

Non-technical summary
Astrocytes have been shown to release transmitters by vesicle fusion, in a manner similar to that of neuronal exocytosis. The details of this process in astrocytes are not well understood, so we used a fluorescently labelled vesicle protein, synapto-pHluorin (spH), to track how these fusions occurred. When astrocytes were mechanically stimulated we saw a slow burst of fusions, while other stimuli caused a relatively even sustained rate of fusion. We observed two distinct types of events, transient and full fusions, the proportion of which was stimulus dependent. Similarly, stability of the vesicle fusion pore with the plasma membrane varied with the stimulus. We describe the effects on fusion events resulting from expressing variants of exocytotic proteins, synaptotagmin 1 and SNAP25B. Studying the characteristics of astrocytic exocytosis will aid in the general understanding of this process and also events at the tripartite synapse, both in health and disease.
Abstract
Abstract
Astrocytes can release various gliotransmitters in response to stimuli that cause increases in intracellular Ca2+ levels; this secretion occurs via a regulated exocytosis pathway. Indeed, astrocytes express protein components of the vesicular secretory apparatus. However, the detailed temporal characteristics of vesicular fusions in astrocytes are not well understood. In order to start addressing this issue, we used total internal reflection fluorescence microscopy (TIRFM) to visualize vesicular fusion events in astrocytes expressing the fluorescent synaptobrevin 2 derivative, synapto-pHluorin. Although our cultured astrocytes from visual cortex express synaptosome-associated protein of 23 kDa (SNAP23), but not of 25 kDa (SNAP25), these glial cells exhibited a slow burst of exocytosis under mechanical stimulation; the expression of SNAP25B did not affect bursting behaviour. The relative amount of two distinct types of events observed, transient and full fusions, depended on the applied stimulus. Expression of exogenous synaptotagmin 1 (Syt1) in astrocytes endogenously expressing Syt4, led to a greater proportion of transient fusions when astrocytes were stimulated with bradykinin, a stimulus otherwise resulting in more full fusions. Additionally, we studied the stability of the transient fusion pore by measuring its dwell time, relation to vesicular size, flickering and decay slope; all of these characteristics were secretagogue dependent. The expression of SNAP25B or Syt1 had complex effects on transient fusion pore stability in a stimulus-specific manner. SNAP25B obliterated the appearance of flickers and reduced the dwell time when astrocytes were mechanically stimulated, while astrocytes expressing SNAP25B and stimulated with bradykinin had a reduction in decay slope. Syt1 reduced the dwell time when astrocytes were stimulated either mechanically or with bradykinin. Our detailed study of temporal characteristics of astrocytic exocytosis will not only aid the general understanding of this process, but also the interpretation of the events at the tripartite synapse, both in health and disease.
Introduction
Astrocytes can release gliotransmitters using various mechanisms, which can result in signalling to neurons (Ni et al. 2007; Malarkey & Parpura, 2008, 2009). Exocytosis is one of the prominent mechanisms underlying gliotransmitter release from astrocytes (Parpura et al. 2010; Parpura & Zorec, 2010). Much effort has been applied to the study of this process in electrically non-excitable astrocytes with comparisons of astrocytic exocytosis to that occurring in electrically excitable cells, such as neurons and chromaffin cells (reviewed in Lee & Parpura, 2007). Astrocytes exhibit a form of excitability based on intracellular Ca2+ elevations, which can stimulate gliotransmitter release from astrocytes. Indeed, astrocytes express protein components of exocytotic secretory machinery, including the core fusion complex as well as transporters and pumps necessary for filling astrocytic vesicles with gliotransmitter. The characteristics of exocytosis in astrocytes appear different to those observed in neurons; e.g. gliotransmission is markedly slower than neurotransmission (reviewed in Lee & Parpura, 2007; Parpura et al. 2010). These glial cells can release gliotransmitters from their somata and processes (reviewed in Montana et al. 2006). Thus far, much attention has been devoted to events occurring at astrocytic processes. Indeed, the morphological arrangements of exocytotic secretory machinery and functional transmitter receptors in astrocytic processes enable them to receive signals, focally, from adjacent synaptic terminals and respond back to terminals/dendrites via exocytotic gliotransmitter release (reviewed in Montana et al. 2006). This bi-directional neuron–astrocyte signalling in synaptic transmission is referred to as the tripartite synapse (Araque et al. 1999). However, there are still many issues that need to be addressed in order to better understand the exocytotic process in astrocytes; this includes the temporal characteristics of vesicular fusion occurring at somata, investigated here.
Since astrocytes can express a variety of exocytotic proteins (reviewed in Montana et al. 2006), it seems likely that many intracellular interactions between exocytotic proteins mediating docking/priming and fusion could occur with some redundancy and promiscuity (Liu et al. 2006; Montana et al. 2009). For example, astrocytes isolated from visual cortex in our culture system express the components of exocytotic machinery: syntaxin 1A and SNAP23A (synaptosome-associated protein of 23,000 Da) at the plasma membrane, as well as the vesicular proteins synaptotagmin 4 (Syt4), synaptobrevin 2 and its homologue cellubrevin (Parpura et al. 1995a; Montana et al. 2004). Others have reported the additional expression of SNAP25 or Syt1 in astrocytes (Jeftinija et al. 1997; Maienschein et al. 1999; Wilhelm et al. 2004; Stigliani et al. 2006). Thus, in some conditions, co-expression of SNAP23/25 and/or Syt1/4 could differentially regulate vesicular fusion in astrocytes. From single-molecule measurements, it is evident that the presence of SNAP25B in the ternary SNARE (the soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor; Sollner et al. 1993) complex instead of SNAP23A gives this complex an enhanced stability (Montana et al. 2009). Furthermore, in chromaffin cells, SNAP25 affects the temporal characteristics of exocytosis leading to the display of exocytotic ‘bursts’, while in its absence only sustained exocytosis was observed (Sorensen et al. 2003). Moreover, in chromaffin and PC12 cells, Syt1/4 can affect the temporal characteristics of exocytosis, leading to a change in the proportion of transient vs. full vesicular fusions (Wang et al. 2003; Zhang & Jackson, 2010; Zhang et al. 2010). Yet, how the expression of these exocytotic protein variants affects the temporal characteristics of vesicular fusion in astrocytes has not been determined. Therefore, we used total internal reflection fluorescence microscopy (TIRFM) to visualize the fusion of vesicles containing the fluorescently labelled synaptobrevin 2, super-ecliptic synapto-pHluorin (spH) (Sankaranarayanan et al. 2000), in an attempt to define these temporal characteristics of vesicular fusion in astrocytes. We also investigated the effects that exogenous expression of SNAP25B or Syt1, as additions to the endogenous repertoire of exocytotic proteins in our culture system, exerts on the temporal characteristics of astrocytic exocytosis.
Bursts of exocytotic fusion events have been seen in hippocampal astrocytes within ∼0.5 s after stimulation of metabotropic glutamate (mGlu) (Bezzi et al. 2004; Marchaland et al. 2008), metabotropic purinergic (P2Y1) (Domercq et al. 2006) or chemokine (CXC4) receptors (Cali et al. 2008). In contrast to such explosive patterns of vesicular fusions, others showed slower temporal patterns of fusions, both brief and sustained, in astrocytes after mechanical stimulation or agonist-induced activation of metabotropic and ionotropic glutamate receptors (mGluRs and iGluRs) (Pasti et al. 2001; Chen et al. 2005). There could be various explanations for the variability in these findings. Since the above studies did not disclose what exocytotic proteins were present in their cultured astrocytes, the role that differential expression of exocytotic proteins plays in these rates of exocytosis is not clear. Here, we report that, although astrocytes mainly display a relatively even sustained rate of exocytosis, in some cases slow bursts of fusions occur within 6 s of the initiation of a stimulus in astrocytes for which we report expression of the same exocytotic proteins as those displaying sustained exocytosis. We find that the stimulus that is applied to the cell is what determines which temporal pattern will occur, and not the expression of specific exocytotic proteins, such as SNAP25B.
Two distinct types of fusion events, full and transient, can occur in astrocytes (Bezzi et al. 2004; Chen et al. 2005; Bowser & Khakh, 2007; Cali et al. 2008; Marchaland et al. 2008). However, how these different modes of fusion events can be modulated in astrocytes has not been systematically studied. We report that the preferred fusion type was dependent on the stimulus that was applied. Additionally, we studied the stability of the transient fusion pore. We measured its dwell time, relation to vesicular size, flickering and decay slope. All of these characteristics were stimulus dependent in astrocytes with the endogenous expression of exocytotic proteins. The expression of exogenous SNAP25B or Syt1 in astrocytes had complex effects on the proportion of transient fusions and on the above temporal characteristics in a stimulus-specific manner.
Our in-depth description of the temporal characteristics of vesicular fusion in astrocytes sheds new light on exocytotic processes in astrocytes. The reported effects that expression of SNAP25B and Syt1 has on astrocytic exocytosis contribute additional facets to the complexity of this process. Changes in expression of SNAP25 and Syt1 have been associated with various conditions, including attention deficit hyperactivity disorder, schizophrenia and epilepsy (Vician et al. 1995; Tocco et al. 1996; Corradini et al. 2009). Thus, our findings extend the interpretation of events occurring at the tripartite synapse, both in health and disease. Some of these data have appeared in preliminary form (Malarkey & Parpura, 2006).
Methods
Cell culture
All procedures were in strict accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the University of California, Riverside and University of Alabama, Birmingham Institutional Animal Care and Use Committees. The procedures conform to the principles of UK regulations, as descried in The journal of Physiology (Drummond, 2009). We prepared enriched astrocytic cultures using a modification (Parpura et al. 1995b; Montana et al. 2004) of the originally described shaking procedure (McCarthy & deVellis, 1980). Briefly, 0- to 2-day-old Sprague–Dawley rats were killed by decapitation. Their visual cortices were dissected out and treated with papain (20 IU ml−1; Sigma) in Hank's balanced salt solution (HBSS; Invitrogen, Carlsbad, CA, USA) containing l-cysteine (0.2 mg ml−1) for 1 h at 37°C. The tissue was washed with HBSS and then incubated with trypsin inhibitor (type II-O, 10 mg ml−1; Sigma) in HBSS for 5 min at room temperature. After an additional wash with HBSS, the tissue was triturated in culture medium containing α-minimum essential medium (α-MEM, without phenol red; Invitrogen) supplemented with fetal bovine serum (10%; HyClone, Logan, UT, USA), 20 mm d-glucose, 2 mm l-glutamine, 1 mm sodium pyruvate, 14 mm sodium bicarbonate, penicillin (100 IU ml−1) and streptomycin (100 μg ml−1) (pH 7.35). The resulting cell suspension was applied to culture flasks (25 cm2) and maintained in culture medium at 37°C in a 5% CO2–95% air atmosphere incubator. After 6–12 days, the cells were submitted to a procedure for purification of astrocytes. At that time, flasks were shaken on a horizontal orbital shaker at 260 rpm and 37°C for 1.5 h, and after changing the medium twice, shaken again for 18 h. The cells that remained attached to the bottom of the flask were then returned to the incubator to be submitted to the transfection protocol (see below) or plated on coverslips as follows. Cells were detached using trypsin (10,000 Nα-benzoyl-arginine ethyl ester hydrochloride (BAEE) units ml−1; Sigma–Aldrich). After inhibition of trypsin activity by addition of complete culture medium, cells were pelleted using centrifugation at 100 g for 10 min. The resulting cell pellet was resuspended in complete medium and plated onto glass coverslips (12 mm in diameter, thickness no. 1, 0.13–0.16 mm; D-263 glass, Erie Scientific Company; purchased via Fisher Scientific, cat. no. 12-545-82-12CIR-1D) pre-coated with polyethyleneimine (PEI, 1 mg ml−1). Purified astrocytes (>99%) were used in experiments after 4–12 days. The purity of astrocytic culture was confirmed by indirect immunocytochemistry using anti-glial fibrillary acidic protein (GFAP) antibody and visualization of accumulation of a dipeptide β-Ala-Lys conjugated to 7-amino-4-methylcoumarin-3-acetic acid as we previously described (Montana et al. 2004; Malarkey et al. 2008). Astrocytes in our culture system are flat polygonal cells having less complex morphological appearance than astrocytes in situ (Hua et al. 2004; Montana et al. 2004; Malarkey et al. 2008).
Cell transfection
Transfection was done using purified astrocytic culture and a transfection reagent (TransIT-293, Mirus). In a subset of experiments using the light chain of tetanus toxin (lcTeTx), 1 h prior to the transfection procedure, the medium was completely changed and fresh medium was applied to 25 cm2 culture flasks containing astrocyte cultures. Plasmids encoding the lcTeTx (12 μg per flask; provided by Dr Urlich Eisel, University of Groningen, Haren, the Netherlands; Eisel et al. 1993) and superecliptic spH (6 μg per flask; provided by Dr James E. Rothman, Yale University, New Haven, CT, USA (Miesenbock et al. 1998; Sankaranarayanan et al. 2000) or spH alone, were incubated for 10 min at room temperature with the TransIT-293 reagent in α-MEM medium without additives. This mixture (400 μl) was evenly dispersed into a culture flask of confluent astrocytes in complete medium (4 ml) which was incubated for 3–4 h at 37°C in a 5% CO2–95% air incubator. Following incubation and wash out of transfection mixture, fresh complete medium was applied and cells were returned to the incubator for 36–48 h, after which they were detached from the flask and plated onto PEI-coated coverslips. For all other transfections we used an alternate protocol that was similarly effective. One hour prior to the transfection procedure, the medium was completely changed and fresh medium was applied to 35 mm Petri dishes containing astrocyte cultures grown on four PEI-coated coverslips. The transfection reagent was prepared by mixing α-MEM with no additives and 2 μl of TransIT-293 reagent for each microgram of plasmid to be used, followed by vortexing and incubating for 10 min at room temperature. At this time a plasmid encoding spH (0.5–1 μg per dish), rat synaptobrevin 2 (also referred to as vesicle-associated protein 2)–green fluorescence protein chimera (VAMP2-GFP; 0.5 μg per dish; provided by Dr Richard H. Scheller, Stanford University, Palo Alto, CA, USA; Ahmari et al. 2000) or enhanced green fluorescent protein (pEGFP-N1; 0.5 μg per dish; Clontech, Mountain View, CA, USA) was added to the mixture, alone or in combination (for spH or EGFP) with a 1:2 weight ratio of plasmid encoding mouse SNAP25B (1 μg per dish; provided by Dr Michael C. Wilson, University of New Mexico, Albuquerque, NM, USA) or rat synaptotagmin 1 (1–2 μg per dish; provided by Dr Thomas C. Südhof, Stanford University, Palo Alto, CA, USA; Shin et al. 2003) and incubated for 10 min at room temperature. One hundred microlitres of the complexed plasmid(s)–transfection reagent mixture was added to the medium (1 ml) in each dish which was then incubated for 3–4 h at 37°C in a 5% CO2–95% air incubator. Following incubation and wash out of transfection mixture, fresh complete medium was applied and cells were returned to the incubator for 2–5 days before being used in experiments. We previously reported that based on the EGFP expression ∼18% of astrocytes were successfully transfected using this procedure; co-expression of co-complexed plasmids resulted in all astrocytes expressing both proteins of interest (Ni & Parpura, 2009).
Reverse transcription-PCR
Total RNA was extracted from cortical tissue of post-natal Sprague–Dawley rats (0–2 days old) using TRIzol Reagent (Invitrogen) and protocols provided by the manufacturer. Five micrograms of total RNA were used for reverse transcription using Oligo(dT)12–18 and superscript II reverse transcriptase (Invitrogen). Published primers for SNAP23 were used to amplify DNA using PCR (35 cycles); the different isoforms of rat SNAP23 can be determined by the size of the PCR product (SNAP23A at 392 bp and SNAP23B at 236 bp) (Grant et al. 1999). We used a plasmid encoding rat SNAP23A (provided by Dr Paul A. Roche, National Institutes of Health, Bethesda, MD, USA; Sadoul et al. 1997) as a template for PCR amplification and serving as a positive control.
Indirect immunocytochemistry
To confirm astrocytic expression of SNAP23, cells were exposed to Dent's fixative at room temperature for 30 min (Parpura & Haydon, 2000; Montana et al. 2004). Cells were then permeabilized using 0.25% Triton-X 100 phosphate-buffered saline (PBS, pH 7.4) for 10 min. PBS supplemented with 10% goat serum was used to block non-specific binding and cells were then incubated with primary antibodies. A rabbit polyclonal antibody against SNAP23 (Synaptic Systems, Cat. no. 104 111 202; 1:50 dilution; overnight at 4°C) was used. Following washout of primary antibodies, cells were incubated with tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibody for 1 h.
For verifying the expression of endogenous synaptotagmin 4, as well as exogenous SNAP25B or synaptotagmin 1, cultured astrocytes were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature (Montana et al. 2004; Ni & Parpura, 2009). Following permeabilization and blocking steps, cells were incubated with primary antibodies overnight at 4°C. Polyclonal rabbit antibody against synaptotagmin 4 (Immuno-Biological Laboratories, Inc., Minneapolis, MN, USA; 5 μg ml−1) and monoclonal antibodies against SNAP25 (Cat. no. 111 011; 1:500 dilution; Synaptic Systems) or synaptotagmin 1 (Cat. no. 105 011; 1:500 dilution; Synaptic Systems) were used. Following washout of primary antibodies, cells were incubated with TRITC-conjugated secondary antibody for 1 h.
In parallel, control experiments were performed in which primary antibodies were omitted to test for the non-specific binding of secondary antibodies. All imaging data were background subtracted using fluorescence emission from a region of the coverslip containing no cells.
Ca2+ imaging
The intracellular Ca2+ levels in astrocytes were recorded using the Ca2+ indicator fluo-3. Cells were loaded with the acetoxymethyl (AM) ester of fluo-3 (10 μg ml−1; Invitrogen) for 30 min at 37°C at room temperature; the ester's dispersion in aqueous medium was aided by 0.025% w/v Pluronic F-127 (Invitrogen). After wash, the indicator was permitted to de-esterify for 30 min at room temperature in external solution before starting the experiment. The external solution contained 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 5 mm glucose and 10 mm Hepes (pH 7.4). The fluorescent signal was background subtracted using regions of the field containing no cells. Data are expressed as dF/Fo (%) in which dF represents the change in fluorescence, while Fo represents the initial fluorescence of the cell.
Imaging acquisition and processing
All experiments were done at room temperature (20–24°C). Live astrocytes were bathed in external solution. We used an inverted microscope (IX71 or IX81; Olympus) equipped with wide-field epifluorescence and total internal reflection fluorescence (TIRF) illumination. Visualization of spH and VAMP2-GFP was accomplished using a TIRF-488 laser filter set (Chroma Technology, Rockingham, VT, USA); the light source was a 10 mW 488 nm argon laser (Melles Griot, Carlsbad, CA, USA). For epifluorescence visualization of fluo-3, spH and EGFP, we used a standard fluorescein/FITC filter set; a xenon arc lamp (100 W) was used as a light source. For time-lapse image acquisition, an electronic shutter (Vincent Associates, Rochester, NY, USA) inserted in the excitation pathway was controlled by software. Images were captured through a 60× PlanApo oil-immersion TIRFM objective (numerical aperture (NA), 1.45; Olympus) and 1.6× relay lens using a Cascade 650 cooled charge-coupled device (CCD) camera (Roper Scientific, Tucson, AZ, USA) driven by Metamorph imaging software ver. 6.1 (Molecular Devices, Chicago, IL, USA). Experiments using lcTeTx were performed on a Nikon TE2000-S microscope with CFI 60× PlanApo oil-immersion TIRFM objective (NA, 1.45; Nikon) and 1.5× relay lens, similarly equipped for total internal reflection fluorescence. The objectives we used (NA, 1.45) along with refractive indices of borosilicate glass coverslips (1.5252), immersion oil (1.515; Nikon, type NF) and the cytoplasmic components of live cells (1.35–1.37; Liang et al. 2007) yield a calculated penetration depth (see, for example, Steyer & Almers, 2001) of the evanescent wave of 70–77 nm. The laser light was brought to the correct angle for internal reflection by focusing it on the outer edge of the objective's back aperture. For visualization of vesicular fusion events, TIRF imaging frames were streamed directly to a hard drive at 30 or 60 frames per second (33.3 or 16.6 ms per frame, respectively) using a 600 × 450 or 200 × 200 pixel region of the camera chip (41.5 × 31.1 or 13.8 × 13.8 μm area of the coverslip, respectively; the pixel size was 69.1 nm). For immunocytochemistry in astrocytes, we used a Nikon TE 300 equipped with wide-field epifluorescence illumination. As a light source we used a xenon arc lamp (100 W) with an electronic shutter inserted in the excitation pathway. Images were captured through a 60× PlanApo oil-immersion objective (NA, 1.4; Nikon) using a CoolSNAP-HQ CCD camera driven by V++ imaging software (Digital Optics, Auckland, New Zealand) or Metamorph; the pixel size was 106 nm. TRITC-conjugated antibody staining was visualized using a standard rhodamine/TRITC filter set (Chroma Technology, Rockingham, VT, USA), while for EGFP we used a standard fluorescein/FITC filter set (Chroma Technology). For analysis, the fluorescent signal was background subtracted using regions of the field containing no cells. Data are expressed as fluorescence in intensity units (i.u.) or as dF/Fo (%). In experiments using bafilomycin A1 (1 μm, 30 min; Sigma, cat. no. 11711; Fig. 2C and D), pixels that had intensity greater than 3 SD above the mean, baseline subtracted, fluorescence were classified as spH positive. All images shown in the figures represent raw data with their pixel intensities without saturation and within the camera's dynamic range (0–4095 for CoolSNAP-HQ and 0–65535 for Cascade 650).
Figure 2. Synapto-pHluorin reveals exocytotic events in astrocytes.
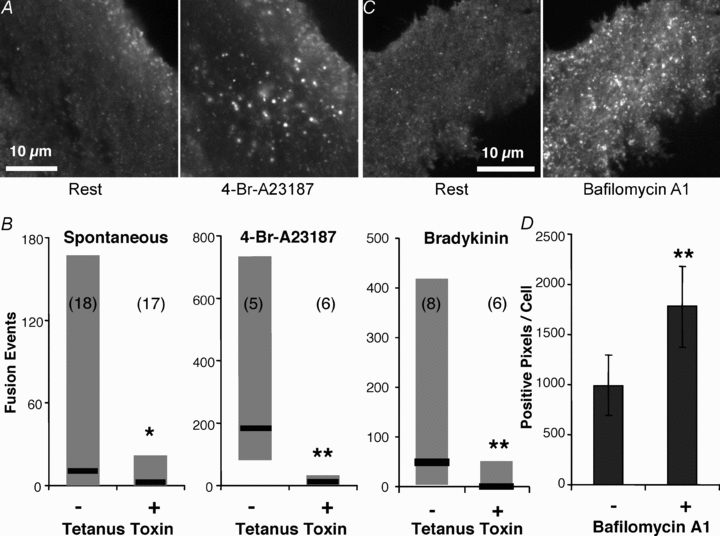
A, TIRF image of a spH-expressing astrocyte showing vesicular fusion events before and after stimulation with the Ca2+ ionophore 4-Br-A23187 (20 μm). B, co-expressing the light chain of tetanus toxin with spH in astrocytes resulted in a decrease in the number of fusions observed, spontaneously or stimulated by 4-Br-A23187 or bradykinin (*P < 0.05, **P < 0.01; Mann–Whitney U test). Graphs represent the median and the range of the number of fusions counted. Numbers in parentheses indicate the number of cells in each group. C, TIRF images of an astrocyte expressing spH before (left) and after (right) incubation with 1 μm bafilomycin A1 for 30 min. This ‘alkaline trapping’ dispels the proton gradient inside vesicles, removing the quench of spH. D, quantification of the bafilomycin A1 effect on spH fluorescence, shown as mean ± SEM of positive pixels per cell (n = 6; paired t test; **P < 0.01).
Generation of standard curve for vesicle diameter approximation
Fluorescent beads of 40, 100, 200, 500 and 1000 nm in diameter (Invitrogen, FluoSpheres size kit, cat. no. F8888) were dispersed in water (18 MΩ, MilliQ, Millipore, Billerica, MA, USA) and placed on glass coverslips. These coverslips were allowed to dry out in order for the beads to adhere to the glass. For imaging, external solution was applied to the coverslip and beads that remained attached to the glass were imaged. Each field of view was imaged with the laser in TIRF mode and also when obliquely illuminated by the laser. Oblique illumination was achieved by focusing the laser through the centre of the objective. The fluorescence intensity of each bead was measured in both modes and an average ratio of oblique/TIRF was generated for each size group (Bowser & Khakh, 2007). These values were used to fit a standard curve providing an exponential relationship between the size of the bead to this fluorescence ratio. The ratio of oblique/TIRF for astrocytic vesicles containing VAMP2-GFP was plotted on the standard curve to obtain an estimate of vesicle diameter.
Stimulation of astrocytes
To stimulate astrocytes, we pressure ejected agents dissolved in external solution from a puffer pipette (70 kPa, 4.5 min) (Parpura et al. 1994): 4-Br-A23187 (20 μm), bradykinin (1 μm), ATP (50 μm), α-latrotoxin (3 nm) or sucrose (300 mm; thus, increasing osmolarity of the external solution by ∼300 mosmol kg−1), after acquisition of a baseline sequence (30 s). In sham-run experiments, reporting on spontaneous events, we ejected external solution alone. Alternatively, we mechanically stimulated astrocytes using patch pipettes filled with external solution (Hua et al. 2004). To control the establishment of the contact between the pipette and an astrocyte, we monitored pipette resistance during delivery of −20 mV, 10 ms square pulses by a patch-clamp amplifier (PC-ONE; Dagan, Minneapolis, MN, USA) equipped with a whole-cell headstage (PC-ONE-30; 1 GΩ). Cell contact was determined by an increase in pipette resistance and was maintained for ∼1 s. The initiation of the contact occurred immediately after the acquisition of a 30 s baseline.
Analysis of fusions
To detect fusion events in the acquired 5 min movies we used Metamorph to mark all sites in the cell in each frame that had an intensity greater than 3 SD above the mean, baseline subtracted, fluorescence. Spots larger than 100 pixels or less than 8 pixels were discarded; this approach was also used to detect VAMP2-GFP-positive vesicles. For analysis, only astrocytes displaying more than 15 fusions were selected. However, in experiments assessing the effect of the lcTeTx (data shown in Fig. 2B) all cells were included. In those experiments, expression of lcTeTx caused an increase in the proportion of cells that have less than 15 fusion events throughout the time-course of the experiments in various conditions (spontaneous, 15 out of 17; 4-Br-A23187, 3 out of 6; bradykinin, 5 out of 6) when compared to cells expressing spH alone (spontaneous, 11 out of 18; 4-Br-A23187, 0 out of 5; bradykinin, 1 out of 8). Slow bursts of exocytosis were declared if the rate of fusions within 6 s of the initiation of the stimulus exceeded the mean + 6 SD of the fusion rate over the whole course of the experiment; out of 11 bursts detected 8 occurred within 3 s, while the remaining 3 were within 6 s of the stimulus onset.
Two different methods were used to determine the type of fusion that had occurred at each site: fusion with vesicle collapse (full fusion) or without collapse (transient fusion) into the plasma membrane. One method determined fusion type by detecting the outward spread of fluorescence from the initial fusion site. At each site we placed two concentric circles of 10 and 20 pixels (0.691–1.382 μm) in diameter. The average intensity of the fluorescence signal was recorded in the inner circle and outer ring (area delineated by inner and outer circle perimeters) over the course of the experiment/movie. The intensity time-course for each region was exported to Microsoft Excel XP and a formula was used to detect all fusions, determine the type of fusion and the time each fusion occurred. The time of each event was determined by detecting when the fluorescence intensity rapidly (within 333 ms) increased above the mean + 3 SD. A fluorescence peak in the central circle, followed shortly (within 333 ms) by an increase in fluorescence in the surrounding concentric ring indicated a full fusion, while an increase in the central circle alone with no subsequent increase in the outer ring indicated a transient fusion. The other method for detecting fusion type was based on the characteristics of the fluorescence intensity in a single (central) circle. The type of fusion was determined by detecting whether the fluorescence signal stayed elevated (plateaued) in the circle after reaching the peak; staying within 10% of the peak fluorescence for more than 166 ms, indicated a transient fusion. If the fluorescence intensity rapidly decayed immediately after reaching the peak, the event would be considered a full fusion. For horizontal (x-axis) line-scan plots, a line 20 pixels long (1.382 μm; corresponding to diameter of the outer circle) was drawn through the centres of potential fusion sites and the fluorescence intensity at each pixel along this line was recorded for each frame of the fusion event.
Statistical analysis
The effect of lcTeTx on rate of fusions was assessed using a Mann–Whitney U test. The difference in observed vesicles before and after bafilomycin A1 treatment was tested using a paired t test. The expression of SNAP25B and synaptotagmin 1 compared to control cells as visualized by immunostaining, as well as effects of their expression on the proportion of fusion types, were determined using one-way ANOVA followed by Fisher's least significant difference (LSD) test. Unimodality of the distribution of individual vesicle fluorescent intensity was confirmed by the dip test of unimodality (Hartigan, 1985; Hartigan & Hartigan, 1985) using R 2.8.1 program with dip-test package installed (the R Foundation for Statistical Computing). The difference in pore open times and decay slopes under various conditions, as well as the effect of SNAP25B on the proportion of transient fusion flickering, were determined using Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test. The significance of the relationship between normalized/relative intensity and dwell time was established at P < 0.0001 using regression ANOVA.
Results
Size estimate of synaptobrevin 2-containing vesicles in astrocytes
To estimate the size of the synaptobrevin 2-containing vesicles in cortical astrocytes in our experimental conditions, we exploited the sectioning ability of TIRFM. Since under total internal reflection, the evanescent field of the laser only penetrates a short distance into the sample, we could use this property to estimate the size of the vesicles in the live cultured astrocytes (Fig. 1A). Using fluorescent beads of known diameters (40, 100, 200, 500 and 1000 nm) we observed their intensities when illuminated by the laser in TIRF mode and compared this to the same beads fully illuminated by the laser penetrating the sample at an oblique angle (Fig. 1B, top) (Bowser & Khakh, 2007). With the laser adjusted at an oblique angle the entire sample is illuminated for its entire depth so larger or deeper objects will fluoresce, making deep objects brighter than when illuminated in TIRF mode. Taking the average ratio (oblique/TIRF) of these intensities for each bead size (n = 404 for 40 nm, n = 348 for 100 nm, n = 139 for 200 nm, n = 112 for 500 nm, and n = 92 for 1000 nm beads) we constructed a standard curve (Fig. 1C) which we fitted to an exponential equation (r = 0.99). We then repeated the same procedure on astrocytes expressing GFP appended to the C-terminus of synaptobrevin 2 (VAMP2-GFP) to label vesicles (Fig. 1B, bottom). We obtained the mean ratio of individual cell averages for all vesicles within them (23 astrocytes containing 8,042 synaptobrevin 2-laden vesicles within the TIRF field). Plotting the vesicular mean ratio of oblique to TIRF on the standard curve gives us an estimated average vesicle diameter of 312 nm (range, 161–422 nm).
Figure 1. Size estimate of synaptobrevin 2-containing vesicles in live astrocytes.
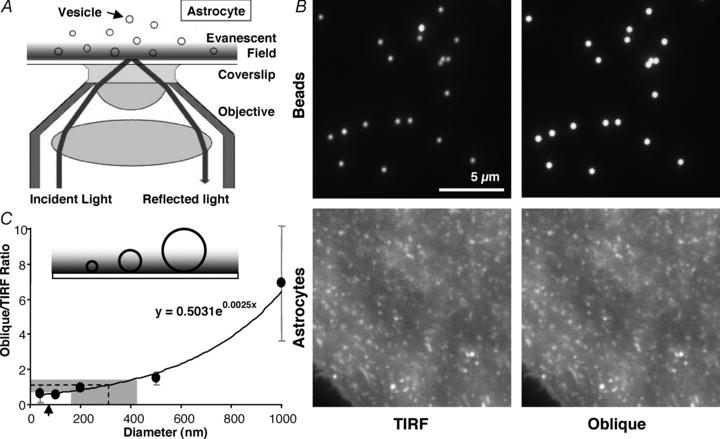
A, total internal reflection fluorescence microscopy (TIRFM). A laser beam is totally reflected off the interface between the coverslip and cell when the incident angle (73 deg in our experiments) exceeds a critical angle (62–66 deg in our experiments). A portion of the radiation, called the evanescent wave, continues across the interface but its intensity decays exponentially (a penetration depth of 70–77 nm in our experiments). TIRFM takes advantage of the evanescent field to illuminate fluorophores only within ∼100 nm of the coverslip, while fluorophores above this range will not be excited. Angles and objects are not to scale. B, images of 500 nm fluorescent beads (upper) and vesicles in astrocytes labelled with GFP appended to synaptobrevin 2 (VAMP2-GFP, lower) viewed with the illuminating laser in TIRF mode (left) and obliquely adjusted to penetrate the entire sample depth (right). C, inset illustrates how only portions of the larger beads/objects would be illuminated in TIRF mode. Using the ratio of the intensity of beads of various sizes, imaged when obliquely and TIRF illuminated, a standard curve was constructed which is described by an exponential equation (x, diameter; y, ratio). The same ratio was obtained for VAMP2-GFP-containing vesicles in astrocytes; plotting this vesicle ratio onto the curve gives an estimate of the diameter of the vesicles in astrocytes of ∼312 nm on average (dotted line). Points indicate means ± SD. The shaded area represents the range of vesicle sizes observed. Arrow indicates calculated penetration depth of TIRF evanescent field.
Synapto-pHluorin reports on vesicular fusions in astrocytes
To visualize vesicle fusion events in cultured astrocytes we expressed superecliptic spH (Miesenbock et al. 1998; Sankaranarayanan et al. 2000), a fusion protein consisting of synaptobrevin 2 with a modified GFP, pHluorin, fused to its C-terminus which is inside the vesicle lumen. This optical reporter is drastically quenched at the resting pH of acidic vesicles (pH ∼5.5), so that there is very little observable fluorescence. However, when a vesicle fuses with the plasma membrane the pH equalizes with that of the external medium (pH 7.4) and a dramatic increase in fluorescence can be observed. Expressing spH in astrocytes labels any vesicles that normally contain synaptobrevin 2; therefore, different vesicles bearing a variety of cargoes would be labelled (reviewed in Montana et al. 2006). Astrocytes expressing spH were identified using wide-field epifluorescence or laser illumination at an oblique angle. We were able to distinguish spH-expressing astrocytes in our culture by a relatively dim, diffuse fluorescence throughout the entire cell, presumably due to spH incorporating into the plasma membrane after vesicle fusions. Also, we would often observe several bright puncta, presumably representing vesicles in which the pH was less acidic. Upon switching to TIRF illumination, which allows us to view a thin section of the cell adjacent to the glass coverslip and eliminates fluorescence from deeper within the cell so events at the plasma membrane can be resolved clearly (Axelrod, 2001), we often observed a rugose pattern of spH fluorescence, presumably representing folding of the plasma membrane. To avoid associated possible artifacts, we chose regions of cells devoid of such pattern within the field of view of the camera for experiments. Using TIRFM, we saw that astrocytes at rest showed fine puncta throughout the cell (Fig. 2A, left). We also often observed sporadic, small punctate flashes of spH fluorescence occurring throughout the cell presumably due to spontaneous vesicular fusions. After stimulation, by applying the Ca2+ ionophore 4-Br-A23187, for example, which allows Ca2+ to enter the cell and promote vesicular fusion, we observed a large increase in the number of spH flashes that occurred (Fig. 2A, right).
To verify that the spH events we observed were truly vesicle fusions, we attempted to block exocytosis using the light chain of tetanus toxin (lcTeTx) which cleaves synaptobrevin 2 and, thus, prevents SNARE-mediated exocytosis (reviewed in Montana et al. 2006; Malarkey & Parpura, 2008). Astrocytes were co-transfected with plasmids encoding spH and lcTeTx, in a 1:2 ratio. In these astrocytes, we saw a significant reduction in the number of spontaneous spH events taking place (n = 17) when compared to time-course matched controls expressing spH alone (n = 18) (Fig. 2B; P < 0.05, Mann–Whitney U test). Similarly, astrocytes expressing lcTeTx that were stimulated by 4-Br-A23187 (20 μm, n = 6) or bradykinin (1 μm, n = 6) displayed reduced numbers of fusions when compared to their controls (n = 5 and 8, respectively; Fig. 2B; P < 0.01 Mann–Whitney U test). Further, we performed ‘alkaline trapping’ (Sankaranarayanan et al. 2000) using bafilomycin A1, which inhibits the vacuolar type proton ATPase (V-ATPase) responsible for maintaining the proton gradient inside vesicles. Blocking V-ATPase function would reduce the proton gradient in the vesicles and result in increased vesicular spH fluorescence (Montana et al. 2004; Reyes & Parpura, 2008). Indeed, the application of bafilomycin A1 (1 μm, 30 min) resulted in an increase of positive fluorescent spH puncta throughout astrocytes (n = 6, P < 0.01, paired t test; Fig. 2C and D). Taken together, lcTeTx and bafilomycin A1 experiments confirm that spH in astrocytes is reporting on vesicular activity.
Temporal pattern of vesicular fusions in astrocytes: sustained vs. burst
Having determined that spH fluorescence is localized to acidic vesicles and that sudden changes of intensity report on vesicular fusions, we more closely examined the temporal characteristics of exocytotic events. We systematically monitored astrocytes for 5 min and were able to observe spH fusion events occurring throughout this entire period. These fusions would occur spontaneously, without an applied stimulus, at an average rate of 14 ± 1.2 fusions per minute (n = 26 cells; average cell area, 1115 ± 34 μm2) (see Methods for exclusion criteria). However, for a more relevant comparison with stimulated cells in respect to the mode of fusion (see below), we chose several of the more active cells (n = 4) that displayed spontaneous vesicular fusions, averaging 21 fusions per minute (Fig. 3A, top). These fusions occurred with no change in global cytoplasmic Ca2+ levels as determined, in parallel, by monitoring the fluorescent Ca2+ indicator fluo-3 in cells (n = 28) exposed to the same treatment and illuminated using epifluorescence (Fig. 3A, bottom).
Figure 3. Time course of vesicular fusions and global intracellular Ca2+ in astrocytes.
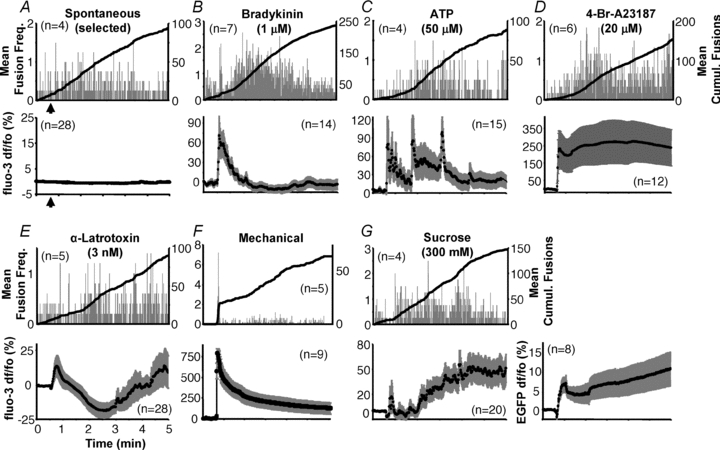
Astrocytes (illuminated using TIRF/evanescence wave) display spontaneous fusion events (A) over an extended period with fusions occurring relatively consistently throughout the period. Similar time courses were observed with the majority of stimuli (B–E, G). While stimulation with bradykinin showed an initial acceleration of vesicle fusion (B), mechanical contact was the only stimulus that elicited a bona fide exocytotic burst (F). Upper plots of each panel (A–G) show average number of spH fusions detected per second (grey bars) and average cumulative fusions over the entire period (black trace) that occurred under the various conditions. Note that for a more relevant comparison with stimulated cells in respect to the mode of fusion (see below), we selected a subset of the more active cells to display spontaneous vesicular fusions in A. Lower plots illustrate average of global cytoplasmic Ca2+ activity measured within astrocytes (illuminated by epi-fluorescence) in response to the same stimulation used to elicit fusions; traces show mean fluo-3 fluorescence ± SEM as dF/Fo (%). The fluorescence intensity of EGFP was monitored in a set of cells exposed to sucrose stimulation to account for any change in fluo-3 fluoresence due to cell volume change because of hypertonicity (G, lower right chart). The number of cells in each group is indicated on each chart (n). All stimuli (or sham in A) were delivered starting 30 s after the beginning of recording (arrows indicated only in A, and subsequently omitted for simplicity) and lasting until the end of the experiment (4.5 min), with the exception of mechanical stimulation that was delivered transiently (lasting ∼1 s).
Astrocytes were stimulated to promote vesicular fusion using stimuli that caused an increase in cytoplasmic Ca2+ concentration. Again, we observed vesicular fusions using TIRF illumination, while bulk cytoplasmic Ca2+ dynamics were monitored in parallel using epifluorescence illumination. Application of bradykinin (1 μm, 4.5 min), which induces Ca2+ elevation by activating metabotropic, G-protein-coupled receptors (GPCRs) and stimulating the release of Ca2+ from internal endoplasmic reticulum (ER) stores via inositol 1,4,5-trisphosphate receptors (IP3Rs) (reviewed in Verkhratsky, 2009), caused a transient cytoplasmic Ca2+ elevation (Fig. 3B, bottom) lasting ∼1.5 min. Consistent with the findings that bradykinin leads to exocytotic gliotransmission (reviewed in Malarkey & Parpura, 2008, 2009), we observed an increased rate of 53.9 fusions per minute (Fig. 3, top). However, unlike the transient Ca2+ dynamics, the fusion events induced by this stimulation occurred in a sustained pattern and lasted throughout the course of the experiment (Supplemental Movie 1). We also noticed some acceleration in the fusion rate immediately after the addition of bradykinin to the astrocytes, but this feature did not differentiate itself from the average rate of fusions enough to be considered a burst of exocytosis (compare Fig. 3B, top to Fig. 3F, top; see Methods for definition of burst).
ATP induces Ca2+ elevation in astrocytes through both ionotropic receptor activation, allowing Ca2+ to enter from outside the cell, and release of Ca2+ from the ER store by activating metabotropic receptors (reviewed in Verkhratsky, 2009). This stimulus also leads to exocytotic gliotransmission (reviewed in Malarkey & Parpura, 2008, 2009). Applying ATP (50 μm, 4.5 min) to astrocytes resulted in oscillatory cytoplasmic Ca2+ elevations, as previously reported (Lee & Parpura, 2009; Lee et al., 2008), along with an increased rate of 31.4 fusions per minute over the course of the experiment (Fig. 3C). These data indicate that receptor agonist stimulation results in sustained rates of vesicular fusion.
We next induced Ca2+ elevation throughout the astrocyte, bypassing receptor mechanisms, by exposing cells to the Ca2+ ionophore 4-Br-A23187 (20 μm, 4.5 min), which allows Ca2+ to directly enter the cell from the extracellular space. This stimulus can lead to the exocytotic release of glutamate from astrocytes (Innocenti et al. 2000). 4-Br-A23187 induced a large and sustained increase in cytoplasmic Ca2+. It promoted a similar increase in fusion rate to that seen with agonists, of 35.9 fusions per minute (Fig. 3D). Taken together, three different stimuli that caused three different patterns of global intracellular Ca2+ elevation had remarkably similar time courses of fusion events. It should be noted, however, that we have monitored whole cell Ca2+ changes and not Ca2+ events occurring immediately adjacent to the plasma membrane that could differentially govern exocytotic events (Marchaland et al. 2008).
We next utilized α-latrotoxin and hyperosmolarity (sucrose), stimuli that can tamper with secretory machinery, to study the time-course of vesicular fusion in astrocytes. At low concentrations, α-latrotoxin induces vesicle fusion via direct interaction with secretory machinery employing a Ca2+-independent mechanism with CIRL/latrophilins as receptors (Deak et al. 2009); in astrocytes, it can cause glutamate release with only minor changes in cytosolic Ca2+ (Parpura et al. 1995b; reviewed in Malarkey & Parpura, 2008). Consequently, application of α-latrotoxin (3 nm, 4.5 min) caused slight changes in cytosolic Ca2+ when compared to agonist and ionophore-induced stimulation (compare Fig. 3E, bottom to Fig. 3B–D, bottom) and also an increased fusion rate which was only slightly above that seen during spontaneous fusion, 20.5 fusions per minute (Fig. 3E), similar to that of the selected, more highly active astrocytes exhibiting spontaneous vesicle fusions (Fig. 3A).
Fusions that occur as a result of hypertonicity induced by sucrose have been defined as part of the readily releasable pool of vesicles: those vesicles are already docked/primed and ready for exocytosis (Rosenmund & Stevens, 1996). Applying sucrose (300 mm, 4.5 min) caused an increase in the rate of fusion to 31.4 fusions per minute, sustained over the course of the experiment (Fig. 3G, top). This stimulation mechanism is reported to operate independently of Ca2+ (Delaney et al. 1991; Basarsky et al. 1994). When initially applying sucrose we did not see much of a change in fluo-3 fluorescence, but as the experiment progressed we observed a steady increase (Fig. 3G, bottom). We surmised that this change might be only an apparent increase in fluorescence due to cell shrinkage after being exposed to the high osmolarity sucrose solution, in effect, concentrating the indicator dye. To determine if this was the case, we expressed EGFP in astrocytes and exposed them to the same sucrose stimulation. We saw a similar steady increase in fluorescence intensity over the course of the experiment (Fig. 3G, right). Since EGFP does not change its fluorescent intensity with regard to Ca2+ concentration, the increase in EGFP/fluo-3 fluorescence was due to the fluorophore being concentrated in a smaller cell volume.
Stimulation by mechanically contacting the plasma membrane of astrocytes, which may represent a physiological event (see discussion in Reyes et al. 2011), induces cytoplasmic Ca2+ increases. The source of this Ca2+ elevation arises from both the ER stores and store-operated Ca2+ entry from the extracellular space, and this stimulus has been shown to induce exocytotic glutamate release (Hua et al. 2004; Malarkey et al. 2008; also reviewed in Reyes & Parpura, 2009). Indeed, mechanically contacting an astrocyte with a glass pipette for ∼1 s caused a large increase in cytoplasmic Ca2+ concentration (Fig. 3F, bottom). While there was only a slight increase in the average rate of fusion for the entire experiment, 15.4 fusions per minute (Fig. 3F), stimulation by mechanical contact caused an initial large burst of fusions (3 out of 5 cells) followed by a consistent rate of fusions over the rest of the experiment. These data indicate that astrocytes can display either a burst or a sustained rate of vesicular fusions depending on the conditions to which the cells are exposed.
Expression of SNAP25B in astrocytes is not the determinant of an exocytotic burst
In chromaffin cells, exocytotic bursts have been reported to rely on the involvement of SNAP25, while SNAP23 did not support a burst (Sorensen et al. 2003); also, SNAP25B was shown to be much more effective at promoting an exocytotic burst than SNAP25A. Astrocytes in our culture express SNAP23, but not SNAP25, as a component of their ternary SNARE complex (Montana et al. 2004) as confirmed by immunocytochemistry (Supplemental Fig. S1 and Fig. 4B, respectively). Using PCR we determined that the isoform expressed in our cultures is SNAP23A (Supplemental Fig. S1). It should be noted, however, that astrocytes can also express SNAP25 (reviewed in Montana et al. 2006). Consequently, we postulated that expressing SNAP25B in astrocytes from the visual cortex might be able to induce exocytotic bursts with stimuli other than that of mechanical contact, more specifically, bradykinin. We initially co-transfected astrocytes with plasmids encoding EFGP and SNAP25B, while in the control group we only transfected astrocytes with a plasmid encoding EGFP. The expression of SNAP25B in astrocytes was verified by immunocytochemistry with an antibody against SNAP25 (Fig. 4A and B). In a subset of experiments where the primary antibody was omitted, only a faint diffuse non-specific signal was observed (n = 46), while in astrocytes transfected with SNAP25B along with EGFP, we saw specific punctate immunofluorescence with a distinct appearance around the periphery of the cell (n = 22). There was also dense staining next to the nucleus, consistent with the site of protein synthesis and initiation of trafficking through the Golgi apparatus. This specific staining was absent in control cells transfected with EGFP alone (n = 35; Fig. 4A). Having performed immunocytochemistry to confirm the lack of endogenous expression and expression of the exogenous SNAP25B, we co-expressed SNAP25B and spH in astrocytes to monitor fusion events. We stimulated the astrocytes co-expressing SNAP25B and spH with bradykinin, which had not caused a distinct exocytotic burst in astrocytes expressing spH. Under these conditions we did not find that SNAP25B causes a burst of fusions when expressed in astrocytes, while mechanical stimulation of these astrocytes still resulted in exocytotic bursts (2 out of 5 cells; Fig. 4C; compare to Fig. 3B and F, respectively). This suggested that while bursts in astrocytes are possible, SNAP25B is not a sole determinant of this ability.
Figure 4. Expression of SNAP25B in astrocytes does not induce an exocytotic burst.
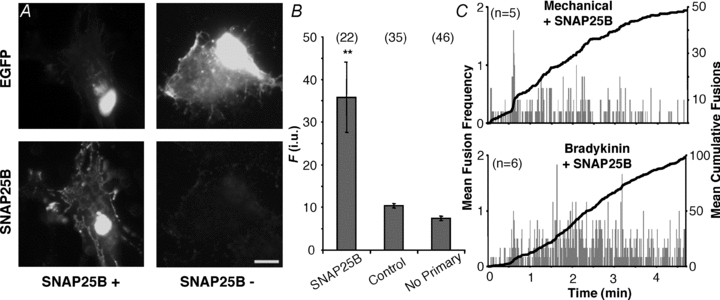
A, verification of the expression of exogenous SNAP25B and its localization in cultured astrocytes. Transfected cells were identified by the expression of EGFP (top, FITC), while the expression of SNAP25B was assessed by labelling with an antibody (bottom, TRITC). It should be noted that due to the permeabilization procedure for indirect immunocytochemistry in some cells EGFP fluorescence can be diminished from the cytosolic compartment, leaving mainly perinuclear stain (top, compare left and right). Staining by the SNAP25 antibody was absent when cells were transfected with EGFP alone (SNAP25B −), but readily detectable in cells expressing exogenous SNAP25B (SNAP25B +). Scale bar: 10 μm. B, quantification of immunoreactivity expressed in fluorescence (F) intensity units (i.u.). We found no difference between TRITC fluorescence levels in cells transfected with EGFP alone regardless whether primary antibody was present. Bars represent means ± SEMs of measurements. Numbers in parentheses indicate the number of cells in each group. Asterisks indicate a significant change of measurements compared with the control group (one-way ANOVA followed by post hoc Fisher's LSD test; **P < 0.01). C, plots showing average number of spH fusions detected per second (grey bars) and average cumulative fusions over the entire period (black trace) reveal that the expression of SNAP25B in astrocytes does not induce a bursting phenotype in cells stimulated with bradykinin, while bursting was still seen with mechanical contact. The number of astrocytes in each group is indicated on each chart (n).
Detection of two forms of vesicular fusion in astrocytes: transient vs. full fusions
Two types of vesicle fusion have been described, mostly from work in neurons and chromaffin cells: fusions where the vesicle collapses into the plasma membrane upon fusion, termed full fusion, and fusions where vesicles remain intact and open a transient fusion pore. These transient events are referred to as either ‘kiss-and-run’ if the vesicle, following fusion, undocks and locally recycles or ‘kiss-and-stay’ if the vesicle, following fusion, remains in the releasable pool of vesicles without undocking from the membrane (for review see Sudhof, 2004; Harata et al. 2006). Since our approach cannot assess functional docking (i.e. the formation of stable SNARE complexes) we shall refer to fusion events as either full or transient events. Indeed, such events have been described in astrocytes (Bezzi et al. 2004; Chen et al. 2005; Bowser & Khakh, 2007; Cali et al. 2008; Marchaland et al. 2008). To investigate the occurrence of the two types of fusion in astrocytes under various conditions, some of which are comparable to those used in previously published work, we acquired time lapse video of cells at 60 frames per second. By implementing a commonly used method (Zenisek et al. 2000; Bezzi et al. 2004; Bowser & Khakh, 2007) where we placed two concentric circles of 0.691 and 1.382 μm in diameter around each potential fusion site (Fig. 5A), we could detect any spread of fluorescence away from the initial point of fusion in the inner circle. From this we were able to resolve two distinct types of fusion. One type of fusion displayed a large increase of fluorescence in the inner circle but no subsequent increase in the outer ring; also the intensity of the inner circle reached a plateau which was maintained for a brief period before subsiding to the pre-event levels (Fig. 5B). This time-course is indicative of transient fusion where the vesicle would fuse for a brief period with the plasma membrane, before retracting and re-acidifying. The other type of fusion was distinguished by a similar large increase in fluorescence in the inner circle followed by a gradual increase in fluorescence in the outer ring, presumably as the vesicle fully collapsed and spH spread out into the plasma membrane (Fig. 5C).
Figure 5. Detection of transient and full forms of vesicular fusion in astrocytes.
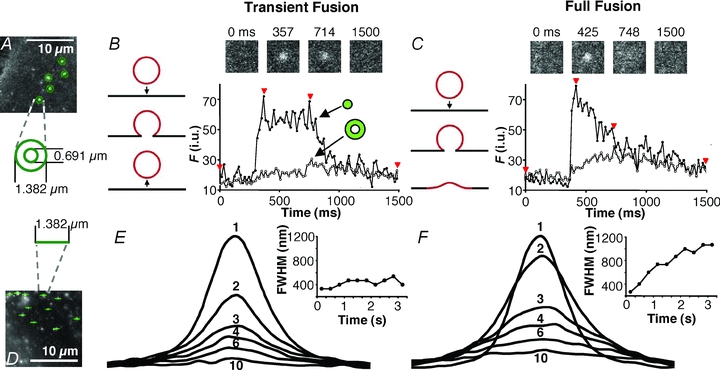
A, monitoring fusion events at 16.6 ms acquisition rate. Potential fusions were detected as bright spots where the intensity exceeded 3 standard deviations (SD) above the mean intensity of the image. The average intensities of 2 concentric circular regions with diameters of 1.382 μm and 0.691 μm (20 and 10 pixels, respectively) centred over each site were measured. A fusion was counted when the intensity in the centre circle exceeds the mean +3 SD. If the outer ring surpassed the mean + 3 SD, the fusion was considered a full fusion, otherwise it was considered a transient fusion. In subsequent experiments the profile of intensity of the centre region alone was used to determine fusion type (see text). B, representative trace of a fusion event displaying transient (kiss-and-run) characteristics. The intensity increases predominately in the inner region (trace, black circles) while intensity in the outer ring (trace, white circles) remains relatively unchanged. This is consistent with the vesicle having a transient fusion pore with the plasma membrane (drawing; arrow pointing down, fusion; arrow pointing up, vesicle retrieval from the plasma membrane). Across the top are images of this region: before fusion, when fusion initiated, during fusion, and afterwards. The times for each image are indicated by arrowheads on the trace. C, representative trace of a fusion event displaying full fusion characteristics. The intensity increases initially in the inner circle (trace, black circles) followed by an increase of intensity in the outer ring (trace, white circles). This is consistent with the vesicle fully collapsing into the plasma membrane (drawing, arrow) and spH fluorescence diffusing over a large area of the plasma membrane. Across the top are images of this region: before fusion, when fusion initiated, during the fusion, and afterwards. The times for each image are indicated by arrowheads on the trace. D, line-scan plots showing spread of fluorescence during fusion events. A 1.382 μm line was drawn through the centres of potential fusion sites and the fluorescence intensity at each pixel along this line was recorded for each frame of the fusion event. E, averaged line-scan traces from 131 transient fusion events from an astrocyte stimulated with 4-Br-A23187. Each trace (1–10) was taken 333 ms apart; some have been omitted for clarity. The corresponding chart shows the full-width half-maximum (FWHM) for each trace over time. The peak fluorescence (trace 1; initial point) occurred immediately after fusion. Subsequent traces do not get wider, nor does the FWHM increase appreciably, indicating that the fluorescence is contained after fusion, consistent with transient fusion type. F, averaged line-scans through the centres of 347 full fusions from the same cell as in E. Traces and charts made as in E. Traces after the peak (trace 1) become wider indicating an outward spread of fluorescence from the site of fusion, as quantified by the increasing values of FWHM in the chart, consistent with full fusion type.
We found, however, that using the two-circle method to distinguish fusions was not ideal in astrocytes. The apparent size of the fluorescently labelled vesicles was not uniform, so larger or more intensely labelled vesicles would extend fluorescence into the outer ring, regardless of what form of fusion occurred. Inversely, the fluorescence of smaller vesicles would not reach the outer ring even when full fusion occurred. Also, we saw that some vesicles would wobble about after fusion which tended to cause fluorescence to occur in the outer ring for transient events. To circumvent these issues and avoid false classification, we developed an alternate means of determining fusion type based on the shape of the fluorescence time-course of the fusion event. We classified events as full fusion events if they displayed a peak increase in fluorescence followed immediately by an exponential decay in intensity (Fig. 5C), whereas in transient events, the initial increase displayed a plateau in intensity with variable duration and was followed by a decay that was variable in rate (Fig. 5B). Since the time-courses of fusion events were much longer (see below) than our acquisition rate, we were able to decrease sampling rate to 30 frames per second in order to increase the field of view we could capture, allowing for a larger portion of the cell to be monitored. We confirmed this alternative method of detecting fusion type by making line-scan intensity plots of each fusion in a cell over time (similar to the ‘radial sweep’ approach described in Cali et al. 2008). A 20 pixel line (1.382 μm; diameter of outer circle) was placed through the centre of each fusion event (Fig. 5D) and the fluorescence intensity along the line was recorded for each frame. In both types of fusion, the line-scans displayed Gaussian distributions with decreasing amplitude over time (Fig. 5E and F). However, in events determined to be transient fusions, distributions did not become wider than the initial peak line-scan (Fig. 5E, inset), while in full fusion events intensity distributions from times after the peak line-scan were wider (Fig. 5F, inset). This indicates that fluorescence spread out after fusion in full fusion events, but remained confined in events labelled as transient fusion. When we compared this alternative intensity profile approach to the two-circle method, we found that in astrocytes, the two-circle method favoured detection of transient fusions and underrepresented full fusions by 34–59% depending on the stimulus applied (Table 1). Subsequently, we report on fusion type using the intensity profile method.
Table 1.
Comparison between 2-circle and intensity profile methods for fusion type detection in astrocytes based on spH fluorescence
| Transient (%) | Full (%) | ||||
|---|---|---|---|---|---|
| Condition* | 2-circle | Profile | 2-circle | Profile | d (%) |
| Spontaneous | 84.1 | 50.1 | 15.9 | 49.9 | 34.0 |
| Bradykinin | 71.4 | 28.0 | 28.6 | 72.0 | 43.3 |
| ATP | 94.3 | 35.6 | 5.7 | 64.4 | 58.7 |
| 4-Br-A23187 | 75.7 | 45.5 | 24.3 | 54.5 | 30.2 |
Note: averages of the per cent of fusions detected as either transient or full fusion events using the two concentric circle method of analysis or the intensity profile method. The per cent difference (d) between the two methods for various stimuli is indicated. Asterisk denotes number of astrocytes used in each condition as reported in upper charts of Fig. 3A–D, respectively.
Preferred vesicle fusion type in astrocytes is stimulus dependent
Previous work in astrocytes has indicated that astrocytes can display distinct types of vesicle fusion depending on the stimulation applied (Chen et al. 2005; Bowser & Khakh, 2007). Thus, we further investigated how astrocytes responded to an expanded repertoire of different stimuli. When we allowed fusions to occur spontaneously in astrocytes, we saw that over the course of 5 min, the two types of fusion were represented equally, each accounting on average for 50% of the total number of fusions (Fig. 6A). When the astrocytes were stimulated with 4-Br-A23187, ATP or bradykinin, which increased cytoplasmic Ca2+ concentration, full fusions were favoured over transient fusion (Fig. 6A). Bradykinin stimulation resulted in the largest shift toward full fusions (on average 72% were full fusions; Fig. 6A). The application of ATP displayed more full fusions (on average 64%) but was not as shifted as bradykinin (Fig. 6A). Stimulation with 4-Br-A23187 showed a shift in fusion type (on average 54% full fusions), not very different than that of spontaneous events. Two compounds that cause vesicular fusion by more Ca2+-independent mechanisms had the opposite effect, shifting events towards transient fusions. Stimulation of astrocytes with hyperosmotic (300 mm sucrose) saline resulted in slightly more transient events (on average 58%) than full fusions (Fig. 6A). Also, inducing fusion by applying α-latrotoxin (3 nm) produced a ratio of the two types of fusion similar to that seen with spontaneous events (on average 54% transient fusions). Interestingly, mechanical stimulation, which causes large cytoplasmic Ca2+ elevations in astrocytes, resulted in a stark preference for transient type fusions (76% transient).
Figure 6. Preferred fusion type in astrocytes is stimulus dependent.
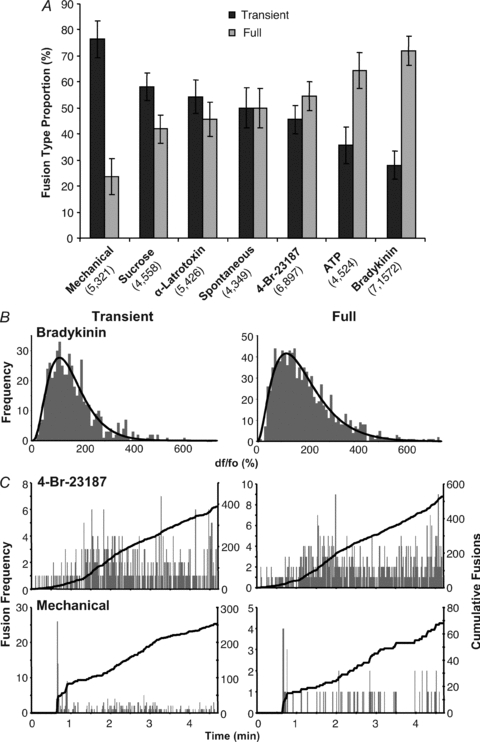
A, the number events categorized as either transient or full fusions were plotted as a percentage of the total number of fusions that occurred, either spontaneously or when astrocytes were stimulated as in Fig. 3A. Bars represent means ± SEM of the proportion (in percentage) of each fusion type in individual astrocytes; values in parentheses are: (number of cells, total number of events). B, the peak fluorescence intensities of transient (left) and full fusion (right) events in astrocyte stimulated with bradykinin are shown as dF/Fo plotted in histograms. The intensity distributions of both fusion types are unimodal (back curve) indicating that the vesicles involved belong to a single population. C, plots showing the total number of fusions per second (grey bars) and the total cumulative fusions (black trace) occurring over the course of the experiment using cells in A; transient (left) and full fusion events (right) are plotted separately. Both fusion types occur throughout the entire time of the experiment.
We confirmed that the detection of fusion events and fusion types were obtained from measuring the spH fluorescence that originated from single vesicles (Aravanis et al. 2003). An example of analysis is shown in Fig. 6B, measuring the change in intensities of all vesicles in astrocytes stimulated with bradykinin. Peak intensities of transient and full fusion events showed unimodal distributions (P < 0.05 for both, dip test), indicating that a single vesicle is participating in each event. Additionally, there was no temporal preference in the occurrence of the two types of fusion, since both happened throughout the time-course of the experiment, as shown in examples of astrocytes stimulated either by 4-Br-A23187 or mechanically (Fig. 6C; for a closer look at the mechanically induced exocytotic burst see Supplemental Fig. S2).
Type and timing of recurrent fusions occurring in the same location
We observed that more than one fusion could occur in the same spot at the plasma membrane over time. Consequently, we counted how often these recurring fusions happened, what types of fusion occurred and the time interval between individual events over the course of the experiments (Table 2). In most cases (78.0%) locations on the plasma membrane displayed a single fusion event regardless of conditions (Table 2), while the remaining loci received two (19.7%) or more (2.4%) fusions; we noted variability in these observations associated with different stimuli (Table 2). When more than one fusion occurred at the same spot, the most common pattern was two transient fusions in succession (14.1%) regardless of whether the stimulus used led to preferentially transient (e.g. mechanical) or full fusion (e.g. bradykinin) overall. Other combinations, transient followed by full, full followed by transient or full followed by full fusion had much lower incidence (1.4%, 2.2% and 1.9%, respectively). The interval between fusions was the shortest in each category when mechanical stimulation was used, which is perhaps consistent with the presence of bursts with this stimulus. In general, the longest time was recorded for the interval between two full fusions (Table 2). On very rare occasions (∼0.5% of total fusions; i.e. 8 out of 1572 bradykinin-induced fusions) we observed full fusions reminiscent of compound exocytosis in which vesicles fuse to each other before they fuse to the plasma membrane (reviewed in Smith et al. 2008) (Supplemental Fig. S3); these events were not studied further.
Table 2.
Fusions recurring in the same cellular location
| Total | Single | T–T | T–F | F-T | F-F | >2 fusions | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Condition | n | n | % | n | % | t (s) | n | % | t (s) | n | % | t (s) | n | % | t (s) | n | % |
| Spontaneous | 349 | 271 | 77.7 | 54 | 15.5 | 1.28 | 5 | 1.4 | 11.55 | 7 | 2.0 | 11.57 | 5 | 1.4 | 14.57 | 7 | 2.0 |
| Bradykinin | 1572 | 1315 | 83.7 | 106 | 6.7 | 1.95 | 33 | 2.1 | 7.70 | 34 | 2.2 | 12.10 | 55 | 3.5 | 16.71 | 29 | 1.8 |
| ATP | 524 | 378 | 72.1 | 68 | 13.0 | 0.88 | 17 | 3.2 | 0.16 | 20 | 3.8 | 5.05 | 23 | 4.4 | 14.47 | 18 | 3.4 |
| 4-Br-A23187 | 897 | 775 | 86.4 | 78 | 8.7 | 1.86 | 9 | 1.0 | 5.13 | 12 | 1.3 | 7.97 | 13 | 1.4 | 17.58 | 10 | 1.1 |
| α-Latrotoxin | 426 | 346 | 81.2 | 58 | 13.6 | 1.66 | 3 | 0.7 | 13.51 | 9 | 2.1 | 1.04 | 3 | 0.7 | 19.53 | 7 | 1.6 |
| Mechanical | 321 | 244 | 76.0 | 59 | 18.4 | 0.20 | 4 | 1.2 | 0.13 | 3 | 0.9 | 0.23 | 2 | 0.6 | 18.85 | 9 | 2.8 |
| Sucrose | 558 | 383 | 68.6 | 129 | 23.1 | 0.86 | 2 | 0.4 | 20.50 | 16 | 2.9 | 7.22 | 7 | 1.3 | 18.71 | 21 | 3.8 |
| Average | 78.0 | 14.1 | 1.24 | 1.4 | 8.38 | 2.2 | 6.45 | 1.9 | 17.21 | 2.4 | |||||||
Locations in astrocytes where two or more fusions occurred in the same spot were counted and the type of fusion (T, transient; F, full) and order they occurred were recorded. The number of fusions (n) along with the per cent (%) of the total fusions are shown in each column. As most repeat fusions consisted of 2 fusions in the same spot we show only the first 2 fusions in the sequence. The number of repeat fusions greater than 2 events is shown to the far right. We also measured the time interval (t), in seconds, between recurring fusions. The bottom row reports the average proportion of total fusions that occurred in each particular order, and the average time between two fusions.
Probability of vesicular fusion in astrocytes
To determine the proportion of the total alkaline-trapped spH-laden vesicles within the TIRF field, that undergo fusion when astrocytes are stimulated, we applied bafilomycin A1 (1 μm, 30 min) which should reveal all the spH-containing vesicles (see Fig. 6C of Reyes & Parpura, 2008). This blockade of the V-ATPase leads to a depletion of glutamate from astrocytic vesicles and results in grossly reduced exocytotic release of transmitter from these cells when exposed to a variety of stimuli (reviewed in Malarkey & Parpura, 2008, 2009). Here, full fusions of spH-containing vesicles could be observed upon stimulation as punctate fluorescence that suddenly decayed away as spH diffused throughout the plasma membrane. These data indicate that astrocytic vesicles depleted of transmitter can fuse, a finding that has been described in neurons (Fremeau et al. 2004; Wojcik et al. 2004; commented in Schuske & Jorgensen, 2004). Transient fusions could not be observed as the vesicles were already fluorescent, displaying no detectable change over the time-course of the experiment. We determined the total number of fusions that should have occurred by extrapolating from the number of full fusions using the ratio of transient to full fusions, calculated from the source data for Fig. 6A. In this manner we determined that on average 17.7% of alkaline-trapped vesicles are fusogenic when astrocytes (n = 10) were stimulated (Table 3). This probability of vesicular fusion in astrocytes is in good agreement to that found at presynaptic terminals (Rosenmund & Stevens, 1996).
Table 3.
Proportion of the total alkaline trapped vesicles that undergo fusion in astrocytes
| Stimulus1 | Vesicles (all) | F | Ratio (T/F) | F+T | Fusion (%) |
|---|---|---|---|---|---|
| 4-Br-A23187 (4) | 469 | 55 | 0.84 | 100 | 20.1 |
| Bradykinin (2) | 315 | 13 | 0.39 | 18 | 6.5 |
| Mechanical (3) | 281 | 12 | 3.24 | 52 | 24.5 |
| Sucrose (1) | 322 | 17 | 1.39 | 41 | 12.6*17.7 |
| Average proportion of fusogenic vesicles (%) | |||||
Note, the total number of spH-containing vesicles in astrocytes (number of cells in parentheses) was revealed by ‘alkaline trapping’ using bafilomycin A1 (1 μm, 30 min), which caused them to become fluorescent in the TIRF field. Upon stimulation, full fusions (F) were observed as a sudden disappearance of fluorescent vesicles. The total number of fusions, including transient (F+T), was estimated by adjusting the number of full fusions detected by the ratio of transient to full fusions (mean of individual cell T/F ratios) measured previously under each stimulus (Fig. 6A). The proportion of the total vesicles in astrocytes that undergo fusion (fusogenic vesicles) is given as percentage.
Experiments stimulating astrocytes exposed to bafilomycin, especially when using sucrose, for unknown reasons turn out to have a very low success rate. Thus, due to restrictive group size, the comparison between various groups should not be made, even though we disclose, for completeness, the results obtained for each stimulus. Asterisk denotes the size of ready-releasable pool of vesicles.
Synaptotagmin 1, but not SNAP25B, modulates the ratio between fusion types in a stimulus-dependent manner
Different isoforms of synaptotagmin (Syt) have been reported to favour one type of fusion vs. the other. For instance, Syt1 modulates the ratio between kiss-and-run and full fusion (Wang et al. 2003). Astrocytes express Syt4, and the decrease of it by RNA interference, as well as expression of a mutated form, reduce Ca2+-dependent glutamate release (Zhang et al. 2004a); we confirmed Syt4 expression in our cells by immunocytochemistry (Supplemental Fig. S1). In our culture system, astrocytes do not express Syt1 (Parpura et al. 1994; also see Fig. 7A), albeit in some conditions astrocytes may express this isoform as well (reviewed in Montana et al. 2006). Thus, we expressed Syt1 in astrocytes to test whether this protein would affect the ratio of vesicular fusion types. Initially, astrocytes were co-transfected with EGFP and Syt1 (1:2 ratio). Verification of Syt1 expression was accomplished by immunolabelling in the same manner as described for SNAP25B. In astrocytes transfected with Syt1 along with EGFP, we saw punctate immunofluoresence of Syt1 (n = 22). In the control group where we transfected astrocytes only with the plasmid encoding EGFP (n = 34), we observed a lack of staining comparable to the low level signals seen in experiments where the primary antibody was omitted (n = 46) (Fig. 7A and B).
Figure 7. Expression of Syt1 in astrocytes does not affect the temporal pattern of fusions.
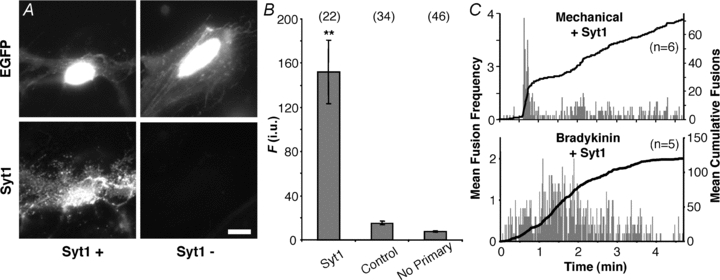
A, verification of the expression of synaptotagmin 1 (Syt1) and its localization in cultured astrocytes. Transfected cells were identified by the expression of EGFP (top), while the expression of exogenous Syt1 (Syt1+) was confirmed by labelling with an antibody (bottom). Staining by the Syt1 antibody was absent when cells were transfected with EGFP alone (Syt1−). Scale bar, 10 μm. B, quantification of immunoreactivity expressed in fluorescence (F) intensity units (i.u.). Bars represent means ± SEMs of measurements. We found no difference between TRITC fluorescence levels in cells transfected with EGFP alone (control) regardless of whether primary antibody was present. Asterisks indicate a significant change of measurements compared with the control group (**P < 0.01, one-way ANOVA followed by post hoc Fisher's LSD test). Numbers in parentheses indicate the number of cells in each group. C, plots showing average number of spH fusions detected per second (grey bars) and average cumulative fusions over the entire period (black trace) reveal that the expression of Syt1 in astrocytes does not affect bursting with mechanical contact or the sustained phenotype in cells stimulated with bradykinin.
Having demonstrated our ability to express Syt1 in astrocytes, we then co-transfected astrocytes with plasmids encoding spH and Syt1. Cells expressing both constructs were chosen by observing the fluorescence of spH. Expression of Syt1 in astrocytes did not grossly affect the temporal pattern of spH fusions; mechanically stimulated cells (6 out of 6) displayed bursts, while those stimulated by bradykinin did not (Fig. 7C). Next, we studied the effects that Syt1 expression exerts on the mode of vesicular fusion. In parallel, we also assessed whether SNAP25B could modulate the ratio between the two fusion types. We chose to use the two stimuli that fell farthest apart on the ‘spectrum’ of full to transient fusion ratios seen in astrocytes (Fig. 6A): mechanical contact that heavily favours transient fusions (76%) and bradykinin stimulation, which produces more full fusion events (28% transient). In mechanically stimulated astrocytes expressing either Syt1 or SNAP25B, we saw a small and statistically insignificant shift towards full fusions (64% or 65% transient, respectively; Fig. 8; one-way ANOVA, F(2,13) = 0.41). However, when we stimulated astrocytes expressing Syt1 or SNAP25B with bradykinin, we saw a larger shift in fusion type towards transient fusions (54% or 44% transient, respectively), which reached marginal significance only when Syt1 was expressed (Fig. 8; one-way ANOVA followed by post hoc Fisher's LSD test, P < 0.05). These results may indicate that under specific stimulation conditions, the isoform of synaptotagmin expressed in astrocytes can influence the form of fusion a vesicle will undergo.
Figure 8. The effects of expression of exogenous SNAP25B or synaptotagmin 1 on vesicle fusion type in astrocytes.
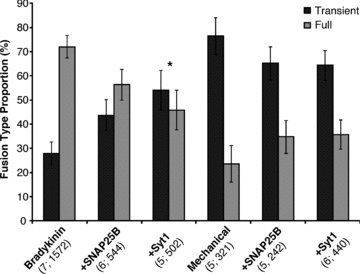
The events categorized as either transient or full fusions were calculated as the average proportion (%) of the total number of fusions that occurred with individual astrocytes stimulated by either bradykinin (1 μm) or mechanical contact (replotted from Fig. 6A), or when these two stimuli were used in cells expressing (+) either SNAP25B or Syt1. SEMs are the same for both types of fusion. *P < 0.05, one-way ANOVA followed by post hoc Fisher's LSD test.
Transient fusion pore stability: dwell time, its relation to vesicular size, flickering and decay slope
The two different modes of exocytosis have distinct characteristics (reviewed in Smith et al. 2008). A better understanding of the dynamics of these events in astrocytes might provide clues to why the two distinct modes might be employed in different situations (Shigetomi et al. 2008). Initially, we measured how long the plateau phase, also referred to as dwell time, of the transient events lasted for each stimulus. This was done by determining the full-width half-maximum value for the trace of each event (Fig. 9A). The plateau corresponds to the time, after fusion, when a vesicle is in very close apposition to the plasma membrane and has not internalized/re-acidified. It gives a close approximation of the length of time the fusion pore remains open. The plateau lasted approximately 1 s for spontaneous events and was not significantly different for most stimuli (Fig. 9A), but in the case of sucrose and mechanical stimulation, it was nearly doubled (P < 0.01, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test). These two stimuli also had the largest proportion of transient events, although this might only be a coincidence. We observed modulation of dwell-time when astrocytes were transfected to express SNAP25B or Syt1. This modulation was stimulus dependent, as expression of SNAP25B marginally decreased the duration of transient fusion dwell time when astrocytes were mechanically stimulated, while expression of Syt1 was marginally effective in decreasing dwell duration when cells were stimulated either mechanically or with bradykinin (Fig. 9B; P < 0.05, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test).
Figure 9. The open time of the fusion pore (dwell time) of transient fusion events in astrocytes.
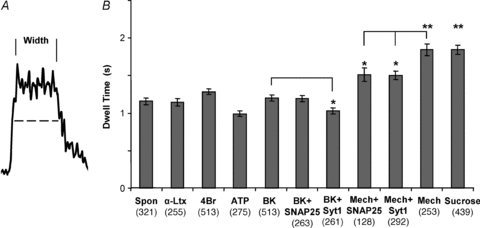
A, the time each transient fusion event spent in close proximity to the plasma membrane after fusion, without vesicular internalization/re-acidification, was estimated by measuring the full-width half-maximum intensity (dwell time, dashed line) for each event (number of events shown in parentheses). This approximates the open time of the fusion pore during these events. B, average duration of dwell time. Mechanical and sucrose stimulation resulted in significantly longer pore open times. The expression of SNAP25B or Syt1 resulted in a shortening of the open time during mechanical stimulation. However, under bradykinin stimulation only Syt1 shortened the open time. *P < 0.05, **P < 0.01; Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test. Abbreviations: Spon, spontaneous; α-Ltx, α-latrotoxin; 4Br, 4-Br-A231887; BK, bradykinin; Mech, mechanical.
We next assessed the relationship between the size of individual vesicles and the stability of their fusion pores (Zhang & Jackson, 2010). Here, we used dwell time as one of the parameters of fusion pore stability. As indicated earlier, the distribution of peak fluorescence intensities detected during transient fusions is unimodal (Fig. 6B, left), indicating that only a single vesicle of a certain size participates in each event. Assuming that an abundant vesicle protein, such as synaptobrevin 2/spH (Takamori et al. 2006), would be proportionally distributed among vesicles of different sizes, we can then use normalized fluorescence as an estimate of vesicular size. To avoid the variability of spH expression in different astrocytes, we normalized the fluorescence intensity of each transient event to the transient event with the greatest fluorescence intensity in each cell. When we compared the normalized/relative intensity of each vesicle to its dwell time we could only establish a significant linear correlation for events occurring during stimulation with bradykinin or sucrose (regression ANOVA, P < 0.0001); however, this correlation was not very strong (r = 0.251 and 0.246, respectively) (Fig. 10). Interestingly, in astrocytes stimulated with bradykinin but also expressing SNAP25B, the relationship between normalized intensity and dwell time did not reach significance. In contrast, when astrocytes expressed Syt1, we not only observed a significant relationship when cells were stimulated with bradykinin, but also when they were mechanically stimulated, even though in the absence of Syt1, this latter stimulus did not yield a significant relationship. These data indicate that the size of the vesicle only has a weak influence on the open time of its pore and only under certain conditions; in general, the larger the vesicle, the longer the duration of the open pore of its transient fusion is. SNAP25B and Syt1 perhaps act as modifiers of this relationship (see Discussion).
Figure 10. The relationship between the relative vesicle size and the duration (dwell time) of transient fusions.
Graphs of the normalized fluorescence intensity of spH-containing vesicles that underwent transient fusion vs. the (dwell) time the vesicle maintained an open pore after fusion with the plasma membrane. The intensities are normalized to the brightest vesicle in each cell/experiment. This relative intensity is used as an estimate of the relative vesicle size. The time is measured as the full-width half-maximum intensity for each transient event as in Fig. 9. Lines represent significant linear relationships (significance established using regression ANOVA at P < 0.0001) given by the equation (dwell time (y) = slope* normalized (relative) spH fluorescence (x) + intercept); r, correlation coefficient of a given relationship. Abbreviations as in Fig. 9.
We further investigated the stability of transient fusion pores by looking for ‘flickering’ (Zhou et al. 1996; Ales et al. 1999; Klyachko & Jackson, 2002; Staal et al. 2004; Chen et al. 2005; Vardjan et al. 2007). By examining the intensity of transient fusions more closely, we found that in some cases the fluorescence intensity of the plateau phase did not remain stably elevated the entire time that the vesicle was associated with the membrane (Fig. 11). This most probably indicates that fusion pore instability, flickering, had caused a fluctuation in the spH signal. Such flickers were seen on average in ∼15.7% of spontaneously occurring transient fusions (Table 4). Stimulated astrocytes also displayed flickering of transient fusions in different proportions ranging from 2.3% when astrocytes were challenged with hypertonicity/sucrose to 16.0% when stimulated with bradykinin (Table 4). However, the differences between flicker proportions under the various conditions was statistically insignificant (Kruskal–Wallis one-way ANOVA, P = 0.33). The expression of Syt1 or SNAP25B did not significantly affect the occurrence of transient pore flickers when astrocytes were stimulated with bradykinin (Kruskal–Wallis one-way ANOVA, P = 0.325). However, when astrocytes were mechanically stimulated, expression of SNAP25B, but not Syt1, completely abolished flickers (Table 4; P < 0.05, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test). Taken together, these results show that the stability of the transient fusion pore can be improved in a stimulus-specific context by the presence of SNAP25B.
Figure 11. Transient fusions can display flickering.
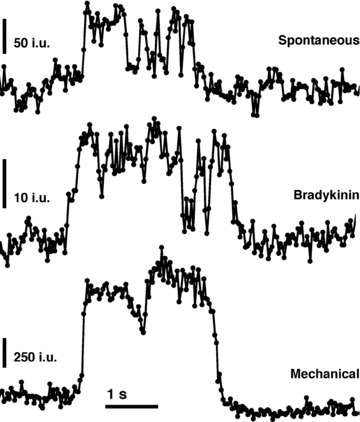
Traces represent examples of transient fusions that displayed variability in the spH fluorescence intensity (i.u.) of their plateau phase. This change in intensity probably reflects temporal variations in the fusion pore size, and hence its instability, while the vesicle is still associated with the membrane. These flickering events were observed in spontaneous transient fusions, as well as those evoked with all the different stimuli (see Table 4).
Table 4.
Proportion of the transient fusions displaying flickering
| Flickers | |||
|---|---|---|---|
| Condition (cells; T) | Count | (%) | SEM |
| Spontaneous (4; 188) | 26 | 15.7 | 10.5 |
| α-Latrotoxin (5; 214) | 8 | 2.5 | 1.7 |
| 4-Br-A23187 (6; 380) | 17 | 9.0 | 5.0 |
| ATP (4; 181) | 19 | 15.0 | 6.2 |
| Bradykinin (7; 469) | 67 | 16.0 | 5.9 |
| +SNAP25B (6; 246) | 11 | 4.2 | 1.8 |
| +Syt1 (5; 234) | 21 | 6.4 | 3.5 |
| Mechanical (5; 253) | 12 | 5.0 | 1.2 |
| +SNAP25B (5; 136) | 0 | 0.0 | 0.0* |
| +Syt1 (6; 299) | 11 | 9.1 | 3.7 |
| Sucrose (4; 362) | 9 | 2.3 | 0.8 |
The number of events (count) categorized as flickers were calculated as the average proportion (%) of the total number of transient fusions (T) that occurred with individual astrocytes (cells) in particular condition. SNAP25B expression completely abolishes flickers when astrocytes are mechanically stimulated.
P < 0.05, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test.
We next studied the decay of fluorescence after the plateau phase of transient fusions. The kinetics of this parameter can give insight into the process of vesicle retrieval from the plasma membrane (endocytosis/internalization) and/or re-acidification of the vesicular lumen (Gandhi & Stevens, 2003; Atluri & Ryan, 2006). Unlike the very regular decay observed with full fusion (see below), the fluorescence decay to baseline of transient events was variable and could not be reliably fitted to an equation for the entire time-course of the decay. Consequently, to assess the rate of spH fluorescence decay of transiently fused vesicles, we measured the portion of signal that could be reliably fitted to a linear function. Hence, we analysed the average slope of the fluorescence decay falling within a window of 333 ms centred at the half-maximal fluorescence intensity of the decay phase of the transient fusion (Fig. 12A). We found that the slope was significantly reduced (shallower) when astrocytes were stimulated with 4-Br-A23187, hypertonicity (sucrose) or mechanically compared to the slope of spontaneously fusing vesicles (Fig. 12B; P < 0.05, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test); the slopes on average ranged from −0.82 ± 0.04 i.u. per 333 ms for spontaneous to −0.49 + 0.03 i.u. per 333 ms for 4-Br-A23187 stimulation. Furthermore, we found that the expression of SNAP25B or Syt1 did not significantly affect decay slopes when astrocytes were mechanically stimulated (Kruskal–Wallis one-way ANOVA, P = 0.208). However, there was a significant reduction in slope when SNAP25B-, but not Syt1-, expressing astrocytes were stimulated with bradykinin (Fig. 12; P < 0.05, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test). These data indicate that vesicular internalization/re-acidification seems to vary under different stimulation conditions and that it can be modulated by SNAP25B in a stimulus-specific manner.
Figure 12. Decay slope of transient fusion events in astrocytes.
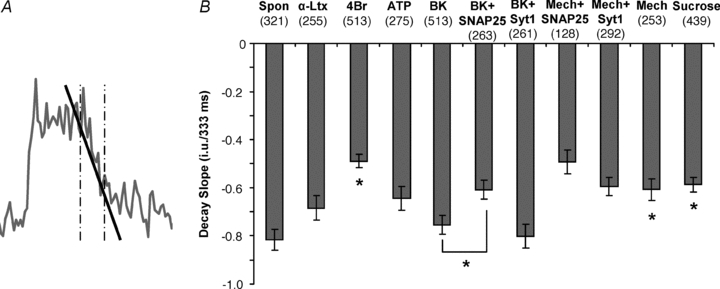
A, the average slope (black line) of fluorescence decay within a time window of 333 ms (vertical dotted lines) centred at half-maximal fluorescence intensity of the decay phase of each transient fusion was measured. This approximates the rate of the vesicular internalization/re-acidification process in transiently fusing vesicles. B, the average slope is given as the change in fluorescence intensity units (i.u.) per 333 ms time widow. 4-Br-A23187, mechanical and sucrose stimulation resulted in significantly reduced slopes. The expression of SNAP25B resulted in a reduction of the slope during stimulation with bradykinin (*P < 0.05, Kruskal–Wallis one-way ANOVA followed by post hoc Dunn's test). Abbreviations as in Fig. 9.
Full fusion decay: estimate of synaptobrevin 2 diffusion in the plasma membrane
The fluorescence decay for full fusion events is governed by the diffusion of spH into the plasma membrane (Sankaranarayanan & Ryan, 2000). We determined the time constant for this decay and calculated the diffusion coefficient of spH, a synaptobrevin 2 derivative. Since the loss of fluorescence in full fusion events is a result of spH diffusing out into the surrounding plasma membrane, we should be able to fit the fluorescence decay to Fick's equation for diffusion from a point source in two dimensions and get an estimate for the time constant:
| (1) |
where I is the spH fluorescence intensity, t is the time and τ is the time constant. We averaged the fluorescence decay following the peak fluorescence for all full fusion events in various conditions and found it to decay exponentially with a time constant of about 600 ms (Fig. 13A). Spontaneous fusions had the shortest time constant (∼534 ms), which is comparable to that measured for fusions caused by stimulation with ATP (∼538 ms), while the longest time constant was recorded when cells were exposed to sucrose (∼673 ms). The expression of SNAP25B or Syt1 did not affect (within 10%) the time constant when astrocytes were stimulated with bradykinin (Fig. 13B). However, there was some increase in the decay time, compared to control, when astrocytes expressing Syt1 or SNAP25B (33% and 23% increase, respectively) were stimulated mechanically.
Figure 13. Estimate of post-fusion diffusion of vesicular synaptobrevin 2 within the plasma membrane.

A, averaged time course of full fusion spH fluorescence intensity decay; astrocytes were stimulated with bradykinin. When fitted to eqn (1), the time constant, τ, of the decay rate can be estimated. The diffusion coefficient, D, of the movement of spH, a synaptobrevin 2 derivative, in the plasma membrane can be estimated using eqn (2) (see text for details). B, table shows the time constant and diffusion coefficient for each condition; number of full fusion events used in calculations is shown in parentheses.
We also estimated the diffusion coefficient, D, of spH in the plasma membrane. Since the initial fluorescence increase in full fusion is contained within a circular patch of membrane from the vesicle collapse, we can assume a Gaussian profile for this fluorescence intensity (as seen in Fig. 5F). The diffusion constant D can be calculated from eqn (2):
| (2) |
where w is the width of the inner circle (0.691 μm in diameter) and t1/2 is the time required for the intensity in this area to lose half of its initial intensity (Sprague et al. 2004). We found that, for all stimuli, the diffusion coefficient was in the order of 10−9 cm2 s−1 (10−1μm2 s−1) (Fig. 13B), a value consistent with other calculations of the diffusion of spH (Sankaranarayanan & Ryan, 2000) and various receptors (Barak et al. 1997; Schwille et al. 1999; Serge et al. 2002; Shrivastava et al. 2010; Oddi et al. 2011) in the plasma membrane.
Discussion
The intent of this study was to obtain detailed temporal characteristics of synaptobrevin 2-containing vesicle fusion in astrocytes at the single vesicle level using spH and TIRFM. Since astrocytes can express a variety of exocytotic proteins (reviewed in Montana et al. 2006), it appears that there could be many intracellular interactions between exocytotic proteins that would occur with some redundancy and promiscuity (Liu et al. 2006; Montana et al. 2009). Astrocytes isolated from visual cortex in our culture system express SNAREs; their ternary SNARE complexes are composed of: syntaxin 1A, synaptobrevin 2 or its homologue cellubrevin, and SNAP23 (Parpura et al. 1995a; Montana et al. 2004). Cellubrevin appears functionally interchangeable with synaptobrevin 2 in Ca2+-dependent exocytosis (Bhattacharya et al. 2002; Borisovska et al. 2005; Deak et al. 2006). In addition to SNAP23, astrocytes in culture and in vivo can express SNAP25 (Jeftinija et al. 1997; Maienschein et al. 1999; Wilhelm et al. 2004; Stigliani et al. 2006). The presence of SNAP25B instead of SNAP23A in the ternary SNARE complex gives the complex enhanced stability, as determined in isolation from any other protein/lipid components using a single-molecule approach (Montana et al. 2009). Furthermore, in chromaffin cells, SNAP25 affects the temporal characteristics of exocytosis (Sorensen et al. 2003). In terms of synaptotagmins, which interact with the SNARE complex, astrocytes in vivo and in our culture express Syt4 (Zhang et al. 2004a). However, astrocytes can additionally express Syt1 (Maienschein et al. 1999), although probably not together with SNAP25 (Wilhelm et al. 2004). These synaptotagmins can differentially interact with various combinations of proteins within the ternary SNARE complex (Chieregatti et al. 2004). In chromaffin and PC12 cells, Syt4 can affect the temporal characteristics of exocytosis (Wang et al. 2003; Zhang & Jackson, 2010; Zhang et al. 2010). Therefore, we also characterized the effects that exogenous expression of SNAP25B or Syt1, as additional players to the endogenous cast of exocytotic proteins, have on the temporal characteristics of synaptobrevin 2-laden vesicle fusion in cultured astrocytes from rat visual cortex.
There have been a large variety of sizes reported for secretory vesicles in astrocytes (reviewed in Montana et al. 2006; Parpura & Zorec, 2010). Immunoelectron microscopy (IEM) of astrocytes demonstrated that synaptobrevin 2 can be associated with electron-lucent (clear) and some dense core vesicular structures, with diameters ranging from 100 to 700 nm (Maienschein et al. 1999). EM of immunoisolated synaptobrevin 2-containing vesicles have measured diameters of mainly clear vesicles from 30 to over 100 nm (Crippa et al. 2006). The IEM of cellubrevin and VGLUT1 or 2 in astrocytes in situ showed association of these proteins with small clear vesicles with a mean diameter of ∼30 nm (Bezzi et al. 2004). However, EM of photoconverted FM1-43-labelled glutamatergic vesicles resulted in a measured diameter of 310 nm, which was similar to estimates of their sizes in live cultured astrocytes (340 nm) using differential interference contrast microscopy (Chen et al. 2005). Visualization of vesicles expressing the synaptobrevin 2 fluorescent derivative, spH, in astrocytes using TIRFM gave an estimated diameter of under ∼100 nm (Bowser & Khakh, 2007). Similarly, diameters of VGLUT2 or VGLUT1-pHluorin expressing vesicles in astrocytes using TIRFM were estimated to be ∼40 nm based on comparison to 40 nm diameter fluorescent beads (Cali et al. 2008; Marchaland et al. 2008). Glutamatergic vesicles labelled by capturing an extracellular antibody against VGLUT1 in a Ca2+-dependent manner while recycling with the plasma membrane are electron-lucent and have diameters of ∼50 nm (Stenovec et al. 2007). Vesicles found within gliosomes, subcellular components of astrocytic processes isolated from brain by fractionation (Nakamura et al. 1993), express synaptobrevin 2 and VGLUT1 and measure ∼30 nm in diameter (Stigliani et al. 2006). Using TIRF and oblique illumination to visualize vesicles containing synaptobrevin 2-/VAMP2-GFP in astrocytes, we obtained an estimated vesicle diameter of ∼312 nm (range, 161–422 nm). This measurement fits well with previous reports of vesicle size in astrocytes under physiologically relevant conditions, where synaptobrevin 2 and VGLUTs are found in association with relatively small, clear vesicles (reviewed in Parpura & Zorec, 2010).
To visualize vesicle fusion events, we expressed superecliptic spH in cultured astrocytes. Thus, we labelled any synaptobrevin 2-laden vesicles that might contain a variety of cargoes. It has been shown that synaptobrevin 2-containing vesicles in astrocytes co-localize with VGLUT1, 2 and 3 (Montana et al. 2004; Bowser & Khakh, 2007) and isolated vesicles containing synaptobrevin 2 are able to take up glutamate (Crippa et al. 2006). However, not all synaptobrevin 2-containing vesicles were positive for VGLUTs, indicating that subpopulations of vesicles are likely to contain other transmitters, such as d-serine, which has been shown to reside in synaptobrevin 2-positive vesicles in astrocytes (Martineau et al. 2008).
Astrocytes in our culture system appear as flat polygonal cells. They have less complex morphological features than astrocytes in situ which display elaborate processes. Because of this limitation of our culture system, we were not able to study the fusion kinetics of spH at the somatic region vs. astrocytic processes. The vast majority of exocytotic release of transmitters in neurons, and perhaps in astrocytes, occurs at processes. Astrocytes in culture can exocytotically release glutamate from their bodies (Innocenti et al. 2000; Bezzi et al. 2004; Hua et al. 2004; Montana et al. 2004) and such release can affect the excitability of nearby neurons which can be also observed in situ (reviewed in Ni et al. 2007). Is should be noted that somatic exocytosis and transmitter release has been demonstrated in neurons (Chen & Ewing, 1995; Chen et al. 1995; Huang & Neher, 1996; Parpura et al. 1998). Interestingly, in a culture system where astrocytes had some processes, astrocytic vesicles containing Sb2-EGFP preferentially fused in a Ca2+-dependent manner at processes (Crippa et al. 2006). Furthermore, vesicular age plays a role in determining the intracellular location of vesicles in astrocytes, as determined using cultured astrocytes and expression of a chimeric form of synaptobrevin 2 having a fluorescent ‘timer’ protein appended (Loson et al. 2011). Hence, younger vesicles predominately localized at the periphery of astrocytic somata and processes, while older vesicles predominately located at the central portion of the astrocyte body. Thus, studying temporal (and other) characteristics using astrocytes in culture might yield important general information that could then be applied to further studies of astrocytes in situ.
The advantage of using spH to detect vesicular exocytotic events, compared to other methods such as FM1-43 (Chen et al. 2005; Xu et al. 2007) or acridine orange (AO) (Bezzi et al. 2004), is that spH can disclose repeated fusion events, regardless of whether vesicles are filled with or depleted of transmitter (Sankaranarayanan et al. 2000). Amperometry partly overcomes the problem of detecting repeated fusions, but an oxidizable transmitter must be released. To achieve this in astrocytes, a pharmacological dose of a surrogate transmitter such as dopamine (70 mm, 45 min) needs to be pre-loaded into glutamatergic vesicles (Chen et al. 2005). Capacitance measurements (Kreft et al. 2004; Zhang et al. 2004b) report over an extended period, but presently there are no published data to distinguish individual vesicle fusions in astrocytes. Indeed, each approach contributes a unique perspective/information to aid in the understanding of exocytosis.
TIRFM combined with spH and VGLUT1-pHluorin have recently been used to disclose vesicular fusions in astrocytes (Bowser & Khakh, 2007; Cali et al. 2008; Marchaland et al. 2008). Spontaneous vesicle fusions in astrocytes were reported to occur at a rate of 0.26 events per minute within a ∼1000 μm2 area (Bowser & Khakh, 2007), which is a substantially lower rate than the average rate of 14 ± 1.2 fusions per minute within a similar astrocytic area (∼1100 μm2) that we have measured. However, this is expected, since we excluded (with the exception of Fig. 2) cells that had less than 15 fusions during the 5 min course of an experiment from our analysis (see Methods). When we used the Ca2+ ionophore 4-Br-A23187 to induce a large and sustained increase of cytoplasmic Ca2+, it promoted an increase in fusion rate of 35.9 fusions per minute, (Fig. 3D), fairly similar to the 52 per minute seen elsewhere in astrocytes stimulated using a different ionophore, ionomycin (Bowser & Khakh, 2007). However, the time-course of the fusion events we observed was strikingly different from that seen when ionomycin was applied to cells using AO to monitor events (Bezzi et al. 2004). In this case, almost all events occurred within 30 s. Although this discrepancy may be explained by the use of a different ionophore it seems likely that it is due to the loss of the indicator dye, preventing further fusions from being disclosed. Similarly, when using α-latrotoxin (3 nm) we observed a sustained fusion rate over 5 min, while the addition of α-latrotoxin by Bezzi et al. (2004), albeit at a much higher concentration (12 nm), caused almost all events to occur within ∼30 s. These differences might be explained by the loss of AO and/or by α-latrotoxin possibly acting as a Ca2+ ionophore when applied at high concentrations (Surkova, 1994; Sudhof, 2001; Rohou et al. 2007).
Exocytotic bursts of fusion events were seen in hippocampal astrocytes within ∼0.5 s after the application of the metabotropic glutamate receptor (mGluR) agonist, dihydroxyphenylglycine (DHPG) using AO-loaded VGLUT1/2-positive vesicles (Bezzi et al. 2004) or VGLUT1-pHluorin (Marchaland et al. 2008) and TIRFM. Such exocytotic bursts were also observed when metabotropic purinergic (P2Y1) or chemokine (CXC4) receptors in hippocampal astrocytes were stimulated while monitoring AO or FM4-64-loaded VGLUT2-positive vesicles, respectively, using TIRFM (Domercq et al. 2006; Cali et al. 2008). This is in apparent contrast to findings by other groups showing sustained temporal patterns of fusions occurring over prolonged periods in astrocytes. Using cultured astrocytes, Chen et al. (2005) recorded amperometric spikes from the exocytosis of dopamine-loaded vesicles occurring over minutes, while Pasti et al. (2001), using a ‘sniffer’ cell approach, detected quantal release of glutamate occurring over several minutes. There could be various explanations for these seemingly disparate findings, including that different culturing conditions in these studies may lead to expression of different sets of exocytotic proteins (reviewed in Montana et al. 2006). However, our data reveal slow bursts occurring within 6 s of the initiation of a stimulus as well as sustained/prolonged fusions both taking place in astrocytes for which we report the same SNARE protein expression. We find that the temporal pattern which occurs is dependent upon the stimulus that induces exocytosis.
Mechanical contact was able to cause a burst of exocytosis in a subset of astrocytes, an effect that we did not observe with any other stimulus. This could be due to the fact that mechanical stimulation causes larger and quicker intracellular Ca2+ loads than achieved with any other stimulus studied. Nonetheless, the burst in astrocytes can occur in the absence of SNAP25, a SNARE shown to govern exocytotic bursts in chromaffin cells (Sorensen et al. 2003). Consequently, we attempted to induce bursts under different conditions by expressing exogenous SNAP25B in astrocytes. This approach did not affect the astrocytic (dis)ability to display bursts. Mechanical contact caused burst, while bradykinin led to a sustained pattern of vesicular fusions, both patterns matching findings in control astrocytes lacking SNAP25B. This finding implies that SNAP25B alone does not promote exocytotic bursts in astrocytes. Interestingly, expression of Syt1 in astrocytes led to an increased proportion of cells displaying bursts when mechanically stimulated. Again, stimulation with bradykinin did not induce bursting. Thus, at present, the underlying mechanism that governs exocytotic bursts in astrocytes remains elusive.
Consistent with previous studies (Bezzi et al. 2004; Chen et al. 2005; Bowser & Khakh, 2007; Cali et al. 2008; Marchaland et al. 2008) we were able to resolve two distinct types of fusion events, full and transient (‘kiss-and-run’ and ’kiss-and-stay’), occurring in astrocytes. We report that spontaneously occurring events had an equal ratio of transient to full fusions, which is similar to the 40/60% ratio seen by Bowser & Khakh (2007). Any change in preference for one fusion type over the other was dependent on the stimulus used. It appeared that agonists that caused increases in cytoplasmic Ca2+ concentration, bradykinin and ATP, caused more full fusions while stimuli that induced fusions via Ca2+-independent means, such as sucrose and α-latrotoxin, caused more transient events. Mechanical stimulation was unusual in this regard since it caused large increases in cytoplasmic Ca2+ concentration yet induced the highest proportion of transient events. This finding is in contrast to amperometric recordings of dopamine release as a ‘surrogate’ transmitter for glutamate, where stimulation of astrocytes with glutamate resulted only in kiss-and-run events, while mechanical stimulation elicited mainly full fusion events (Chen et al. 2005). However, it was estimated elsewhere that kiss-and-runs represent ∼60%, while full fusions represent ∼40% of the total number of exocytotic VGLUT-positive vesicle events in astrocytes stimulated with an mGluR agonist (Bezzi et al. 2004). A possible reconciliation of these findings is that dopamine-loaded vesicles might represent only a subpopulation of VGLUT vesicles. When we stimulated astrocytes mechanically or with sucrose, the length of the plateau phase, or pore open time, became almost doubled in transient fusions. This is drastically different from what was seen with the other stimuli. This led us to speculate that mechanical contact induces fusions by a different or a more complex mechanism than other Ca2+-elevating stimuli. Interestingly, the hypertonicity caused by sucrose is believed to induce exocytosis by mechanical means (Rosenmund & Stevens, 1996; Grinnell et al. 2003). Therefore, it is possible that mechanical contact induces exocytosis both by actions involving a Ca2+-dependent pathway and via directly affecting secretory machinery similar to hypertonicity. Some support for this notion might be shown in our previous reports where we preincubated astrocytes with a cell-permeant from of the Ca2+-chelator BAPTA-AM. This treatment greatly reduced the glutamate release from astrocytes that were mechanically stimulated, but spared about ∼23–35% of the release (Hua et al. 2004; Montana et al. 2004), although this could also be a consequence of the BAPTA-AM concentration used (50 μm). Another interpretation of this doubled dwell time finding is the possibility that the vesicles recruited by sucrose and mechanical stimulation are of a larger size. However, since we determined that there is only a significant relationship between vesicular size and the dwell time duration for sucrose, but not for mechanical stimulation, this explanation seems unlikely.
Investigation of the relationship between vesicle size and the duration of its fusion pore displayed a significant relationship only in conditions where astrocytes were stimulated by bradykinin or sucrose. Presently, it is hard even to speculate on any common denominator underlying this effect. Although we had to approximate the size of the vesicle using its normalized intensity, it is unlikely that our methodological approach caused this effect. Interestingly, it appears that the docking proteins SNAP25B and Syt1 (de Wit et al. 2009), can modify this vesicle size–dwell time relationship. The expression of the plasma membrane-associated SNAP25B was found to impair the vesicle size–dwell time relationship in astrocytes stimulated with bradykinin. However, expression of Syt1 in astrocytes resulted in the enhancement of this relationship; astrocytes displayed a significant relationship, not only when stimulated with bradykinin, but also when they were stimulated mechanically. These findings may point to a novel function of the plasma membrane and vesicular docking proteins as modifiers, suppressors or enhancers of mechanisms underlying the relationship between vesicle size and the duration of a transient fusion pore. Also, one could speculate that if SNAP25B and Syt1 were co-expressed in astrocytes that this might lead to some sort of ‘occlusion’ and a lack of any modulation. Whether this speculation can be extended to SNAP23 or Syt4 would need to be assessed in future work by over-expression of these proteins.
In PC12 cells, expression of Syt4 favoured kiss-and-run fusion events, while Syt1 favoured full fusion events (Wang et al. 2003). However, in experiments using liposomes and synthetic membranes in the presence of neuronal SNAREs, Syt1 could promote fusion events, but hemi-fusions, where only one leaflet of the membrane fused, were more favoured (Lu et al. 2006). Furthermore, a significant fraction of such hemi-fusions were transient events, referred to as a ‘hemi-and-run’. When we expressed Syt1 in astrocytes we saw an insignificant shift in fusions from transient to full fusions when under mechanical stimulation. However, when astrocytes were stimulated with bradykinin we saw a significant shift in the opposite direction; Syt1 favoured transient fusion. This is contrary to observation in PC12 cells, but PC12 cells express a different set of exocytotic protein machinery than astrocytes. For instance, PC12 cells endogenously express both Syt1 and Syt4 (Zhang et al. 2010) as well as SNAP23 and SNAP25 (Salaun et al. 2005). It is possible that interactions between these different Syts and various components of the ternary SNARE complex and its associated proteins could account for these discrepancies. Meanwhile, it appears that Syt1 might be serving as a ‘stabilizer’ promoting both types of fusion, but keeping the ratio within a certain range, and thus preventing extremes of either case.
We have determined that the dwell time of transient fusions in astrocytes expressing Syt1 was reduced regardless of whether these cells were stimulated mechanically or with bradykinin. This is consistent with finding in PC12 cells where kiss-and-runs (seen as stand alone feet in amperometric recordings) were significantly longer when these cells were transfected to over-express Syt4 as opposed to Syt1 (Wang et al. 2003). Interestingly, when SNAP25B was expressed in astrocytes which were mechanically stimulated, but not when challenged with bradykinin, we recorded shorter dwell times. This is an apparent disagreement with data obtained from chromaffin cells originating from a Snap25 knock-out mouse (Sorensen et al. 2003). Chromaffin cells lacking SNAP25 (presumably still expressing endogenous SNAP23; Grant et al. 1999) had significantly reduced durations of amperometric feet (transmitter pre-release from flickering transient fusion pores) preceding spikes (full fusion) when compared to control cells from wild-type animals. Additionally, in astrocytes SNAP25B abolished flickers in transient fusions when astrocytes where stimulated mechanically, but not with bradykinin. It should be noted that we report a substantially higher number of flickers (range, 2.3–16% of all transient fusions for various stimuli; Table 4) than has been observed in astrocytes releasing dopamine (only ∼0.4% of amperometic spikes preceded by feet). Nonetheless, our data indicate that the presence of SNAP25B in astrocytes differentially regulates the stability of the transient fusion pore, making it more rigid (absence of flicker) but shorter lasting. Since membrane fusion and stability of a pore inherently depends on an interplay between proteins and lipids (Tamm et al. 2003), it is tempting to speculate that differential lipid partitioning for SNAP25 vs. SNAP23 may play a role in the observed effects (Salaun et al. 2005).
Transient fusion is usually considered advantageous because the same vesicle can be reused repeatedly without having to be reformed and refilled. A short fusion time-course and small pore opening would allow for a fraction of the vesicle contents to be released during each fusion event. However, from what we have observed in astrocytes, transient events have a prolonged time-course which, even with a very small pore diameter, would result in complete emptying of the vesicle amino acid transmitter, as indicated by simulations of glutamate release from vesicles with various pore diameters (Rabie et al. 2006). Also experiments with FM1-43 indicate that even a slow leak of this dye from transient fusion events through small pores would allow for complete emptying of the vesicle of the order of a few milliseconds (Richards et al. 2005). Nonetheless, it will be necessary to actually measure the size of these fusion pores in astrocytes using a capacitance approach. We determined the time in between recurrent transient–transient fusion events in the same cellular location. We assume that these recurrent events were due to fusion of the same vesicle. Thus, conditions that would lead to shorter dwell-time (Fig. 9), quicker vesicular internalization/re-acidification (Fig. 13) and longer time inbetween recurrent transient–transient fusion events (Table 2) would allow more complete vesicle refilling with transmitter, under an assumption that recruited vesicles would have a similar content of vesicular transmitter transporters; for glutamatergic vesicles in astrocytes, the amount of VGLUT3 is a key limiting factor in regulating their (re)filling with glutamate (Ni & Parpura, 2009). Consequently, for example, spontaneous or bradykinin-induced transient fusions would lead to more transmitter release in recurrent events than if such fusions would occur due to mechanical or sucrose stimulation. Indeed, full fusion with collapse of the vesicle into the membrane would result in complete release of the vesicular contents almost instantly. This may be beneficial when large amounts of transmitter must be released quickly. The two forms of vesicular release could have profound differences when computing astrocytic contributions to the tripartite synapse (Araque et al. 1999). A glimpse at the possible differential effects of transient vs. full vesicular fusions, due to P2Y1 and PAR-1 receptor activation (Bowser & Khakh, 2007), respectively, have been reported on extra-synaptic glutamate-mediated slow-inward currents (Shigetomi et al. 2008). Our report that the expression of various isoforms of proteins affects the time-course of vesicular fusion in astrocytes expands the complexity of astrocyte–neuron signalling. These fusion differences could also be influenced by region-specific and developmental changes, as well as the expression of alternative/additional proteins in certain pathophysiological conditions. For instance, Syt1 and Syt4 are differentially regulated in a region- and developmentally-dependent manner following kainic-induced seizures (Vician et al. 1995; Tocco et al. 1996). Such changes in expression in neurons and astrocytes could have profound effects on synaptic transmission and plasticity that would affect cognitive abilities. Indeed, SNAP25B chronic over-expression in adult rat dorsal hippocampus affects paired-pulse facilitation and mediates cognitive deficits (McKee et al. 2010). Furthermore, changes in SNAP25B expression have been associated with various conditions, including attention deficit hyperactivity disorder, schizophrenia and epilepsy, that involve cognitive dysfunction (reviewed in Corradini et al. 2009).
Acknowledgments
We thank Victoria Kang and Vice Šunjara for providing the experimental data set for PCR on SNAP23 isoform, and for immunocytochemistry of SNAP23, respectively. This work is supported by the National Institute of Mental Health (MH 069791) and the National Science Foundation (CBET 0943343).
Glossary
Abbreviations
- AO
acridine orange
- GFP
green fluorescence protein
- PEI
polyethyleneimine
- SNAP23
synaptosome-associated protein of 23 kDa
- SNAP25
synaptosome-associated protein of 25 kDa
- spH
synapto-pHluorin
- Syt
synaptotagmin
- TIRFM
total internal reflection fluorescence microscopy
- TRITC
tetramethylrhodamine isothiocyanate
- V-ATPase
vacuolar type proton ATPase
Author contributions
Both authors have contributed to the conception and design of experiments, collection, analysis and interpretation of the data and writing the article; we both approved this final version of the manuscript for publication. The experiments were done at University of California, Riverside, CA and University of Alabama, Birmingham, AL, USA.
Supplementary material
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Movie 1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Ales E, Tabares L, Poyato JM, Valero V, Lindau M, Alvarez de Toledo G. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat Cell Biol. 1999;1:40–44. doi: 10.1038/9012. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Aravanis AM, Pyle JL, Tsien RW. Single synaptic vesicles fusing transiently and successively without loss of identity. Nature. 2003;423:643–647. doi: 10.1038/nature01686. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Ryan TA. The kinetics of synaptic vesicle reacidification at hippocampal nerve terminals. J Neurosci. 2006;26:2313–2320. doi: 10.1523/JNEUROSCI.4425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Martenson C, Meyer T, Caron MG. Internal trafficking and surface mobility of a functionally intactβ2-adrenergic receptor-green fluorescent protein conjugate. Mol Pharmacol. 1997;51:177–184. doi: 10.1124/mol.51.2.177. [DOI] [PubMed] [Google Scholar]
- Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Stewart BA, Niemeyer BA, Burgess RW, McCabe BD, Lin P, Boulianne G, O'Kane CJ, Schwarz TL. Members of the synaptobrevin/vesicle-associated membrane protein (VAMP) family in Drosophila are functionally interchangeable in vivo for neurotransmitter release and cell viability. Proc Natl Acad Sci U S A. 2002;99:13867–13872. doi: 10.1073/pnas.202335999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Sudhof T, Bruns D. v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 2005;24:2114–2126. doi: 10.1038/sj.emboj.7600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc Natl Acad Sci U S A. 2007;104:4212–4217. doi: 10.1073/pnas.0607625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali C, Marchaland J, Regazzi R, Bezzi P. SDF 1-α (CXCL12) triggers glutamate exocytosis from astrocytes on a millisecond time scale: imaging analysis at the single-vesicle level with TIRF microscopy. J Neuroimmunol. 2008;198:82–91. doi: 10.1016/j.jneuroim.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Chen G, Ewing AG. Multiple classes of catecholamine vesicles observed during exocytosis from the Planorbis cell body. Brain Res. 1995;701:167–174. doi: 10.1016/0006-8993(95)00989-9. [DOI] [PubMed] [Google Scholar]
- Chen G, Gavin PF, Luo G, Ewing AG. Observation and quantitation of exocytosis from the cell body of a fully developed neuron in Planorbis corneus. J Neurosci. 1995;15:7747–7755. doi: 10.1523/JNEUROSCI.15-11-07747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang L, Zhou Y, Zheng LH, Zhou Z. ‘Kiss-and-run’ glutamate secretion in cultured and freshly isolated rat hippocampal astrocytes. J Neurosci. 2005;25:9236–9243. doi: 10.1523/JNEUROSCI.1640-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieregatti E, Chicka MC, Chapman ER, Baldini G. SNAP-23 functions in docking/fusion of granules at low Ca2+ Mol Biol Cell. 2004;15:1918–1930. doi: 10.1091/mbc.E03-09-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini I, Verderio C, Sala M, Wilson MC, Matteoli M. SNAP-25 in neuropsychiatric disorders. Ann N Y Acad Sci. 2009;1152:93–99. doi: 10.1111/j.1749-6632.2008.03995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa D, Schenk U, Francolini M, Rosa P, Verderio C, Zonta M, Pozzan T, Matteoli M, Carmignoto G. Synaptobrevin2-expressing vesicles in rat astrocytes: insights into molecular characterization, dynamics and exocytosis. J Physiol. 2006;570:567–582. doi: 10.1113/jphysiol.2005.094052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyas-Kovacs A, Riedel D, Sorensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Deak F, Liu X, Khvotchev M, Li G, Kavalali ET, Sugita S, Sudhof TC. Alpha-latrotoxin stimulates a novel pathway of Ca2+-dependent synaptic exocytosis independent of the classical synaptic fusion machinery. J Neurosci. 2009;29:8639–8648. doi: 10.1523/JNEUROSCI.0898-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak F, Shin OH, Kavalali ET, Sudhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney K, Tank DW, Zucker RS. Presynaptic calcium and serotonin-mediated enhancement of transmitter release at crayfish neuromuscular junction. J Neurosci. 1991;11:2631–2643. doi: 10.1523/JNEUROSCI.11-09-02631.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem. 2006;281:30684–30696. doi: 10.1074/jbc.M606429200. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisel U, Reynolds K, Riddick M, Zimmer A, Niemann H, Zimmer A. Tetanus toxin light chain expression in Sertoli cells of transgenic mice causes alterations of the actin cytoskeleton and disrupts spermatogenesis. EMBO J. 1993;12:3365–3372. doi: 10.1002/j.1460-2075.1993.tb06010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature. 2003;423:607–613. doi: 10.1038/nature01677. [DOI] [PubMed] [Google Scholar]
- Grant NJ, Hepp R, Krause W, Aunis D, Oehme P, Langley K. Differential expression of SNAP-25 isoforms and SNAP-23 in the adrenal gland. J Neurochem. 1999;72:363–372. doi: 10.1046/j.1471-4159.1999.0720363.x. [DOI] [PubMed] [Google Scholar]
- Grinnell AD, Chen BM, Kashani A, Lin J, Suzuki K, Kidokoro Y. The role of integrins in the modulation of neurotransmitter release from motor nerve terminals by stretch and hypertonicity. J Neurocytol. 2003;32:489–503. doi: 10.1023/B:NEUR.0000020606.58265.b5. [DOI] [PubMed] [Google Scholar]
- Harata NC, Aravanis AM, Tsien RW. Kiss-and-run and full-collapse fusion as modes of exo-endocytosis in neurosecretion. J Neurochem. 2006;97:1546–1570. doi: 10.1111/j.1471-4159.2006.03987.x. [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Hartigan PM. The dip test of unimodality. Anal Stat. 1985;13:70–84. [Google Scholar]
- Hartigan PM. Computation of the dip statistic to test for unimodality. Appl Stat. 1985;34:320–325. [Google Scholar]
- Hua X, Malarkey EB, Sunjara V, Rosenwald SE, Li WH, Parpura V. Ca2+-dependent glutamate release involves two classes of endoplasmic reticulum Ca2+ stores in astrocytes. J Neurosci Res. 2004;76:86–97. doi: 10.1002/jnr.20061. [DOI] [PubMed] [Google Scholar]
- Huang LY, Neher E. Ca2+-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–145. doi: 10.1016/s0896-6273(00)80287-1. [DOI] [PubMed] [Google Scholar]
- Innocenti B, Parpura V, Haydon PG. Imaging extracellular waves of glutamate during calcium signaling in cultured astrocytes. J Neurosci. 2000;20:1800–1808. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeftinija SD, Jeftinija KV, Stefanovic G. Cultured astrocytes express proteins involved in vesicular glutamate release. Brain Res. 1997;750:41–47. doi: 10.1016/s0006-8993(96)00610-5. [DOI] [PubMed] [Google Scholar]
- Klyachko VA, Jackson MB. Capacitance steps and fusion pores of small and large-dense-core vesicles in nerve terminals. Nature. 2002;418:89–92. doi: 10.1038/nature00852. [DOI] [PubMed] [Google Scholar]
- Kreft M, Stenovec M, Rupnik M, Grilc S, Krzan M, Potokar M, Pangrsic T, Haydon PG, Zorec R. Properties of Ca2+-dependent exocytosis in cultured astrocytes. Glia. 2004;46:437–445. doi: 10.1002/glia.20018. [DOI] [PubMed] [Google Scholar]
- Lee W, Parpura V. Exocytotic release of glutamate from astrocytes: comparison to neurons. In: Bean A, editor. Protein Trafficking in Neurons. Amsterdam: Elsevier; 2007. pp. 329–365. [Google Scholar]
- Lee W, Parpura V. Micropatterned substrates for studying astrocytes in culture. Front Neurosci. 2009;3:381–387. doi: 10.3389/neuro.01.033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Malarkey EB, Reyes RC, Parpura V. Micropit: A New Cell Culturing Approach for Characterization of Solitary Astrocytes and Small Networks of these Glial Cells. Front Neuroeng. 2008;1:2. doi: 10.3389/neuro.16.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XJ, Liu AQ, Lim CS, Ayi TC, Yap PH. Determining refractive index of single living cell using an integrated microchip. Sensor Actuat a-Phys. 2007;133:349–354. [Google Scholar]
- Liu W, Montana V, Bai J, Chapman ER, Mohideen U, Parpura V. Single molecule mechanical probing of the SNARE protein interactions. Biophys J. 2006;91:744–758. doi: 10.1529/biophysj.105.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loson OC, Ha CM, Parpura V. Age-dependent spatial segregation of synaptobrevin 2-containing vesicles in astrocytes. J Neurochem. 2011;116:909–915. doi: 10.1111/j.1471-4159.2010.07018.x. [DOI] [PubMed] [Google Scholar]
- Lu X, Xu Y, Zhang F, Shin YK. Synaptotagmin I and Ca2+ promote half fusion more than full fusion in SNARE-mediated bilayer fusion. FEBS Lett. 2006;580:2238–2246. doi: 10.1016/j.febslet.2006.03.035. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maienschein V, Marxen M, Volknandt W, Zimmermann H. A plethora of presynaptic proteins associated with ATP-storing organelles in cultured astrocytes. Glia. 1999;26:233–244. doi: 10.1002/(sici)1098-1136(199905)26:3<233::aid-glia5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. doi: 10.1002/glia.20656. [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Monitoring of exocytosis in astrocytes using synaptopHluorins and total internal reflection fluorescence microscopy. J Neurochem. 2006;96:60. [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–154. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of transmitter release from astrocytes. In: Parpura V, Haydon PG, editors. Astrocytes in (Patho)Physiology of the Nervous System. Boston, MA: Springer; 2009. pp. 301–350. [Google Scholar]
- Marchaland J, Cali C, Voglmaier SM, Li H, Regazzi R, Edwards RH, Bezzi P. Fast subplasma membrane Ca2+ transients control exo-endocytosis of synaptic-like microvesicles in astrocytes. J Neurosci. 2008;28:9122–9132. doi: 10.1523/JNEUROSCI.0040-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Galli T, Baux G, Mothet JP. Confocal imaging and tracking of the exocytotic routes for D-serine-mediated gliotransmission. Glia. 2008;56:1271–1284. doi: 10.1002/glia.20696. [DOI] [PubMed] [Google Scholar]
- McKee AG, Loscher JS, O'Sullivan NC, Chadderton N, Palfi A, Batti L, et al. AAV-mediated chronic over-expression of SNAP-25 in adult rat dorsal hippocampus impairs memory-associated synaptic plasticity. J Neurochem. 2010;112:991–1004. doi: 10.1111/j.1471-4159.2009.06516.x. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Montana V, Liu W, Mohideen U, Parpura V. Single molecule measurements of mechanical interactions within ternary SNARE complexes and dynamics of their disassembly: SNAP25vs. SNAP23. J Physiol. 2009;587:1943–1960. doi: 10.1113/jphysiol.2009.168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana V, Malarkey EB, Verderio C, Matteoli M, Parpura V. Vesicular transmitter release from astrocytes. Glia. 2006;54:700–715. doi: 10.1002/glia.20367. [DOI] [PubMed] [Google Scholar]
- Montana V, Ni Y, Sunjara V, Hua X, Parpura V. Vesicular glutamate transporter-dependent glutamate release from astrocytes. J Neurosci. 2004;24:2633–2642. doi: 10.1523/JNEUROSCI.3770-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K. Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia. 1993;9:48–56. doi: 10.1002/glia.440090107. [DOI] [PubMed] [Google Scholar]
- Ni Y, Malarkey EB, Parpura V. Vesicular release of glutamate mediates bidirectional signaling between astrocytes and neurons. J Neurochem. 2007;103:1273–1284. doi: 10.1111/j.1471-4159.2007.04864.x. [DOI] [PubMed] [Google Scholar]
- Ni Y, Parpura V. Dual regulation of Ca2+-dependent glutamate release from astrocytes: vesicular glutamate transporters and cytosolic glutamate levels. Glia. 2009;57:1296–1305. doi: 10.1002/glia.20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi S, Dainese E, Fezza F, Lanuti M, Barcaroli D, De Laurenzi V, Centonze D, Maccarrone M. Functional characterization of putative cholesterol binding sequence (CRAC) in human type-1 cannabinoid receptor. J Neurochem. 2011;116:858–865. doi: 10.1111/j.1471-4159.2010.07041.x. [DOI] [PubMed] [Google Scholar]
- Parpura V, Baker BJ, Jeras M, Zorec R. Regulated exocytosis in astrocytic signal integration. Neurochem Int. 2010;57:451–459. doi: 10.1016/j.neuint.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Parpura V, Fang Y, Basarsky T, Jahn R, Haydon PG. Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 1995a;377:489–492. doi: 10.1016/0014-5793(95)01401-2. [DOI] [PubMed] [Google Scholar]
- Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci U S A. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Liu F, Brethorst S, Jeftinija K, Jeftinija S, Haydon PG. Alpha-latrotoxin stimulates glutamate release from cortical astrocytes in cell culture. FEBS Lett. 1995b;360:266–270. doi: 10.1016/0014-5793(95)00121-o. [DOI] [PubMed] [Google Scholar]
- Parpura V, Tong W, Yeung ES, Haydon PG. Laser-induced native fluorescence (LINF) imaging of serotonin depletion in depolarized neurons. J Neurosci Methods. 1998;82:151–158. doi: 10.1016/s0165-0270(98)00056-9. [DOI] [PubMed] [Google Scholar]
- Parpura V, Zorec R. Gliotransmission: exocytotic release from astrocytes. Brain Res Rev. 2010;63:83–92. doi: 10.1016/j.brainresrev.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabie HR, Rong J, Glavinovic MI. Monte Carlo simulation of release of vesicular content in neuroendocrine cells. Biol Cybern. 2006;94:483–499. doi: 10.1007/s00422-006-0061-0. [DOI] [PubMed] [Google Scholar]
- Reyes RC, Parpura V. Mitochondria modulate Ca2+-dependent glutamate release from rat cortical astrocytes. J Neurosci. 2008;28:9682–9691. doi: 10.1523/JNEUROSCI.3484-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes RC, Parpura V. The trinity of Ca2+ sources for the exocytotic glutamate release from astrocytes. Neurochem Int. 2009;55:2–8. doi: 10.1016/j.neuint.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes RC, Perry G, Lesort M, Parpura V. Immunophilin deficiency augments Ca2+-dependent glutamate release from mouse cortical astrocytes. Cell Calcium. 2011;49:23–34. doi: 10.1016/j.ceca.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Bai J, Chapman ER. Two modes of exocytosis at hippocampal synapses revealed by rate of FM1-43 efflux from individual vesicles. J Cell Biol. 2005;168:929–939. doi: 10.1083/jcb.200407148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Nield J, Ushkaryov YA. Insecticidal toxins from black widow spider venom. Toxicon. 2007;49:531–549. doi: 10.1016/j.toxicon.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sadoul K, Berger A, Niemann H, Weller U, Roche PA, Klip A, Trimble WS, Regazzi R, Catsicas S, Halban PA. SNAP-23 is not cleaved by botulinum neurotoxin E and can replace SNAP-25 in the process of insulin secretion. J Biol Chem. 1997;272:33023–33027. doi: 10.1074/jbc.272.52.33023. [DOI] [PubMed] [Google Scholar]
- Salaun C, Gould GW, Chamberlain LH. The SNARE proteins SNAP-25 and SNAP-23 display different affinities for lipid rafts in PC12 cellsRegulation by distinct cysteine-rich domains. J Biol Chem. 2005;280:1236–1240. doi: 10.1074/jbc.M410674200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- Schuske K, Jorgensen EM. NeuroscienceVesicular glutamate transporter-shooting blanks. Science. 2004;304:1750–1752. doi: 10.1126/science.1100475. [DOI] [PubMed] [Google Scholar]
- Schwille P, Haupts U, Maiti S, Webb WW. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serge A, Fourgeaud L, Hemar A, Choquet D. Receptor activation and homer differentially control the lateral mobility of metabotropic glutamate receptor 5 in the neuronal membrane. J Neurosci. 2002;22:3910–3920. doi: 10.1523/JNEUROSCI.22-10-03910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Rhee JS, Tang J, Sugita S, Rosenmund C, Sudhof TC. Sr2+ binding to the Ca2+ binding site of the synaptotagmin 1 C2B domain triggers fast exocytosis without stimulating SNARE interactions. Neuron. 2003;37:99–108. doi: 10.1016/s0896-6273(02)01145-5. [DOI] [PubMed] [Google Scholar]
- Shrivastava S, Pucadyil TJ, Paila YD, Ganguly S, Chattopadhyay A. Chronic cholesterol depletion using statin impairs the function and dynamics of human serotonin(1A) receptors. Biochemistry. 2010;49:5426–5435. doi: 10.1021/bi100276b. [DOI] [PubMed] [Google Scholar]
- Smith SM, Renden R, von Gersdorff H. Synaptic vesicle endocytosis: fast and slow modes of membrane retrieval. Trends Neurosci. 2008;31:559–568. doi: 10.1016/j.tins.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Sprague BL, Pego RL, Stavreva DA, McNally JG. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal RG, Mosharov EV, Sulzer D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 2004;7:341–346. doi: 10.1038/nn1205. [DOI] [PubMed] [Google Scholar]
- Stenovec M, Kreft M, Grilc S, Potokar M, Kreft ME, Pangrsic T, Zorec R. Ca2+-dependent mobility of vesicles capturing anti-VGLUT1 antibodies. Exp Cell Res. 2007;313:3809–3818. doi: 10.1016/j.yexcr.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Almers W. A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol. 2001;2:268–275. doi: 10.1038/35067069. [DOI] [PubMed] [Google Scholar]
- Stigliani S, Zappettini S, Raiteri L, Passalacqua M, Melloni E, Venturi C, Tacchetti C, Diaspro A, Usai C, Bonanno G. Glia re-sealed particles freshly prepared from adult rat brain are competent for exocytotic release of glutamate. J Neurochem. 2006;96:656–668. doi: 10.1111/j.1471-4159.2005.03631.x. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. α-Latrotoxin and its receptors: neurexins and CIRL/latrophilins. Annu Rev Neurosci. 2001;24:933–962. doi: 10.1146/annurev.neuro.24.1.933. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Surkova I. Can exocytosis induced byα-latrotoxin be explained solely by its channel-forming activity? Ann N Y Acad Sci. 1994;710:48–64. doi: 10.1111/j.1749-6632.1994.tb26613.x. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Tamm LK, Crane J, Kiessling V. Membrane fusion: a structural perspective on the interplay of lipids and proteins. Curr Opin Struct Biol. 2003;13:453–466. doi: 10.1016/s0959-440x(03)00107-6. [DOI] [PubMed] [Google Scholar]
- Tocco G, Bi X, Vician L, Lim IK, Herschman H, Baudry M. Two synaptotagmin genes, Syt1 and Syt4, are differentially regulated in adult brain and during postnatal development following kainic acid-induced seizures. Brain Res Mol Brain Res. 1996;40:229–239. doi: 10.1016/0169-328x(96)00055-1. [DOI] [PubMed] [Google Scholar]
- Vardjan N, Stenovec M, Jorgacevski J, Kreft M, Zorec R. Subnanometer fusion pores in spontaneous exocytosis of peptidergic vesicles. J Neurosci. 2007;27:4737–4746. doi: 10.1523/JNEUROSCI.0351-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. Neurotransmitter receptors in astrocytes. In: Parpura V, Haydon PG, editors. Astrocytes in (Patho)Physiology of the Nervous System. Boston, MA: Springer; 2009. pp. 49–67. [Google Scholar]
- Vician L, Lim IK, Ferguson G, Tocco G, Baudry M, Herschman HR. Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc Natl Acad Sci U S A. 1995;92:2164–2168. doi: 10.1073/pnas.92.6.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Lu JC, Bai J, Chang PY, Martin TF, Chapman ER, Jackson MB. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature. 2003;424:943–947. doi: 10.1038/nature01857. [DOI] [PubMed] [Google Scholar]
- Wilhelm A, Volknandt W, Langer D, Nolte C, Kettenmann H, Zimmermann H. Localization of SNARE proteins and secretory organelle proteins in astrocytes in vitro and in situ. Neurosci Res. 2004;48:249–257. doi: 10.1016/j.neures.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Peng H, Kang N, Zhao Z, Lin JH, Stanton PK, Kang J. Glutamate-induced exocytosis of glutamate from astrocytes. J Biol Chem. 2007;282:24185–24197. doi: 10.1074/jbc.M700452200. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci U S A. 2004a;101:9441–9446. doi: 10.1073/pnas.0401960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pangrsic T, Kreft M, Krzan M, Li N, Sul JY, Halassa M, Van Bockstaele E, Zorec R, Haydon PG. Fusion-related release of glutamate from astrocytes. J Biol Chem. 2004b;279:12724–12733. doi: 10.1074/jbc.M312845200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Jackson MB. Membrane bending energy and fusion pore kinetics in Ca2+-triggered exocytosis. Biophys J. 2010;98:2524–2534. doi: 10.1016/j.bpj.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang Z, Jackson MB. Synaptotagmin IV modulation of vesicle size and fusion pores in PC12 cells. Biophys J. 2010;98:968–978. doi: 10.1016/j.bpj.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Misler S, Chow RH. Rapid fluctuations in transmitter release from single vesicles in bovine adrenal chromaffin cells. Biophys J. 1996;70:1543–1552. doi: 10.1016/S0006-3495(96)79718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.