as central and arterial chemoreceptors (Sharp & Bernaudin, 2004), the molecular cascades involved in oxygen sensing are highly conserved in all cell types (Sharp & Bernaudin, 2004; Semenza, 2009, 2010).
TLRs are an integral part of the innate immune system, providing the first line of defence against pathogens at the cellular level. TLR signalling is an important mediator of ischaemic damage in the brain, but it can also mediate inflammation-induced cross tolerance such as through stimulation with lipopolysaccharide (LPS) or tumour necrosis factor-α (TNF-α) (Marsh et al. 2009). In general, TLRs activate transcription factors through common intracellular pathways, with distinct effects in different cell types or tissues (Marsh et al. 2009). TLRs are a major discriminator between ‘self’ and ‘foreign’ (Akira & Takeda, 2004). A signalling cascade resulting in activation of nuclear factor NF-κB transcription and an inflammatory response are initiated following ligation of pathogen-associated molecular patterns (PAMPs) with TLRs (Liew et al. 2005). Host-derived damage-associated molecular patterns (DAMPs), which are released upon ischaemic injury (Vartanian & Stenzel-Poore, 2010), can also induce inflammatory signalling through the TLR pathway (Seong & Matzinger, 2004) and contribute to ischaemic damage in the brain (Kariko et al. 2004; Lehnardt et al. 2007; Ziegler et al. 2007; Dirnagl et al. 2009). In turn, inflammatory signalling can boost HIF-1 transcription, which controls many genes involved in regulation of inflammation and host defence (Nizet & Johnson, 2009) and which has been found to be essential for the cellular innate immune response in inflammation (Cramer et al. 2003). Furthermore, mitochondria are very sensitive to changes in homeostasis, and are important sensors of cellular stress (see below). TLRs in the brain are constitutively expressed in astrocytes, microglia and endothelial cells. TLRs can be upregulated upon inflammatory stimulation in these cells, but also in neurons and oligodendrocytes (Marsh et al. 2009). Little is known about the differential role of the various TLR types and cell types which express those TLRs in mediating endogenous neuroprotection.
Regulators and effectors
Hypoxia induces substantial changes in gene expression patterns in the brain (Bernaudin et al. 2002). Preconditioning is thought to reprogram the brain's genomic response to a noxious stimulus (Stenzel-Poore et al. 2007). For example, many of the HIF-1 target genes are involved in regulating cellular metabolism, survival, proliferation and angiogenesis (Semenza, 2009). Furthermore, increasing evidence suggests that different epigenetic regulatory mechanisms are activated in the context of conditioning paradigms, and regulate the endogenous protective response. Inhibition of DNA methylation and histone deacetylation reduce ischaemic damage by altering the transcriptional profile (Endres et al. 2000; Meisel et al. 2006; Yildirim et al. 2008). Micro RNAs (miRNAs), small RNA molecules that function as post-transcriptional regulators of gene expression (Lagos-Quintana et al. 2001), are important regulators of diverse aspects of brain function, including development and maintenance of brain plasticity (Saugstad, 2010). Furthermore, miRNAs have emerged as important mediators of endogenous tolerance in the brain (Dharap & Vemuganti, 2010; Lusardi et al. 2010), and both HIF-1 and TLR signalling can be modulated by miRNAs (Crosby et al. 2009; O'Neill et al. 2011).
A variety of kinases involved in proliferation and survival are involved in eliciting an endogenous protective response in the brain. Among others, these include protein kinase C (PKC) (Speechly-Dick et al. 1994), mitogen-activated protein kinase (MAPK)/p38, extracellular signal-regulated kinase (ERK), Akt-kinase (Ruscher et al. 2002; Gao et al. 2008) and mTOR (Pagel, 2008; Swiech et al. 2008). The PI3K–Akt–mTOR pathway senses nutrient availability. Activation of this pathway leads to increased transcription of HIF-1α, further supporting adaptation of metabolism to substrate deprivation (DeBerardinis et al. 2008; Swiech et al. 2008).
Ischaemia is associated with profound metabolic imbalances and much of the cellular response initiated by conditioning events is geared to alter metabolic pathways to maintain basal metabolic integrity. Therefore, under hypoxia, glycolytic flux is diverted from oxidative phosphorylation to glycolysis. HIF-1 changes the expression of genes of the entire glycolytic cascade to adapt metabolism to hypoxic conditions (Iyer et al. 1998; Semenza, 2009). As a consequence, the glycolytic intermediate pyruvate is shunted away from the mitochondrial tricarboxylic acid (TCA) cycle by pyruvate dehydrogenase kinase 1 (PDK1). PDK1 inhibits pyruvate dehydrogenase (PDH), thereby reducing flux through the TCA cycle and ultimately reducing excess production of reactive oxygen species (ROS) (Kim et al. 2006; Papandreou et al. 2006). In addition, lactate dehydrogenase A (LDHA) converts pyruvate to lactate, which in the brain can be rapidly taken up and shuttled away by astrocytes (Gandhi et al. 2009). HIF-1 control over metabolism is not only limited to states of hypoxia, but appears to be of general relevance for survival and proliferation (DeBerardinis et al. 2008). Furthermore, glycolytic enzymes such as mitochondrial hexokinase (HK) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are emerging as important regulators of cell death (Majewski et al. 2004; Kim & Dang, 2005; Colell et al. 2007), with striking mechanistic similarities between neurons and cancer cells (Vaughn & Deshmukh, 2008).
Mitochondria, equipped to efficiently generate ATP through oxidative phosphorylation, also function as oxygen sensors by inhibiting PHD activity through ROS production, thereby stabilizing HIF-1α. Mitochondria are highly susceptible to changes in oxygen concentration and abruptly react by generating ROS (Kaelin, 2005; Kim et al. 2006; Klimova & Chandel, 2008; Semenza, 2010). Importantly, ROS signalling not only contributes to ischaemic damage, but is also involved in endogenous protection evoked by pre-, per-, post- and remote conditioning (Gidday, 2006; Tapuria et al. 2008; Hausenloy, 2009; Ovize et al. 2010; Saxena et al. 2010; Semenza, 2010; Xin et al. 2010).
As described above, inflammation mediated by the innate immune system as well as by the adaptive immune system (Yilmaz et al. 2006; Hurn et al. 2007; Liesz et al. 2011) contributes to brain injury following stroke. However, inflammatory stimulation using LPS can also induce endogenous tolerance (Bastide et al. 2003; Kunz et al. 2007; Orio et al. 2007). Additionally, cerebral ischaemia, as well as other insults to the central nervous system, lead to immunosuppression – a phenomenon termed CNS injury-induced immunosupression (CIDS) (Meisel et al. 2005). CIDS might therefore serve to contain an autoaggressive immune response following stroke (Gee et al. 2007). In humans, ischaemic preconditioning by transient forearm ischaemia changes gene expression patterns in circulating leukocytes, thereby suppressing leukocyte activation and potentially modulating innate and adaptive immune responses (Konstantinov et al. 2004; Saxena et al. 2010).
Clinical applications: expressway or roadblock?
The discovery of ischaemic tolerance, the wealth of knowledge that has subsequently been gathered on mechanisms of endogenous organ protection, and the development of clinically applicable strategies of pre-, per- and postconditioning have precipitated a rush into clinical trials in cardiology, nephrology, anaesthesiology and neurology/neurosurgery, among other disciplines. Some of these rather small proof of concept trials, often using surrogate endpoints, have generated promising results (e.g. Chan et al. 2005; Bøtker et al. 2010; Lonborg et al. 2010; Schäbitz et al. 2010). For the brain, a putative beneficial effect of transient ischaemic attacks (as ‘preconditioning equivalents’) suggested the existence of endogenous neuroprotection in the human brain (Weih et al. 1999; Wegener et al. 2004), although this has been disputed (Johnston, 2004). Meanwhile, however, a number of negative or inconclusive randomized clinical trials (RCT, e.g. Hong et al. 2010; Rahman et al. 2010; Walsh et al. 2010) have been published on various forms of conditioning in several organ systems, including heart and brain, and a less optimistic spirit prevails (Ludman et al. 2010). This is reminiscent of ‘classical’ neuroprotection trials, where promising preclinical and early clinical testing was not followed by evidence for efficacy in large RCTs (Green, 2008; Tymianski, 2010). Nevertheless, for conditioning strategies the jury is still out: the search term ‘preconditioning OR perconditioning OR ‘remote conditioning’ AND brain’ reveals 50 open clinical trials in the trial registry of the National Institute of Health (clinicaltrials.gov). This does not include RCTs testing pharmacological strategies of endogenous neuroprotection such as granulocyte-colony stimulating factor (G-CSF), AX200 for the treatment of ischaemic stroke (AXIS-2, NCT00927836) or erythropoietin (Safety Study of Carbamylated Erythropoietin (CEPO) to Treat Patients With Acute Ischemic Stroke, NCT00756249, publication of results pending). Table 1 lists a selection of currently recruiting RCTs with neurological endpoints in the field of ‘conditioning’ or endogenous neuroprotection.
It has been argued that in contrast to conventional neuroprotection trials, in which neuroprotective drugs are given after the ischaemic event, preconditioning strategies allow baseline assessment of neurological status before patients experience the index event: patients can be functionally tested before preconditioning them against an index event, such as focal neurological deficits after carotid or heart surgery, or delayed neurological deficits after subarachnoid haemorrhage. This may help cut down the variance in results and reduce the number of patients to recruit (Dirnagl et al. 2009), which for a Phase III neuroprotection trial in stroke may run into the thousands. However, the downside of this approach is that only a fraction of patients experience the index event (e.g. around 1% of strokes after coronary bypass surgery), potentially annihilating the advantage of an individual baseline and leading to the exposure of patients to possibly harmful treatments they do not actually need.
Outlook
Basic research on endogenous mechanisms has established a plethora of conditioning strategies and unravelled, among others, neurogenic, immunological, genetic and epigenetic mechanisms of brain protection. Nevertheless, many issues remain unsolved, including questions such as how remote preconditioning exerts its effects (humoral? neuronal?), or whether the dogma that the conditioning stimulus is subthreshold to damage is really true (it has been proposed that in many cases, damage was simply not assessed, or the tools were not sensitive enough; Dirnagl et al. 2003; Sommer, 2008). In the current clinical arena, many teams worldwide are testing the safety and efficacy of such diverse strategies as the prevention or amelioration of CNS damage when it can be anticipated (e.g. delayed vasospasm after subarachnoid haemorrhage), the prevention of CNS damage during potentially harmful interventions (e.g. neurosurgery), the induction of endogenous CNS protection by remote procedures (e.g. repeated limb ischaemia after acute stroke), or the pharmacological induction of endogenous CNS protection (e.g. HIF-1 induction via Desferoxamine) (see Table 1). Over the next few years some of those RCTs may provide evidence not only for the existence of endogenous neuroprotection, but also as to whether related mechanisms can be therapeutically exploited to benefit patients at risk for or with evolving CNS damage. Given the complexities and challenges of the underlying pathophysiology, as well as the design and implementation of clinical trials, and given the frustrating experiences regarding neuroprotection in the stroke field, we are well advised to learn from previous mistakes and to conduct preclinical research of the highest quality (Dirnagl, 2006) and not to prematurely rush into ill-designed and underpowered clinical trials (Weaver et al. 2004).
Acknowledgments
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF), the German Research Foundation (ExcellenceCluster DFG-EXC 257 NeuroCure), and the European Union's Seventh Framework Programme (FP7/2008-2013) under grant agreements No. 201024 and No. 202213 (European Stroke Network).
Glossary
Abbreviations
- Akt
Akt-kinase
- CIDS
CNS injury-induced immunosupression
- DAMP
damage-associated molecular patterns
- EPO
erythropoeitin
- ERK
extracellular signal-regulated kinase
- G-CSF
granulocyte colony stimulating factor
- HIF-1
hypoxia-inducible factor-1
- HK
hexokinase
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- miRNA
micro RNA
- mTOR
mammalian target of rapamycin
- PAMP
pathogen-associated molecular patterns
- PDH
pyruvate dehydrogenase
- PDK1
pyruvate dehydrogenase kinase 1
- PHD
prolyl-hydroxylase
- PI3K
phosphatidylinositol 3-kinases
- RCT
randomized controlled trial
- ROS
reactive oxygen species
- TLR
toll-like receptor
- VEGF
vascular endothelial growth factor
References
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Bastide M, Gele P, Petrault O, Pu Q, Caliez A, Robin E, Deplanque D, Duriez P, Bordet R. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb Blood Flow Metab. 2003;23:399–405. doi: 10.1097/01.WCB.0000050064.57184.F2. [DOI] [PubMed] [Google Scholar]
- Bernaudin M, Tang Y, Reilly M, Petit E, Sharp FR. Brain genomic response following hypoxia and re-oxygenation in the neonatal ratIdentification of genes that might contribute to hypoxia-induced ischemic tolerance. J Biol Chem. 2002;277:39728–39738. doi: 10.1074/jbc.M204619200. [DOI] [PubMed] [Google Scholar]
- Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet. 2010;375:727–734. doi: 10.1016/S0140-6736(09)62001-8. [DOI] [PubMed] [Google Scholar]
- Chan MT, Boet R, Ng SC, Poon WS, Gin T. Effect of ischemic preconditioning on brain tissue gases and pH during temporary cerebral artery occlusion. Acta Neurochir Suppl. 2005;95:93–96. doi: 10.1007/3-211-32318-x_20. [DOI] [PubMed] [Google Scholar]
- Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby ME, Devlin CM, Glazer PM, Calin GA, Ivan M. Emerging roles of microRNAs in the molecular responses to hypoxia. Curr Pharm Des. 2009;15:3861–3866. doi: 10.2174/138161209789649367. [DOI] [PubMed] [Google Scholar]
- Dahl NA, Balfour WM. Prolonged anoxic survival due to anoxia preexposure: brain ATP, lactate, and pyruvate. Am J Physiol. 1964;1:452–456. doi: 10.1152/ajplegacy.1964.207.2.452. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Dharap A, Vemuganti R. Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J Neurochem. 2010;113:1685–1691. doi: 10.1111/j.1471-4159.2010.06735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U. Bench to bedside: the quest for quality in experimental stroke research. J Cereb Blood Flow Metab. 2006;26:1465–1478. doi: 10.1038/sj.jcbfm.9600298. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55:334–344. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi GK, Cruz NF, Ball KK, Dienel GA. Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J Neurochem. 2009;111:522–536. doi: 10.1111/j.1471-4159.2009.06333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee JM, Kalil A, Shea C, Becker KJ. Lymphocytes: potential mediators of postischemic injury and neuroprotection. Stroke. 2007;38:783–788. doi: 10.1161/01.STR.0000248425.59176.7b. [DOI] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Gidday JM. Pharmacologic preconditioning: translating the promise. Transl Stroke Res. 2010;1:19–30. doi: 10.1007/s12975-010-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br J Pharmacol. 2008;153:S325–S338. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ. Signalling pathways in ischaemic postconditioning. Thromb Haemost. 2009;101:626–634. [PubMed] [Google Scholar]
- Hong DM, Mint JJ, Kim JH, Sohn IS, Lim TW, Lim YJ, et al. The effect of remote ischaemic preconditioning on myocardial injury in patients undergoing off-pump coronary artery bypass graft surgery. Anaesth Intensive Care. 2010;38:924–929. doi: 10.1177/0310057X1003800518. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SC. Ischemic preconditioning from transient ischemic attacks? Data from the Northern California TIA Study. Stroke. 2004;35:2680–2682. doi: 10.1161/01.STR.0000143322.20491.0f. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr ROS: really involved in oxygen sensing. Cell Metab. 2005;1:357–358. doi: 10.1016/j.cmet.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling – a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- Keep RF, Wang MM, Xiang J, Hua Y, Xi G. Is there a place for cerebral preconditioning in the clinic? Transl Stroke Res. 2010;1:4–18. doi: 10.1007/s12975-009-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005;30:142–150. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- Konstantinov IE, Arab S, Kharbanda RK, Li J, Cheung MM, Cherepanov V, Downey GP, et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics. 2004;19:143–150. doi: 10.1152/physiolgenomics.00046.2004. [DOI] [PubMed] [Google Scholar]
- Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- Lønborg J, Holmvang L, Kelbæk H, Vejlstrup N, Jørgensen E, Helqvist S, et al. ST-segment resolution and clinical outcome with ischemic postconditioning and comparison to magnetic resonance. Am Heart J. 2010;160:1085–1091. doi: 10.1016/j.ahj.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Ludman AJ, Yellon DM, Hausenloy DJ. Cardiac preconditioning for ischaemia: lost in translation. Dis Model Mech. 2010;3:35–38. doi: 10.1242/dmm.003855. [DOI] [PubMed] [Google Scholar]
- Lusardi TA, Farr CD, Faulkner CL, Pignataro G, Yang T, Lan J, Simon RP, Saugstad JA. Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J Cereb Blood Flow Metab. 2010;30:744–756. doi: 10.1038/jcbfm.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. 2009;158:1007–1020. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel A, Harms C, Yildirim F, Bosel J, Kronenberg G, Harms U, Fink KB, Endres M. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem. 2006;98:1019–1031. doi: 10.1111/j.1471-4159.2006.04016.x. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- Orio M, Kunz A, Kawano T, Anrather J, Zhou P, Iadecola C. Lipopolysaccharide induces early tolerance to excitotoxicity via nitric oxide and cGMP. Stroke. 2007;38:2812–2817. doi: 10.1161/STROKEAHA.107.486837. [DOI] [PubMed] [Google Scholar]
- Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, et al. Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406–423. doi: 10.1093/cvr/cvq129. [DOI] [PubMed] [Google Scholar]
- Pagel PS. Postconditioning by volatile anesthetics: salvaging ischemic myocardium at reperfusion by activation of prosurvival signaling. J Cardiothorac Vasc Anesth. 2008;22:753–765. doi: 10.1053/j.jvca.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Rahman IA, Mascaro JG, Steeds RP, Frenneaux MP, Nightingale P, Gosling P, et al. Remote ischemic preconditioning in human coronary artery bypass surgery: from promise to disappointment? Circulation. 2010;14, 122:S53–S59. doi: 10.1161/CIRCULATIONAHA.109.926667. [DOI] [PubMed] [Google Scholar]
- Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, et al. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci. 2002;22:10291–10301. doi: 10.1523/JNEUROSCI.22-23-10291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad JA. MicroRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. J Cereb Blood Flow Metab. 2010;30:1564–1576. doi: 10.1038/jcbfm.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena P, Newman MA, Shehatha JS, Redington AN, Konstantinov IE. Remote ischemic conditioning: evolution of the concept, mechanisms, and clinical application. J Card Surg. 2010;25:127–134. doi: 10.1111/j.1540-8191.2009.00820.x. [DOI] [PubMed] [Google Scholar]
- Schäbitz WR, Laage R, Vogt G, Koch W, Kollmar R, Schwab S, et al. AXIS: a trial of intravenous granulocyte colony-stimulating factor in acute ischemic stroke. Stroke. 2010;41:2545–2551. doi: 10.1161/STROKEAHA.110.579508. [DOI] [PubMed] [Google Scholar]
- Schurr A, Reid KH, Tseng MT, West C, Rigor MB. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res. 1986;374:244–248. doi: 10.1016/0006-8993(86)90418-x. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta. 2010;1813:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- Sommer C. Ischemic preconditioning: postischemic structural changes in the brain. J Neuropathol Exp Neurol. 2008;67:85–92. doi: 10.1097/nen.0b013e3181630ba6. [DOI] [PubMed] [Google Scholar]
- Speechly-Dick ME, Mocanu MM, Yellon DM. Protein kinase C. Its role in ischemic preconditioning in the rat. Circ Res. 1994;75:586–590. doi: 10.1161/01.res.75.3.586. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007;38:680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR. Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury – a review. J Surg Res. 2008;150:304–330. doi: 10.1016/j.jss.2007.12.747. [DOI] [PubMed] [Google Scholar]
- Tymianski M. Can molecular and cellular neuroprotection be translated into therapies for patients?: yes, but not the way we tried it before. Stroke. 2010;41:S87–S90. doi: 10.1161/STROKEAHA.110.595496. [DOI] [PubMed] [Google Scholar]
- Vartanian K, Stenzel-Poore M. Toll-like receptor tolerance as a mechanism for neuroprotection. Transl Stroke Res. 2010;1:252–260. doi: 10.1007/s12975-010-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SR, Sadat U, Boyle JR, Tang TY, Lapsley M, Norden AG, Gaunt ME. Remote ischemic preconditioning for renal protection during elective open infrarenal abdominal aortic aneurysm repair: randomized controlled trial. Vasc Endovascular Surg. 2010;44:334–340. doi: 10.1177/1538574410370788. [DOI] [PubMed] [Google Scholar]
- Weaver CS, Leonardi-Bee J, Bath-Hextall FJ, Bath PM. Sample size calculations in acute stroke trials: a systematic review of their reporting, characteristics, and relationship with outcome. Stroke. 2004;35:1216–1224. doi: 10.1161/01.STR.0000125010.70652.93. [DOI] [PubMed] [Google Scholar]
- Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, et al. MRI in Acute Stroke Study Group of the German Competence Network Stroke. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke. 2004;35:616–621. doi: 10.1161/01.STR.0000115767.17923.6A. [DOI] [PubMed] [Google Scholar]
- Weih M, Kallenberg K, Bergk A, Dirnagl U, Harms L, Wernecke KD, Einhäupl KM. Attenuated stroke severity after prodromal TIA: a role for ischemic tolerance in the brain? Stroke. 1999;30:1851–1854. doi: 10.1161/01.str.30.9.1851. [DOI] [PubMed] [Google Scholar]
- Xin P, Zhu W, Li J, Ma S, Wang L, Liu M, Wei M, Redington AN. Combined local ischemic postconditioning and remote perconditioning recapitulate cardioprotective effects of local ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2010;298:H1819–H1831. doi: 10.1152/ajpheart.01102.2009. [DOI] [PubMed] [Google Scholar]
- Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol. 2008;210:531–542. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab. 2009;29:873–885. doi: 10.1038/jcbfm.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]