

Abstract
The bacterial histidine autokinase CheA contains a histidine phosphotransfer (Hpt) domain that accepts a phosphate from the catalytic domain and donates the phosphate to either target response regulator protein, CheY or CheB. The Hpt domain forms a helix-bundle structure with a conserved four-helix bundle motif and a variable fifth helix. Observation of two nearly equally populated conformations in the crystal structure of a Hpt domain fragment of CheA from Thermotoga maritima containing only the first four helices suggests more mobility in a tightly packed helix bundle structure than previously thought. In order to examine how the structures of Hpt domain homologs may differ from each other particularly in the conformation of the last helix, and whether an alternative conformation exists in the intact Hpt domain in solution, we have solved a high-resolution, solution structure of the CheA Hpt from T. maritima and characterized the backbone dynamics of this protein. The structure contains a four-helix bundle characteristic of histidine phosphotransfer domains. The position and orientation of the fifth helix resembles those in known Hpt domain crystal and solution structures in other histidine kinases. The alternative conformation that was reported in the crystal structure of the CheA Hpt from T. maritima missing the fifth helix is not detected in the solution structure, suggesting a role for the fifth helix in providing stabilizing forces to the overall structure.
Keywords: Two component, CheA, Histidine phosphotransfer, Chemotaxis, Four helix bundle, Histidine kinase
Introduction
The bacterial chemotaxis system regulates the movement of the motile bacteria by controlling the switching the rotary motion of the flagella motor between smooth swimming (counterclockwise)and tumbling states (clockwise) in response to temporal changes in the concentrations of attractants and repellants. Many bacteria rely on a two—component signal transduction system to modulate the motor activity and other cellular responses. These systems consist of a histidine autokinase that facilitates an ATP-dependent autophosphorylation of a histidine residue and a response regulator that is activated as it receives a phosphoryl group from the kinase (Adler 1975; Baker et al. 2005; Borkovich et al. 1989; Sourjik 2004). The histidine kinase CheA plays a central role in bacterial chemotaxis by coupling its activity with the ligand-binding or methylation state of a family of chemotaxis receptors. As a multi-functional modular protein, CheA sequentially encodes five domains (designated as P1–P5 from N- to C- terminus) connected by linkers. The P1 domain is an independent phosphotransfer domain containing the site of phosphorylation histidine and is separated from the ATP-binding and catalytic domain, P4. Phosphorylated P1 transfers its phosphate to the response regulator CheY or CheB that is bound to the P2 domain. P3 is a dimerization domain, resembling the histidine phosphorylation domain in an orthodox histidine kinase. CheA activity is modulated through P5 that interacts with both the receptor and the receptor-coupling protein CheW (Baker et al. 2005).
The P1 domain is a member of the histidine phosphotransfer (HPt) family of proteins. It provides an intermediate for the phosphoryl group to be transferred from ATP to the response regulator. The structures of P1 from E. coli (Zhou et al. 1995) and S. typhimurium (Mourey et al. 2001) show that the P1 domains from these closely related organisms are composed of five α-helices in which four helices form an up and down helix bundle (HA–HD) and the last helix (HE) packs against HC and HD. The essential four-helix bundle structure was also found in the HPt domain of ArcB, a sensor kinase involved in anaerobic response (Kato et al. 1997; Ikegami et al. 2001) and Ypd1, a hyperosmotic stress response protein in S. cerevisiae (Xu and West 1999) although both proteins also contain additional structural elements. The structural data support a suggestion based on sequence alignment that the four-helix bundle is the conserved motif of the HPt domains and the last helix is a variable element (Zhou et al. 1995).
In enteric bacteria such as E. coli and S. typhimurium, the amino acid residues corresponding too much of the fifth helix in those P1 domains play a second functional role. An alternative translation initiation site results in a shortened form of CheA in which the first 97 residues (including the phosphoaccepting His48) of the P1 domain is deleted. The remaining residues of the P1 domain are involved in binding of the CheY phosphate phosphatase, CheZ. It appears that this binding ensures that the phosphatase and the kinase are localized to signaling complexes involving the chemoreceptors. The functional role of the C-terminal helix of the P1 domain in bacteria lacking CheZ remains unclear.
Interestingly, only the N-terminal, four-helix bundle was crystallized for the P1 domain from T. maritima, a species lacking CheZ. This shortened P1 fragment is almost as stable and active as a phosphoryl-acceptor as the full-length P1 (Quezada et al. 2004). The organization of the N-terminal four helices of this P1 fragment shows general agreement with the P1 structures of other enteric bacteria. However, the 0.98 Å resolution crystal structure of this P1 fragment revealed two nearly-equally populated conformations involving a shift of helices HA and HD along the surfaces of the rigid helices they face, HB and HC. Such multiple conformations were not observed in the P1 structures from E. coli and S. typhimurium (Quezada et al. 2004). This observation raises the question whether the last helix in T. maritima P1 is more flexible than in other P1 homologs and if conformational heterogeneity exists in solution in intact P1 from T. maritima. In this paper, we address these questions and present the solution structure and backbone dynamics of intact T. maritima P1.
Materials and methods
Protein expression and purification
Full length His6-tagged P1 construct (residues 1–133) of T. maritima (or TmP1) was subcloned in the vector pET22b(+) (Novagen). Plasmids were transformed into E. coli strain BL21(DE3) (Novagen). The protein was overexpressed by adding 1 mM IPTG during the log phase of bacterial growth. Cells containing isotopically labeled proteins were grown in minimal media which consisted of 1 mM magnesium sulfate, 0.1 mM calcium chloride, 0.5 μg/ml of thiamine, and 100 μg/ml of ampicillin with the addition of 1 g/l 15NH4Cl and 2 g/l 13C glucose (Cambridge Isotope Laboratories) for double labeled (15N/13C) samples or 1 g/l 15NH4Cl for single labeled (15N) samples. His6-tagged TmP1 was purified using Ni–NTA column (QIAGEN) after heat treatment (75°C for 10 min) and centrifugation. Purified protein was dialyzed in 50 mM Na2HPO4, 50 mM NaCl (pH 7.4). All NMR samples contained 0.04% sodium azide and 10% D2O.
NMR data collection and processing
NMR spectra were collected at 25°C on a 600 MHz Varian spectrometer equipped with either a room temperature or cryogenically enhanced 1H[13C/15N/2H] pulsed-field gradient probe. A 15N/13C-labeled sample was used to collect the following spectra: 2D 1H–15N HSQC (Kay et al. 1992), 2D 1H–13C CT–HSQC (Vuister and Bax 1992), 3D HNCACB (Muhandiram and Kay 1994), 3D CBCA(CO)NH, (H)C(CO)NH–TOCSY, H(C)NH–TOCSY (Grzesiek et al. 1993; Logan et al. 1993; Montelione et al. 1992), and 4D 15N, 13C-edited NOESY (Muhandiram et al. 1993) and 4D 13C, 13C-edited NOESY (Vuister et al. 1993). All the spectra except the 4D spectra were used to complete backbone and side-chain assignments and the 4D spectra were used for structure determination. In addition, a 15N-labeled sample was used to collect 3D 15N-edited NOESY (Zhang et al. 1994) and 3D HNHA data (Vuister et al. 1993). All data were processed with nmrPipe (Delaglio et al. 1995). Assignments were done using a modified version of ANSIG3.3 (Kraulis 1989; Kraulis et al. 1994) running under the Linux operating system.
Structure calculations
Structure calculation of TmP1 was carried out by the program X-PLOR 3.851 (Brunger 1992) using distance geometry and dynamical simulated annealing methods (Nilges et al. 1988). Experimental NOE constraints were divided into three classes including weak (5.2–1.8 Å), medium (4.2–1.8 Å), and strong (3.2–1.8 Å) based on NOE intensities. There were 2,759 NOE constraints in which 1,458 were weak, 812 were medium, and 489 were strong. In addition, Cα chemical shift index was used to determine the residues that were likely involved in helical structures. The theoretical dihedral angle 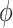 of the helical regions and the H-bond constraints for the amides in the helices were set within the ranges for ideal α-helices. The
of the helical regions and the H-bond constraints for the amides in the helices were set within the ranges for ideal α-helices. The 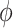 values were restricted to −60° ± 30°. H-bond constraints consisted of two distance restraints: 1.5–2.5 Å between O and HN and 2.4–3.6 Å between O and N.
values were restricted to −60° ± 30°. H-bond constraints consisted of two distance restraints: 1.5–2.5 Å between O and HN and 2.4–3.6 Å between O and N.
A total of 100 structures were generated with only experimental NOE constraints using the distance-geometry substructure-embedding protocol. The distance-geometry simulated-annealing protocol was then used to regularize the structures. These structures were refined with the addition of the dihedral angle and H-bond constraints to gradually improve the qualities of the calculated structures. Twenty-five final structures with the lowest energy were chosen for alignment and determination of root mean square (rmsd) distances using MOLMOL 2K.2 (Koradi et al. 1996).
Backbone 15N relaxation
Spin-lattice and spin–spin relaxation times T 1 and T 2, and {1H}15N NOE measurements were recorded using inverse detected two-dimensional NMR spectroscopy (Kay et al. 1989; Mandel et al. 1995; Palmer et al. 1991). T 1 values were measured using nine relaxation delays: 11.1, 55.5, 111.0, 277.5, 444.0, 721.0, 832.5, 943.5, and 1,110.0 ms. T 2 values were measured using ten relaxation delay values: 16.5, 33.6, 49.4, 65.9, 82.4, 98.9, 115.4, 131.8, 148.3, and 164.8 ms. The relaxation rates R 1 (1/T 1) and R 2 (1/T 2) were then fit using a simple exponential decay function. The {1H}15N NOE was determined by recording two spectra. NOE effect was applied in one spectrum by keeping protons in a saturated state for 3 s during a total of 8 s predelay. The other spectrum was recorded without 1H saturation during the same delay period. The ratio of the peak intensities of the two spectra was calculated as the {1H}15N NOE factor.
The relaxation data were interpreted using the model-free approach (Lipari and Szabo 1982a, b) under the assumption that R 1, R 2 and NOE factor of an amide 15N spin are dictated by dipolar coupling and chemical shift anisotropy (CSA interaction). In addition, the overall molecular motion was assumed to be isotropic and any anisotropy was ignored. This global motion was controlled by the overall rotational correlation time τm. The order parameter S 2 and the effective correlation time τe were used to describe the fast internal motion. The R 1, R 2 and NOE values are related with τm, S 2 and τe values as previously described (Kay et al. 1989). The fact that fast internal motions can be ignored for a first-order approximation allowed us to determined τm from T 1/T 2 ratios. An exchange broadening (R ex) term was also added in an alternative model when we analyzed the relaxation data. The order parameter (S 2), τe, and the additional R ex terms were extracted with the average τm value being constant.
Results
Intact TmP1 is a five-helix bundle
The backbone amide assignments of TmP1 (residues 1–133) were completed with the exception of proline residues and Met1, Met2, Ser82, and Arg113. The 1H–15N correlation spectrum shows good dispersion with a few crowded areas between 7.5 and 8.5 ppm along the 1H dimension (Fig. 1). We expected to see significant chemical shift differences for the residues that are populated in the two conformers seen in the crystal structures. However, only one set of resonances was observed, suggesting structural homogeneity on the microsecond–millisecond or slower time–scale. The Cα chemical shift index clearly shows five helices (HA–HE) joined by turns (Fig. 2c), with HE showing slightly weaker secondary shifts than the first four helices.
Fig. 1.

1H–15N HSQC spectrum and assignments of T. maritima P1 domain. The peaks in the boxes were not assigned. Residues denoted with asterisks are aliased along the 15N dimension. Two weak peaks from Ser127 to Gly128 only appear at much lower contour levels and are indicated by times symbol
Fig. 2.

Five-helix bundle structure of TmP1. a Superposition of the 25 final structures. The alignment was done with the structured parts of the protein, including residues 7–28, 35–52, 59–74, 83–102, and 115–124, giving a rmsd of 0.41 Å for the backbone atoms. b The ribbon diagram of the averaged, minimized TmP1 structure. c The differences between the observed Cα chemical shifts and the random-coil chemical shifts of residues 3–131 are plotted versus residue number. The continuous stretches of positive ΔCα values are indicative of α-helices, while the small stretches with both positive and negative ΔCα values are suggestive of loops
Full resonance assignments of TmP1 were completed using standard triple resonance techniques. The NOESY spectra showed an abundance of well-resolved NOE cross peaks which could be readily assigned. We used these and other restraints to calculate a family of solution structures for TmP1. In the structural calculation, a total of 2,759 NOE constraints were used with the addition of dihedral angle and hydrogen bond constraints. A summary of the statistical results of the structure calculation was shown in Table 1. Twenty-five final structures with the lowest energies were selected from a total of 100 structures, giving an average backbone rmsd of 0.41 Å.
Table 1.
Statistical evaluation of the structure calculation
| Final 25 structures | 2LD6a | |
|---|---|---|
| Rmsd from the distance constraints (Å) and dihedral angle constraints (°) | ||
Long-range NOE 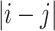 > 4 (462) > 4 (462) |
0.02708 ± 0.0045 | 0.020 |
Medium range NOE 1 ≤ 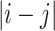 ≥ 4 (1,553) ≥ 4 (1,553) |
0.05088 ± 0.0010 | 0.048 |
| Intraresidue NOE (743) | 0.02256 ± 0.0030 | 0.016 |
| Hydrogen bond (140) | 0.01084 ± 0.0019 | 0.008 |
Dihedral angle 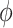 (85) [°] (85) [°] |
0.55 ± 0.09 | 0.541 |
| Rmsd form ideal geometry used in X-PLOR | ||
| Bond lengths (Å) | 0.0044 ± 0.0001 | 0.042 |
| Bond angle (°) | 0.68 ± 0.0077 | 0.682 |
| Improper angle (°) | 0.57 ± 0.0084 | 0.551 |
| Backbone helices | Side-chain | |
|---|---|---|
| Coordinate rmsd (Å) for non-hydrogen atoms in the final structures | ||
| Versus the averaged structure | 0.41 ± 0.033 | 0.94 ± 0.48 |
| Versus the averaged and minimized structure (2LD6) | 0.48 ± 0.28 | 1.08 ± 0.38 |
Numbers in parentheses indicate the total numbers of constraints in the categories
aThe average, minimized structure was derived from the final 25 structures. The thresholds of constraint violations reported here for NOE, dihedral angle and bond length were set at 0.5 Å, 5°, and 0.05 Å, respectively
The TmP1 structure consists of five helices (HD–HE) with the first four forming an antiparallel helix bundle and HE packing against HC and HD (Fig. 2b). Sequential NOEs typical of helical structure were observed between the backbone amides and between the amides and side chain protons for the residues within the five helices, confirming the result from the chemical shift index. Four turn or loop regions joining the helices were defined approximately as Lys29-Met34, Thr53-Ser58, Asn75-Ser82, and Val103-Ile114. The first three turn regions are quite well defined presenting many sequential NOEs and some long-range NOEs. The turn regions Lys29-Met34 and Asn75-Ser82 were positioned next to each other, evidenced by the long-range NOEs between residue pairs Lys29/Leu85, Pro31/Ile80, and Pro31/Leu85, and Glu32/Ile80. The loop region (Val103-Ile114) following HD is the longest and stacks against HD evidenced by the NOEs between residues in HD and the last loop. The loop region is less well defined than other parts of the structure due to the lack of NOEs involving Gly111, Glu112, and Gln113. The fact that there are strong NOEs between CαH of Ile102 and the amide protons of Gly106 and Ser107 suggests that Ile102-Ser107 make a sharp turn around Ser104, leaving Ser104 and Glu105 fully exposed to the solvent.
Backbone dynamics showed that the N-terminal six residues and the C-terminal tail are highly flexible (Fig. 3a). We did not observe any long-range NOEs in these regions.
Fig. 3.

Backbone amide 15N relaxation data. The {1H}15N–NOE a, the order parameters (S 2) b, and relaxation rate R 1 c and R 2 d are shown along with average rmsd values calculated from the backbone non-hydrogen atoms of the 25 final structures e
The alignment of the backbone atoms of the first four helices of our structure against two different conformers of the crystal structure gave similar rmsd values (0.96 and 0.89 Å).
We calculated the surface exposed area of TmP1 in the presence and absence of the fifth helix. We estimate that approximately 790 Å2 of exposed surface of the crystal structure is covered by the fifth helix suggestive of a modeately strong interaction energy of the fifth helix with the other helices.
Backbone dynamics
We examined the dynamics of TmP1 by measuring the relaxation properties (R 1, R 2 and {1H}15N NOE of the backbone 15N nuclei (Fig. 3). A total of 115 R 1 and R 2 values were obtained using well-resolved peaks. The average relaxation rates for TmP1 are 1.25 s−1 for R 1 and 13.7 s−1 for R 2. These data were well fit without the introduction of the chemical exchange (R ex) term on the micro–milli-second time scale for almost all of the backbone amides. These measurements were done at 25°C and gave an overall correlation time for isotropic tumbling of 10.9 ns, consistent with a well folded monomer of this size. The extracted order parameter (S 2) ranges from 0.85 to 0.95 for most residues, especially in the five helical regions, indicating a low degree of fast internal motions in these regions. The loop regions appear to be less rigid than the five helical regions with an S 2 mostly around 0.85 or lower. The N- and C- terminal residues contain low value of S 2 values that fall in the range between 0.27 and 0.61 with the exception of Leu6 with an S 2 value of 0.85. The C terminus appears to have the highest degree of mobility with S 2 values below 0.2, and the lowest {1H}15N NOE values ranging from 0.11 to 0.34. The trend of the order parameter is consistent with the average rmsd of the backbone heavy atom (Fig. 2c).
While the last helix appears to be well structured, its average dynamics deviates slightly from those of the first four helices. Although, its R 2 values are similar to the other regions, HE has a slightly longer R 1 and smaller NOE values. Because both R1 and NOE are sensitive to motions at higher frequencies, this difference indicates that HE is more flexible on the psec–nsec time scale than the first four helices.
Discussion
The T. Maritima P1 domain structure and dynamics in solution show that the protein folds into an overall rigid five-helix bundle, closely resembling the structure and dynamics of the related E. Coli and S. typhimurium P1 domains. We have found no evidence suggesting the existence of alternative, stable conformations that were observed in the crystal structure of the N-terminal four-helix bundle fragment (Quezada et al. 2004). This conclusion is further supported by the lack of chemical exchange contributions to the relaxation of the backbone amide nitrogens. Our data suggest that the two conformations seen in the crystal structure are either inconverting too rapidly in solution to be detected or the presence of the fifth helix provides a stablizing force and reduces the population of the minority conformer to the point that no exchange broadening is observed. The latter possibility corroborates with the sharper thermal melting transition observed in the presence of the fifth helix than the four-helix fragment (Quezada et al. 2004), suggesting that the additional structure interacts in a cooperative fashion with the first four helices of TmP1.
Role of the fifth helix
A direct comparison of the protein containing the first four (P11–104) or all five helices in solution should help us better understand the role of the fifth helix. The shorter TmP1 fragment gave drastically different 1H–15N correlation spectrum than intact TmP1 at millimolar concentrations (data not shown). Many of the resonances shifted significantly and were much broader than in intact TmP1, suggesting possible dimerization of the protein. Measurement of the 15N T 1 and T 2 values yielded a rotational correlation time of ~18 ns at 25°C with the protein at 1.5 mM, indicating the protein is likely a dimer. The resonance position and particularly linewidth displayed strong concentration dependence. At or below approximately 50 μM, the protein displayed mostly sharp resonances as expected from a small monomeric protein. These data strongly suggest that without the fifth helix, TmP1 has exposed surface elements, likely hydrophobic in nature, that can interact with each other and cause the protein to dimerize. Most likley these regions are covered by the binding of the fifth helix. Such an intermolecular interaction might intefere with the phosphate flow in the context of full-length CheA and phosphate transfer to CheY.
Comparisons of P1 domains from various CheA proteins suggest that a number of conserved residues (Leu62, Ile69, Leu84, Ile88, 114, and 121) might be important for some of the interactions between the fourth and fifth helices. The 2.1 Å crystal structure of the P1 domain from S. typhimurium with the fifth helix included did not appear to show an alternate conformer (Mourey et al. 2001), supporting this view. Thus, it appears that the fifth helix plays an important role in the conformation and dynamics of the four helix bundle.
In certain enteric bacteria such as E. coli and S. typhimurium, the C-terminal residues of the P1 domain play an additional role. In these systems an alternative transcriptional start site (residue 98 in the CheA from E. coli) results in a shortened form of CheA called CheAshort. The remaining residues of the domain in CheAshort strongly interact with the CheY phosphate specific phosphatase, CheZ (Cantwell and Manson 2009; Hao et al. 2009; O’Connor et al. 2009). This interaction appears to provide a means to associate the phosphatase with a patch on the inner surface of the cell membrane that contains several hundred chemotaxis receptor molecules as well as the coupling protein CheW and full length CheA. This arrangement of the CheY kinase and phosphatase allows for a more uniform concentration of CheY phosphate in the cell since the source and sink of CheY phosphate are in the same area. This uniform distribution of CheY phosphate provides a mechanism to coordinate the control of the rotation of the six to eight flagellar motors that are themselves relatively uniformly distributed.
Thermotoga maritima and related species do not have CheZ but rather use a completely different protein as a CheY phosphate phosphatase. The absence of this additional functionality may allow for less stringent evolutionary control of the properties of the fifth helix of TmP1. This could then be reflected in the moderate changes we see in fast time scale dynamics in this region as well as the increase in protease sensitivity that resulted in cleavage of the fifth helix during crystalization.
Acknowledgments
This work was supported by NIH grant GM59544.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Adler J. Chemotaxis in bacteria. Annu Rev Biochem. 1975;44:341–356. doi: 10.1146/annurev.bi.44.070175.002013. [DOI] [PubMed] [Google Scholar]
- Baker MD, Wolanin PM, Stock JB. Signal transduction in bacterial chemotaxis. Bioessays. 2005;28:9–22. doi: 10.1002/bies.20343. [DOI] [PubMed] [Google Scholar]
- Bell CH, Porter SL, Strawson A, Stuart DI, Armitage JP. Using structural information to change the phosphotransfer specificity of a two-component chemotaxis signalling complex. PLoS Biol. 2010;8:e1000306. doi: 10.1371/journal.pbio.1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilwes AM, Alex LA, Crane BR, Simon MI. Structure of CheA, a signal-transducing histidine kinase. Cell. 1999;96:131–141. doi: 10.1016/S0092-8674(00)80966-6. [DOI] [PubMed] [Google Scholar]
- Borkovich KA, Kaplan N, Hess JF, Simon MI. Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc Natl Acad Sci USA. 1989;86:1208–1212. doi: 10.1073/pnas.86.4.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT. X-PLOR, version 3.1. New Haven: Yale University Press; 1992. [Google Scholar]
- Cantwell BJ, Manson MD. Protein domains and residues involved in the CheZ/CheAS interaction. J Bacteriol. 2009;191:5838–5841. doi: 10.1128/JB.00280-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic sidechain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J Magn Reson Ser B. 1993;101:114–119. doi: 10.1006/jmrb.1993.1019. [DOI] [Google Scholar]
- Hao S, Hamel D, Zhou H, Dahlquist FW. Structural basis for the localization of the chemotaxis phosphatase CheZ by CheAS. J Bacteriol. 2009;191:5842–5844. doi: 10.1128/JB.00323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JF, Oosawa K, Kaplan N, Simon MI. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell. 1988;53:79–87. doi: 10.1016/0092-8674(88)90489-8. [DOI] [PubMed] [Google Scholar]
- Ikegami T, Okada T, Ohki I, Harayama J, Mizuno T, Shirakawa M. Solution structure and dynamic character of the histidine-containing phosphotransfer domain of anaerobic sensor kinase ArcB from Escherichia coli. Biochemistry. 2001;40:375–386. doi: 10.1021/bi001619g. [DOI] [PubMed] [Google Scholar]
- Kato M, Mizuno T, Shimizu T, Hakoshima T. Insights into multistep phosphorelay from the crystal structure of the C-terminal HPt domain of ArcB. Cell. 1997;88:717–723. doi: 10.1016/S0092-8674(00)81914-5. [DOI] [PubMed] [Google Scholar]
- Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- Kay LE, Keifer EP, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–10665. doi: 10.1021/ja00052a088. [DOI] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14(51–5):29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. ANSIG: a program for the assignment of protein 1H 2D NMR spectra by interactive graphics. J Magn Reson. 1989;84:627–633. doi: 10.1016/0022-2364(89)90130-3. [DOI] [Google Scholar]
- Kraulis PJ, Domaille PJ, Campbell-Burk SL, Van Aken T, Laue ED. Solution structure and dynamics of ras p21. GDP determined by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry. 1994;33:3515–3531. doi: 10.1021/bi00178a008. [DOI] [PubMed] [Google Scholar]
- Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules I. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. doi: 10.1021/ja00381a009. [DOI] [Google Scholar]
- Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules II. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. doi: 10.1021/ja00381a010. [DOI] [Google Scholar]
- Logan TM, Olejniczak ET, Xu RX, Fesik SW. A general method for assigning NMR spectra of denatured proteins using 3D HC(CO)NH-TOCSY triple resonance experiments. J Biomol NMR. 1993;3:225–231. doi: 10.1007/BF00178264. [DOI] [PubMed] [Google Scholar]
- Mandel AM, Akke M, Palmer AG., III Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- Montelione GT, Lyons BA, Emerson SD, Tashiro M. An efficient triple resonance experiment using C-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J Am Chem Soc. 1992;114:10974–10975. doi: 10.1021/ja00053a051. [DOI] [Google Scholar]
- Mourey L, Da Re S, Pedelacq JD, Tolstykh T, Faurie C, Guillet V, Stock JB, Samama JP. Crystal structure of the CheA histidine phosphotransfer domain that mediates response regulator phosphorylation in bacterial chemotaxis. J Biol Chem. 2001;276:31074–31082. doi: 10.1074/jbc.M101943200. [DOI] [PubMed] [Google Scholar]
- Muhandiram DR, Kay LE. An enhanced-sensitivity pure absorption gradient 4D 15N, 13C-edited NOESY experiment. J Magn Reson Ser B. 1994;103:203–216. doi: 10.1006/jmrb.1994.1032. [DOI] [Google Scholar]
- Muhandiram DR, Guang YX, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J Biomol NMR. 1993;3:463–470. doi: 10.1007/BF00176011. [DOI] [Google Scholar]
- Nilges M, Clore GM, Gronenborn AM. Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical simulated annealing calculations. FEBS Lett. 1988;239:129–136. doi: 10.1016/0014-5793(88)80559-3. [DOI] [PubMed] [Google Scholar]
- O’Connor C, Matsumura P, Campos A. The CheZ binding interface of CheAS is located in alpha-helix E. J Bacteriol. 2009;191:5845–5848. doi: 10.1128/JB.00294-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG, III, Rance M, Wright PE. Intramolecular motions of a zinc finger DNA-binding domain from xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J Am Chem Soc. 1991;113:4371–4380. doi: 10.1021/ja00012a001. [DOI] [Google Scholar]
- Quezada CM, Gradinaru C, Simon MI, Bilwes AM, Crane BR. Helical shifts generate two distinct conformers in the atomic resolution structure of the CheA phosphotransferase domain from Thermotoga maritime. J Mol Biol. 2004;341:1283–1294. doi: 10.1016/j.jmb.2004.06.061. [DOI] [PubMed] [Google Scholar]
- Song HK, Lee JY, Lee MG, Moon J, Min K, Yang JK, Suh SW. Insights into eukaryotic multistep phosphorelay signal transduction revealed by the crystal structure of ypd1p from Saccharomyces cerevisiae. J Mol Biol. 1999;293:753–761. doi: 10.1006/jmbi.1999.3215. [DOI] [PubMed] [Google Scholar]
- Sourjik V. Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 2004;12:569–576. doi: 10.1016/j.tim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- Vuister GW, Bax A. Resolution enhancement and spectral editing of uniformly 13C enriched proteins by homonuclear broadband 13C–13C decoupling. J Magn Reson. 1992;98:428–435. doi: 10.1016/0022-2364(92)90144-V. [DOI] [Google Scholar]
- Vuister GW, Bax V. Quantitative J correlation: a new approach for measuring homonuclear three bond J(HNHa) coupling constants in 15N-enriched proteins. J Am Chem Soc. 1993;115:7772–7777. doi: 10.1021/ja00070a024. [DOI] [Google Scholar]
- Vuister GW, Clore GM, Gronenborn AM, Powers R, Garrett DS, Tschudin R, Bax A. Increased resolution and improved spectral quality in four-dimensional 13C/13C separated HMQC-NOESY-HMQC spectra using pulsed field gradients. J Magn Reson Ser B. 1993;101:210–213. doi: 10.1006/jmrb.1993.1035. [DOI] [Google Scholar]
- Xu Q, West AH. Conservation of structure and function among histidine-containing phosphotransfer (HPt) domains as revealed by the crystal structure of YPD1. J Mol Biol. 1999;292:1039–1050. doi: 10.1006/jmbi.1999.3143. [DOI] [PubMed] [Google Scholar]
- Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- Zhou H, Lowry DF, Swanson RV, Simon MI, Dahlquist FW. NMR studies of the phosphotransfer domain of the histidine kinase CheA from Escherichia coli: assignments, secondary structure, general fold, and backbone dynamics. Biochemistry. 1995;34:13858–13870. doi: 10.1021/bi00042a018. [DOI] [PubMed] [Google Scholar]