

Abstract
Heart rate variability (HRV), the quantification of beat-to-beat variability, has been studied as a potential prognostic marker in inflammatory diseases such as sepsis. HRV normally reflects significant levels of variability in homeostasis, which can be lost under stress. Much effort has been placed in interpreting HRV from the perspective of quantitatively understanding how stressors alter HRV dynamics, but the molecular and cellular mechanisms that give rise to both homeostatic HRV and changes in HRV have received less focus. Here, we develop a mathematical model of human endotoxemia that incorporates the oscillatory signals giving rise to HRV and their signal transduction to the heart. Connections between processes at the cellular, molecular, and neural levels are quantitatively linked to HRV. Rhythmic signals representing autonomic oscillations and circadian rhythms converge to modulate the pattern of heartbeats, and the effects of these oscillators are diminished in the acute endotoxemia response. Based on the semimechanistic model developed herein, homeostatic and acute stress responses of HRV are studied in terms of these oscillatory signals. Understanding the loss of HRV in endotoxemia serves as a step toward understanding changes in HRV observed clinically through translational applications of systems biology based on the relationship between biological processes and clinical outcomes.
Keywords: systems biology, inflammation, circadian
heart rate variability (HRV) is generally defined as the quantification of the distribution of time intervals between successive heartbeats. Reduction in HRV, a manifestation of altered autonomic function under stress, is potentially a useful predictor of outcome in myocardial infarction (42), congestive heart failure (63), diabetic neuropathy (58), and neonatal sepsis (46). Diminished HRV has also been observed in critically ill patients in intensive care units (ICU) (52), which motivates interest in HRV as a critical variable in the recovery from critical illness (50). Due to this clinical relevance, dynamic characteristics of HRV have been assessed by time domain, frequency domain (76), and nonlinear metrics (46, 60). The majority of HRV research has thus far focused on the interpretation of the patterns of HRV (45) rather than linking cellular-level mechanisms to patterns (11). The realization that health may be characterized by a certain degree of variability of human heart signals motivates the hypothesis that appropriate physiological variability is the manifestation of robust dynamics of control signals whose fluctuations equip the host with the ability to anticipate external and internal disturbances. We hypothesize that these variable dynamics are driven by the convergence of rhythmic physiological signals on the heart via autonomic modulation.
Studying the effects of critical illness on HRV requires a clinical model that can be experimentally evaluated in great detail. Human endotoxemia, the injection of lipopolysaccharides (LPS or endotoxin, used interchangeable herein) into healthy human subjects, has been extensively used as a model of systemic inflammation due to qualitatively similar responses in systemic physiological and metabolic processes, including changes in leukocyte abundance and behavior, hormonal secretion, and cardiac function (48). Responses observed in human endotoxemia experiments mimic observed responses in systemic inflammation in ICU patients, albeit over different timescales (30), thus making the human endotoxemia model an excellent platform for exploring mechanistic underpinnings of the systemic inflammatory response. A key component in the response to endotoxemia is a decrease in HRV, concomitant with imbalances in autonomic activity reflected by perturbed autonomic oscillatory responses in heart rate (HR) (28, 35, 36, 65).
Despite an understanding of the importance of inflammation in a wide variety of disorders and a large number of experiments elucidating the details of the inflammatory response, novel treatments aimed at controlling inflammation remain elusive (25). The complexity of the interacting, redundant pathways involved in the inflammatory response necessitates a systems-level understanding of inflammation (72, 80), thus leading to interest in the inflammatory response from a systems biology perspective (79). The dynamic signals evoked in an inflammatory response are propagated to the sinoatrial (SA) node of the heart to assess how HRV is perturbed in endotoxemia. Previously, endotoxemia-induced changes in HR and HRV have been described by physicochemical relations that begin to elucidate the signals that give rise to altered phenotypes (22, 23). However, this neglects that HR and HRV are both derived from the same physiological process, the beats of the heart, and that the contraction of the heart as initiated by firing neurons at the SA node is a noisy, discrete process. This motivates the development of a more mechanistic model to produce discrete heartbeat signals that can then be used to calculate HR and HRV, providing a basis for the development of autonomic dysfunction in endotoxemia. This paper proposes a semimechanistic mathematical model linking endotoxemia to cardiac function through an integral pulse frequency modulation (IPFM) model (7) that produces discrete heartbeats as output based on autonomic modulation of the heart. Variability is considered both at high frequencies (autonomic oscillations) and much lower frequencies (circadian rhythms). Outputs of the model, namely HR and HRV, are shown to accurately capture experimentally observed phenomena in human endotoxemia studies. Furthermore, the links between autonomic activity and cardiac function are explored, as well as how these communication links are affected by acute stress. Understanding the loss of variability of cardiac function in endotoxemia serves as a step toward gaining insight into similar changes in HRV observed clinically in response to stress (30). It is important to consider how the communication between the autonomic nervous system and the heart in endotoxemia (68) will affect both measureable parameters of HRV and the mechanistic underpinnings that give rise to altered cardiac function. Thus, connections between processes at the cellular, molecular, and neural levels are quantitatively linked to HRV. This work builds toward translational applications of systems biology (24, 82) by moving toward an understanding of the relationship between fundamental biological processes and clinical outcomes.
METHODS
Human endotoxemia model.
Bacterial endotoxin, a component of the outer cell membrane of gram-negative bacteria, is an important mediator in the pathophysiology of gram-negative bacterial sepsis (57). This complex macromolecule induces its injurious effects by a noncytotoxic interaction with CD14-bearing inflammatory cells, such as macrophage-monocytes, circulating neutrophils, and lung epithelial cells. These effector cells are activated through a family of Toll-like receptors and subsequently release a network of inflammatory products. These host-derived mediators function in concert to induce the systemic inflammatory response syndrome (59) leading to a variety of clinical disorders, including adult respiratory distress syndrome (ARDS) (8, 51). Elective administration of endotoxin to otherwise healthy human volunteers has been used to study systemic inflammation and gain insight into behavior of inflammatory mediators encountered in acute, as well as chronic, inflammatory disease. Human endotoxemia precipitates signs and symptoms characteristic of clinical sepsis (6, 48) and ARDS (15), inducing a reduction in HRV (28, 65). While we do not argue that the human endotoxin (LPS) challenge model precisely replicates an acute infectious or sepsis condition, human endotoxin challenge does serve as a useful model of Toll-like receptor 4 (TLR4) agonist-induced systemic inflammation by providing a reproducible experimental platform tying systemic inflammation to physiological signal generation and alterations in HRV. As an example, it has recently been demonstrated that LPS challenge induces transient dynamic changes in leukocyte gene expression similar to day 1 trauma patients (73).
In an effort to establish quantitative relationships among the components involved in endotoxemia, we developed a mathematical model of human endotoxemia (20, 21). A detailed description of the components of the mathematical model is given in the appendix, and the network structure is displayed in Fig. 1. At the cellular level, recognition of LPS by TLR4 on immune cells leads to the activation of the NF-κB pathway and ultimately the production of both proinflammatory (P) and anti-inflammatory (A) cytokines, which are proximal mediators of the systemic inflammatory response (56) and antagonistically work towards the self-regulation and resolution of inflammation. At the neuroendocrine level, the hypothalamic-pituitary-adrenal axis and the sympathetic nervous system (SNS) are the primary stress response pathways by which the central nervous system (CNS) regulates the immune response (74). This was modeled by assuming that the production and release of counterregulatory anti-inflammatory endogenous hormones cortisol (F) and epinephrine (EPI) respond to proinflammatory cytokines, and then these hormones feed back to modulate the transcriptional response in leukocytes. In addition, circadian rhythms in model components, both at the level of immune cells and CNS activity, were considered by accounting for diurnal patterns in the release of the hormones cortisol and melatonin (M), which then propagate their circadian rhythmicity to other variables (70).
Fig. 1.

Network structure. At the cellular level, LPS is recognized by its receptor, activating the NF-κB signaling cascade that provokes a significant transcriptional response consisting primarily of proinflammatory (P) and anti-inflammatory (A) signaling as well as a decrease in cellular bioenergetic processes (E). Neuroendocrine-immune cross talk results in the secretion of stress hormones cortisol (F) and epinephrine (EPI), which serve as immunoregulatory branches of the central nervous system. They are also centrally regulated to obey circadian dynamics. Finally, these signals propagate to the heart, where heart rate (HR) and heart rate variability (HRV) are modulated in a systemic inflammatory response.
We previously explored the principles of the Warner model (83) to describe the influences of the antagonistic relationship between the sympathetic and parasympathetic branches of the autonomic nervous system on the firing at the SA node of the heart (23). Autonomic activity at the SA node of the heart can be inferred based on blood epinephrine concentration (23), which has a circadian pattern with a peak during the middle of the day, slightly lagging the diurnal behavior of cortisol (17, 44, 70). A1 (Eq. 1a) represents the neurotransmitter concentration at the SNS nerve ending, which is associated with blood norepinephrine concentration and is assumed to be similarly responsive to endotoxemia as epinephrine (69). Plasma norepinephrine ultimately influences the local concentration at the SA node as described by A2 (Eq. 1b). This produces antagonistic changes in effective local sympathetic (Eq. 1c) and parasympathetic (Eq. 1d) activity. These relationships were used to develop a physicochemical model of the effect of endotoxemia on HR (23).
| (1a) |
| (1b) |
| (1c) |
| (1d) |
When combining Eq. 1 with our circadian model (70), described above and in the appendix, we now observe that the effective sympathetic (Tsym) and parasympathetic (Tpar) modulation of HR and HRV exhibit diurnal patterns as imposed by central circadian regulation. Experiments measuring muscle sympathetic nerve activity show that it is responsive to light (67) and that during sleep, sympathetic activity decreases (26). Based on experimental evidence that parasympathetic activity can be estimated by respiratory sinus arrhythmia measured by the spectral analysis of HR, it has been shown that parasympathetic activity also follows circadian dynamics (14), as measured by both time domain (pNN50, the percentage of differences between adjacent normal-to-normal intervals that are >50 ms) and frequency domain metrics (13). These oscillatory dynamics, leading to short-term HRV and long-term circadian rhythms in HR and HRV, have not yet been studied in a model that links autonomic activity to the beating of the heart within the context of an integrated model of inflammation. Below, variability in HR is studied in terms of these rhythmic signals through the development of a model linking the inflammatory response with alterations in the pattern of discrete heart beats.
Modeling autonomic influence on cardiac dynamics.
To describe how internal signals representing cellular and molecular processes responsive to endotoxemia are propagated to the heart, the oscillatory signals giving rise to variability in HR must first be accounted for. We hypothesize that the convergence of these variable autonomic signals, representing both circadian rhythms and higher frequency oscillations, gives rise to the characteristic patterns of variability in HR. Thus, the first step toward developing a more mechanistic model of cardiac function in endotoxemia is describing the nature of autonomic regulation at the SA node of the heart. Three sources of oscillations are considered: sympathetic and parasympathetic oscillations and circadian rhythms.
HRV is typically calculated based on a series of RR intervals, which are generated from electrocardiogram (ECG) signals by measuring the time interval between successive R waves. In the frequency domain of RR intervals, the power spectrum is typically divided into two frequency bands: low frequency (LF, 0.04–0.15 Hz) and high frequency (HF, 0.15–0.4 Hz). While the precise autonomic underpinnings of HF and LF power are unclear and likely indirect (41), HF is related to vagal activity and LF responds to changes in both vagal and sympathetic tone; thus, the ratio LF/HF may give some insight into the relative autonomic control of HR. Incorporating higher-frequency oscillations in autonomic modulation of HR allows for the production of a more biologically realistic heartbeat signal.
Long-term circadian oscillations in autonomic activity at the SA node influence the diurnal pattern of heartbeats. Sympathetic activity increases HR, while parasympathetic activity decreases HR; therefore, combining models that represent autonomic activity in inflammation (23) and circadian rhythms in inflammation (70) generates variables reflecting circadian rhythms in sympathetic and parasympathetic activity at the SA node. These circadian autonomic activities lead to diurnal patterns in both HR and HRV (33, 43, 53). Circadian rhythms are hypothesized to express sympathetic activity as proportional to autonomic modulation and parasympathetic activity as inversely proportional. Circadian rhythms are included to represent autonomic influences on the SA node, specifically slowly evolving circadian rhythms.
The inclusion of autonomic activity through a modified Warner-type model (Eq. 1), stimulated by central hormonal circadian rhythms allow for the assessment of changes in autonomic control of HRV (16). When sympathetic activity increases, the SA autonomic modulation is expected to increase, corresponding to more frequent firing and thus higher HR. When parasympathetic activity increases, the opposite occurs and HR decreases. Our model aims to introduce circadian variability in the autonomic modulation of the SA node, through the connections to our endotoxemia model via Tsym (Eq. 1c) and Tpar (Eq. 1d), which are ultimately linked to circadian rhythms in HR that match well with experimental data showing that HR peaks during the day and is lower at night (53).
HF and LF power have been observed to exhibit circadian rhythms under normal conditions (33, 43, 53). In human endotoxemia, HF and LF power both decrease acutely before recovering (28, 35, 36). Both circadian and acute responses may be explained by the link between vagal activity and HF and LF oscillations. Suppressing vagal activity leads to decreases in both HF and LF power, contrary to the outdated view that HF reflects only vagal activity and LF reflects only sympathetic activity (76). Experimental data of circadian rhythms in HF and LF are in phase with the predicted circadian oscillations in Tpar in Fig. 2. Furthermore, in human endotoxemia, Tpar decreases to reflect diminished parasympathetic activity during the acute systemic inflammatory response (23). Thus, we hypothesize that the variable amplitudes of HF and LF oscillations are governed by parasympathetic activity.
Fig. 2.

Circadian rhythms in the effective sympathetic and parasympathetic activity (Tsym and Tpar) at the sinus node of the heart. Diurnal rhythms from the circadian release of cortisol propagate through epinephrine, ultimately influencing Tsym and Tpar, which oscillate out of phase in homeostasis.
HF and LF oscillations are assumed to contribute in an additive manner to SA node autonomic modulation as two sinusoids. In Ref. 10, HF and LF oscillations are similarly modeled as sinusoids with frequencies 0.334 and 0.025 Hz, respectively. The LF frequency is set so low (below the LF range) because it is meant to also allow for realistic changes in very low frequency (<0.05 Hz) activity. However, this impedes the direct calculation of LF and HF powers because the peak in the power spectrum is so narrow that it hardly influences the power in the LF frequency band. Therefore, in Eq. 2, the mean values of the frequencies in the HF and LF bands were used. Then, the peaks in the power spectrum fall directly within the HF and LF bands, facilitating the use of standard methods of calculating HF and LF power to study model output. The amplitudes of the HF and LF sinusoids depend on Tpar, which produces homeostatic circadian rhythms in HRV (33, 43, 53) as well as acute suppression of HRV in endotoxemia (28, 35, 36).
The aforementioned assumptions are succinctly summarized in the model of Eq. 2. The effective autonomic modulation at the SA node depends on contributions including circadian and higher frequency modulation of the heart as well as a constant activity level which gives rise to the mean resting HR.
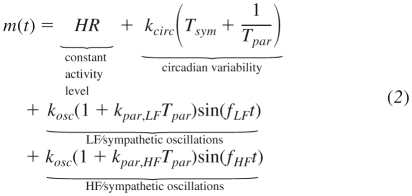 |
Generation of discrete heartbeats.
Autonomic activity influences the heart by modulating the pattern of discrete heartbeats by altering the concentration of neurotransmitters at the SA node. An idealized neuron functions by sensing local neurotransmitter concentration and, when that concentration crosses a threshold, the postsynaptic neuron fires. This type of neural-based discretization process occurs at the SA node of the heart, which normally initiates the electrical impulses that trigger contraction of cardiac tissue. As the SA node is innervated by both sympathetic and parasympathetic branches of the autonomic nervous system and the imbalance between these branches is critical to the loss of HRV, an ideal model would be one that dynamically controls the time interval of integration between successive firings based on autonomic activity as defined in Eq. 2.
A continuous signal can be converted to discrete events via an integrate-and-fire model in which the signal is repeatedly integrated until it reaches a threshold, thus signifying an event. One realization of an integrate-and-fire model that can discretize a continuous signal is an IPFM model. IPFM models allow for the translation of a continuous signal into a discrete series of events, conceptually similar to the behavior of a neuron (7). A continuous input signal m(t) represents modulation of neural firing, such as is defined in Eq. 2 to represent modulation at the SA node. Then, the times of firings are found by repeatedly integrating m(t) until a threshold Δ has been reached:
Δ is set to 1 in all simulations performed here.
This produces a vector t with elements tk to represent the kth discrete event. In our model of heartbeat generation, m(t) represents autonomic modulation of the heart and the discrete events produced through the IPFM model represent heartbeats initiated by the SA node (10, 16). HR is modulated by shifting the mean value of m(t) up (increased HR) or down (decreased HR). And because of variations in autonomic activity, the output of the IPFM model (heartbeats) will contain some variability. There are two primary mechanisms by which HRV is modulated through this model. Most directly, changes in HRV are driven by variable amplitudes of the HF and LF oscillators in Eq. 2. When the amplitude of these oscillators decreases, m(t) becomes more and more flat until there is very little beat-to-beat variability. However, even with constant amplitudes for HF and LF, HRV can still change because RR intervals become shorter as the mean value of m(t)shifts up due to circadian influences from Tsym and Tpar. Thus, the observed decrease in variability as assessed by HRV metrics is partially reflecting the changing mean value of HR (55). This initially may seem analogous to what is observed when sympathetic activity increases, such as in exercise where there seems to be an inverse relationship between HR and HRV (37). However, looking at the raw HR data in these cases makes it clear that the amplitude of oscillations in HR is lost in concert with increased mean HR. Therefore, if the amplitude of these oscillations, and thus HRV, is to be dynamic, it must be represented with a model that has the ability to alter the amplitude of oscillatory components, as in Eq. 2.
Calculation of HRV.
A variety of HRV parameters are assessed, spanning the time domain (SDNN, the standard deviation of normal-to-normal heartbeat intervals), frequency domain (HF, LF, and associated measures), and nonlinear analysis (sample entropy). All parameters are calculated over epochs that are 5 min in length, as is typical in the analysis of HR data (76). Each of the i epochs of RR intervals is denoted by RRi. The time domain measure, SDNN, is simply the standard deviation of interbeat intervals generated by the IPFM model, defined in Eq. 3.
| (3) |
The frequency domain statistics are calculated from mean-subtracted RR interval sequences based on the output of MATLAB's pyulear function with an order of 12, which implements an autoregressive model using the Yule-Walker algorithm to estimate the power spectral density. Then, HF and LF values represent the area under the curve in linear units over the appropriate frequency ranges of 0.15–0.4 and 0.04–0.15 Hz, respectively. HFn and LFn are normalized values, defined as HFn = HF/(HF + LF) and LFn = LF/(HF + LF). The LF/HF ratio is also computed.
Sample entropy (SampEn) (66) is calculated using the implementation available on PhysioNet (http://www.physionet.org/physiotools/sampen/). SampEn is defined as the negative natural logarithm of the estimated conditional probability that two subseries of m points that have all matched within a tolerance r continue to match within that tolerance at the next point. Therefore, a low value of SampEn means that the input series has a very regular structure, and high values correspond with high entropy, irregular signals.
Generation of Poincaré maps and their geometric properties.
Poincaré maps of RR intervals are generated for the scenarios described above. These plots are derived from a time series of RR intervals by plotting each value RR(i) on the x-axis versus its successive value RR(i+1) on the y-axis. Thus, if the system generated two consecutive RR intervals that were identical, that point would lie directly on the 45° diagonal. Variability in the Poincaré map can be quantified by calculating the standard deviation along this diagonal line and perpendicular to the diagonal line. These values, called SD1 and SD2, are visualized by plotting an ellipse whose axes are equal to SD1 and SD2. SD1 and SD2 have been used to roughly represent short-term and long-term variability in HR due to their intuitive, geometric interpretations (9).
RESULTS
Circadian and higher-frequency variability in autonomic modulation at the SA node is taken into account in Eq. 2, allowing for simulation of homeostasis and the biologically rhythms present in homeostasis. Based on this, Fig. 3 shows the homeostatic model output. m(t) has a clear circadian pattern in Fig. 3A and also exhibits higher-frequency variability in Fig. 3B. Both the mean value and the amplitude of variability of m(t) are under diurnal regulation. Circadian rhythms in HR (Fig. 3C) and HRV as assessed by SampEn and SDNN (Fig. 3D) are present in model output. HF and LF power, visualized on the power spectra in Fig. 3, E and F, representing 00:00 and 12:00 respectively, also contain significant diurnal variability. The addition of beat-to-beat variability in RR intervals permits the visualization of the RR interval time series via the Poincaré maps in Fig. 4. Four maps are shown, evenly spaced throughout the day: 00:00, 06:00, 12:00, and 18:00. The inset in each figure is the state of Tpar at the time when the map is generated. The ellipses have axes equal to the standard deviations of the points along the diagonal and perpendicular to the diagonal (9). The mean value of the RR interval (roughly the center of the mass of points) moves, illustrating long-term variability due to circadian changes in the mean value of m(t). Local variability (roughly the spread of points) also undergoes significant changes throughout the day in Fig. 5 where the HF and LF values are plotted over time, exhibiting clear circadian patterns.
Fig. 3.

Autonomic modulation at the sinoatrial (SA) node of the heart, shown at 2 scales (A and B), leading to circadian rhythms in smoothed HR (C) and HRV (D), as assessed by time domain (SDNN, solid line) and nonlinear sample entropy (SampEn, dashed line) metrics. E: a power spectrum calculated from a 5 min window of RR intervals at 00:00. Two peaks, representing low frequency (LF) and high frequency (HF) oscillations, are present. LF and HF values are calculated as the area under this curve, 0.04–0.15 Hz for LF and 0.15–0.4 Hz for HF. F: how that these HF and LF values undergo significant changes throughout the daily circadian cycle.
Fig. 4.

Poincaré maps of RR intervals in homeostasis, at 00:00 (A), 06:00 (B), 12:00 (C), and 18:00 (D). Inset in each figure: the circadian pattern of Tpar and the region that was used to generate the Poincaré map. The ellipses represent the dispersion of points as the axes are equal to the standard deviation of points on each axis. The major and minor axes of the ellipses are drawn on the figure, representing the standard deviations along the y = x diagonal (SD1) and the y = −x diagonal (SD2). A large circadian pattern in the geometry of the Poincaré maps is observed, ranging from a maximum of (SD1, SD2) = (0.13, 0.15) in B to a minimum of (0.027, 0.046) in C.
Fig. 5.

LF and HF values are calculated, as shown in Fig. 3E, at many points throughout the simulation, and these values are plotted as functions of time. A: Circadian rhythms in LF and HF. B: normalized LF and HF [LFn = LF/(LF + HF), HFn = HF/(LF + HF)]. C: the LF/HF ratio. LF and HF are in phase, but their normalized values are out of phase.
An acute dose of LPS is given at 20:00, thus provoking a simulated systemic inflammatory response. As described in detail in the appendix, LPS is recognized by TLR4 on immune cells and instigates a wide range of transcriptional responses, including those that lead to the release of proinflammatory cytokines. These cytokines serve as mediators in neuroendocrine-immune communication, leading to the central release of stress hormones such as cortisol and catecholamines. Figure 6 shows how this acute disturbance propagates through the system, from proinflammatory mediators to anti-inflammatory hormones, finally leading to an increase in Tsym and a decrease in Tpar, which then provoke changes in cardiac dynamics in response to acute stress. In response to changes in Tsym and Tpar, the autonomic modulation of the SA node, m(t) as given in Eq. 2, is shifted up and the amplitudes of its higher-frequency oscillatory components are diminished in Fig. 6E. Figure 7 shows how LF and HF both decrease while the LF/HF ratio increases, in agreement with experimental data (28, 35, 36). The Poincaré maps displayed in Fig. 8 show a significant tightening that begins directly after LPS injection and reaches maximal tightening several hours later. Figure 7D shows the output of two HRV parameters, SDNN and SampEn. Both parameters capture the circadian pattern prior to LPS and both show an acute decrease after LPS treatment, but the decrease in SampEn is much larger than the decrease in SDNN relative to the normal circadian rhythms observed in each parameter. The above results concerning LPS all study the system response to a dose of LPS at the same time point. The computational model presented here allows for a more broad exploration of the circadian influence on the endotoxemia response, shown in Fig. 9 where LPS is given at 5:00 and 12:00, illustrating the maximum differences in responses as quantified by HRV.
Fig. 6.

Response to a dose of LPS given at 20:00. Changes propagate through proinflammatory cytokines secreted by immune cells (A) to neuroendocrine-mediated effects (epinephrine release in B; autonomic activation in C and D) to the activity of the heart, reflected by changes in effective autonomic modulation (E) and HR (F).
Fig. 7.

Changes in HRV in response to a dose of LPS given at 20:00. Both LF and HF are suppressed (A), but relative values (B) and the LF/HF ratio (C) show that HF is more strongly suppressed than LF. SampEn (dashed line) and SDNN (solid line) both decrease in response to LPS, but SampEn decreases more relative to the amplitude of its normal circadian rhythm (D).
Fig. 8.

Poincaré maps showing the response to a dose of LPS at 20:00, showing maps at 20:00 (A), 21:00 (B), 22:00 (C), and 01:00 (D). Inset in each figure: the circadian pattern of Tpar and the region that was used to generate the Poincaré map; in D, the next 24 h are shown. After injection, the points on the map shift down and to the left, reflecting decreased RR intervals and decreased HR. The points also become more tightly distributed, illustrating the loss of HRV in endotoxemia. D: the Poincaré map at 01:00, which is when HRV is most suppressed. The ellipses represent the dispersion of points as the axes are equal to the standard deviation of points on each axis. A change in the geometry of the Poincaré maps is observed, ranging from a maximum of (SD1, SD2) = (0.060, 0.082) at the time of injection in B to a minimum of (0.0060, 0.017) in C. The pre-LPS fitted ellipse from A is shown in C and D to illustrate the difference in both the mean and the distribution of points during the acute response.
Fig. 9.

There is a circadian dependence on the response of the model to a dose of LPS. The maximum difference is observed between LPS given at 5:00 and 12:00, HRV, as quantified by HRV.
To determine the model response to decoupling between the heart and the autonomic nervous system, the amplitude of HF and LF oscillations, kosc in Eq. 2, is halved. Figure 10 shows how SDNN changes under these conditions by showing a 24-h period of diminished kosc in between 1 day on each side of normal conditions. The amplitude of circadian rhythms in SDNN and the magnitude of SDNN are both diminished in the decreased coupling region.
Fig. 10.

Decoupling between the autonomic nervous system and the heart is simulated by decreasing coupling by 50% during the shaded area in the figure. Both the amplitude of circadian rhythms and the magnitude of HRV (assessed by SDNN) are diminished.
DISCUSSION
The components required to link neuroendocrine-immune interactions with circadian and higher-frequency autonomic variability in HR are combined through our proposed model (Eq. 2), which incorporates circadian control of cardiac function via autonomic activity along with HF and LF oscillations. While hormonal circadian rhythms alone produce circadian rhythms in model output, some higher-frequency oscillations are required to produce the local variability that is observed in real heartbeat signals. HF and LF power is dependent on vagal signaling, and more specifically it is known that HF and LF are diminished under human endotoxemia (28, 36). Thus, Eq. 2 represents the dependence of HF and LF oscillatory amplitudes on Tpar. Figure 3A displays a clear change in the shape of m(t) as the local oscillations have very different amplitudes depending on the position in the circadian cycle. Power spectra, taken at 00:00 and 12:00, respectively, which are close to times at which Tpar is at its maximum and minimum values, processed to calculate the HF and LF values shown in Fig. 3F. LF and HF show a significant drop in the area under the curve of the power spectrum over those frequency ranges throughout the normal diurnal cycle. These HF and LF powers, calculated more frequently, are shown in Fig. 5. Though raw LF and HF are in phase, HF is suppressed more strongly so that the normalized values LFn and HFn are actually out of phase (28, 36). A similar pattern is seen in Fig. 7, A–C, after an acute dose of LPS. Both LF and HF decrease, but the decrease in HF is more profound, so that the LFn increases relative to HFn. Thus, the LF/HF ratio remains elevated throughout the recovery phase, illustrating continued imbalance between sympathetic and parasympathetic modulation at the SA node (28, 36).
In Figs. 6 and 7, changes in Tsym, Tpar, and the HRV parameters are persistent for over 24 h after an LPS injection, while all other variables in the model recover to their baseline conditions within 24 h. Run for a longer period of time, these model variables recover within a couple days. Despite this, HRV parameters do recover to within the normal range of circadian variability within 24 h. In studies where LPS is given to humans and ECGs are recorded for 24 h as the inflammatory response is initiated and then resolves in a self-limited response, HRV has generally been observed to recover within 24 h postinjection (5).
The HF and LF sinusoids in Eq. 2 are assumed to mechanistically arise somewhere outside of the model. Biologically, LF and HF oscillations arise largely from vasomotor activity and respiration under control conditions, respectively (47). As m(t) represents only the autonomic modulation specifically at the SA node of the heart, the transduction of the signals producing HF and LF oscillations to the heart must be considered. So, the terms that modulate the amplitude of these sinusoids based on the level of Tpar represent the ability of the oscillatory signals to be reflected in neurotransmitter concentrations at the SA node, based on the observed relationship between vagal signaling and HF and LF components of HR. Vagal activity modulates LF and HF oscillations in HR, and without this vagal activity, LF and HF responses are blunted (41). In other words, the HF and LF peaks that appear in the power spectrum of HR depend on the autonomic nervous system to communicate these signals to the SA node. Thus, LF and HF give some indication as to the coupling between the heart and the autonomic nervous system, which is of particular interest as HR is relatively easy to assess noninvasively. Indeed, in endotoxemia, an increase in regularity is observed in HR, neutrophil function, and plasma cortisol levels (65), in line with theoretical expectations of the response of decoupled biological systems (62) and the results shown in Fig. 10, where variability is lost under decoupling. Although these results show an instant decoupling rather than a gradual process as likely occurs in vivo, decoupling may be important in adverse conditions such as endotoxemia when interorgan communication is diminished (27, 28). Clinically, assessing the interorgan communication by means such as evaluating HF and LF is critical to understanding and assessing the extent of injury in multiple organ dysfunction syndrome and sepsis (71). HF and LF most directly measure cardiac-autonomic coupling, but they can also be used as accessible proxies for measuring general interorgan communication (71). In addition, drugs that normally alter HR by autonomic modulation fail to have an effect in endotoxemia (68). Thus, the effect of endotoxemia on the heart can be viewed as a decoupling between the autonomic nervous system and the SA node. This decoupling represents a potential mechanism for the observed decreased complexity and increased regularity in physiological signals (12, 62). The recovery of HRV following injury can then be viewed as a recoupling of autonomic and cardiac systems and, more generally, a recoupling of organ systems in the recovery phase. The model presented here begins to decipher the nature of this relationship through the variable m(t), which represents the communication link between the autonomic nervous system and the heart.
One of the fundamental contributions of the described modeling work is the incorporation of circadian rhythms in both HR and HRV parameters. This is of particular interest due to the loss of circadian rhythms observed in inflammation (49) and the interplay between inflammatory mediators and molecular circadian machinery both centrally in the suprachiasmatic nucleus and in peripheral tissues (29, 70). The circadian dependence of this model is shown in Fig. 9 where identical doses of LPS are given at two different times, 0:500 and 12:00. These two times produce a maximal difference in responses, as the diurnal peak in cortisol occurs between these time points, and this primes the system for a robust anti-inflammatory response. Thus, before this hormonal priming occurs, a significantly larger decrease in HRV (quantified by SampEn) is observed. However, in both cases, the overall dynamics of the system (an acute response and recovery to baseline) are similar. These computational results match with clinical observations that sepsis patients are at elevated risk of mortality from 02:00 to 06:00, before the circadian peak in cortisol secretion (31). Considering this type of circadian dependence on responses to pathogens and also to therapies is important in optimizing treatment of inflammatory disease (32). In Fig. 6, circadian rhythms in cardiac function are blunted in response to a dose of LPS as the LPS-induced acute increase in Tsym and acute decrease in Tpar overwhelm the normal diurnal pattern of those variables. In Fig. 7A, the decrease in both LF and HF in response to endotoxemia is only slightly larger than the physiologic changes in LF and HF due to circadian rhythms. This is because, in the model, Tpar is predicted to pass relatively close to zero in its diurnal cycle, so in endotoxemia, there is not much further for it to fall. While this result may seem unintuitive, it matches with experimental results showing that the drop from maximum to minimum values during circadian rhythms in HF and LF (33, 43, 53), and the depression in HF and LF due to endotoxemia (28, 35, 36) can both be anywhere from 50 to 90%, depending on experimental protocol. However, by looking at Poincaré maps (compare Figs. 4 and 8) or by assessing HRV by other metrics such as SDNN and SampEn as in Fig. 7D, it is clear that there is a significant increase in regularity in response to LPS that is fundamentally different than what is observed in normal circadian patterns. This illustrates the importance of assessing multiple variability/regularity metrics to tease out subtle patterns in HR data.
An advantage of the model presented here, relative to a continuous physicochemical model of either HR or HRV, is that it produces discrete beats as output. This allows both HR and HRV to be derived from a single variable signal, as they are experimentally, and it allows for comparison of the performance of HRV metrics. Figure 7D is provocative in this regard as it shows SDNN and SampEn, two common HRV parameters, both are able to capture normal circadian dynamics as well as the acute response to LPS; however, when their axes are aligned such as in Fig. 7D so that the amplitude of circadian rhythms is equal for both parameters, it is clear that SampEn is much more significantly suppressed in endotoxemia. Thus, taking a more mechanistic approach that models heartbeats rather than attempting to directly estimate changes in HRV can lead to these types of quantitative differences being discovered.
The representation of neurotransmitter concentration at the SA node (23) is conceptually based on the Warner model (83) of sympathetic and vagal influences of HR. A similar idea is explored in the work of Chiu and Kao (16), in which an IPFM model is modulated by the vagal and sympathetic outputs of the Warner model. The work presented in this paper goes a step further by ultimately linking the model of autonomic modulation at the SA node with a larger, well-established model of human endotoxemia to explore changes in cardiac output specifically within this context. This introduces some additional complexity into m(t) since the amplitude of HF and LF oscillations depend on a nonlinear model, and also circadian influences are directly incorporated through Tsym and Tpar. In general, one cannot assume that this type of multimodal input signal will be effectively transduced through an IPFM model without the addition of significant distortion (54). However, the power spectra in Fig. 3 clearly show that the HF and LF frequency components are strongly present in the short-term variability of IPFM-generated HR.
By linking cardiac dynamics with a detailed model of the inflammatory response, we have begun to explore the mechanistic underpinnings that may underlie the relationship between autonomic dysfunction and modulated HR and HRV in endotoxemia and, by extension, possible decoupling among other organ systems. The mechanism-based approach (81) of the endotoxemia model allows for the future investigation of the relationship between neuroendocrine-immune state and cardiac function. This linking of processes at the molecular and cellular level with outcomes at the systemic level (namely clinically accessible variables such as HR and HRV) is an important step toward developing translational applications of systems biology.
APPENDIX
The model presented in this appendix has been iteratively developed over several publications. Below, the latest form of the model incorporates circadian rhythms (70) and autonomic modulation at the SA node of the heart (23). The unified model combining these two works has not been previously discussed in the literature.
Through the analysis of leukocyte gene expression data, the essential responses characterizing the leukocyte transcriptional dynamics are identified. Specifically, these responses, in the case of transient human endotoxemia, include 1) an early increase in proinflammatory signaling molecule production, 2) an anti-inflammatory response to counter proinflammatory signaling, and 3) an energetic response representing diminished cellular bioenergetic processes. These transcriptional responses are triggered by the activation of critical signaling cascades as a result of the recognition of the extracellular LPS signal. In the endotoxin injury model, the focus has been on NF-κB as the archetypical signaling module that regulates the expression of proinflammatory genes, as provoked by the binding of LPS to its receptor TLR4 (R) (Eq. 4a–d) leading to the activation of the NF-κB, which initiates the transcriptional response to inflammation. NF-κB is normally bound to IκB molecules, which inhibit its translocation to the nucleus, thus inactivating its role as a transcription factor. LPS via TLR4 and adapter molecules stimulates the activation of IKK, which phosphorylates IκBα leading, in turn, to ubiquitination and degradation of IκBα in the proteasome. Then, the free NF-κB can move into the nucleus and stimulate the transcription of a number of genes, including its inhibitor IκBα, thus creating a negative feedback loop. The NF-κB module is based on a reduced model of NF-κB dynamics that includes IKK (Eq. 4e), nuclear (activated) NF-κB (Eq. 4f), and IκBα (Eq. 4, g and h) (34), which allows the model to broadly capture the negative feedback regulatory behavior of NF-κB. The fundamental transcriptional processes found in the gene expression data are the proinflammatory (Eq. 4i), anti-inflammatory (Eq. 4j), and energetic (Eq. 4k) responses. Circadian production of inflammatory mediators is regulated by melatonin (Eq. 4l), which has shown to be correlated to a number of cytokines (61) and thus is used as a proxy for central circadian control of leukocyte transcriptional activity (70).
| (4a) |
| (4b) |
| (4c) |
| (4d) |
| (4e) |
| (4f) |
| (4g) |
| (4h) |
| (4i) |
| (4j) |
| (4k) |
| (4l) |
The interplay between the NF-κB pathway and the pro- and anti-inflammatory responses normally leads to a self-limited inflammatory response that resolves after LPS has been cleared, but high doses of LPS can lead to a state of persistent inflammation. Additionally, corticosteroids (both endogenous and exogenous) play a critical role in modulating the progression of inflammation and significant prior research has elucidated the mechanisms driving corticosteroid activity (1–4, 18, 39, 75, 85). Such studies simulate the pharmacodynamic action of glucocorticoids at the cellular level and the pharmacogenomic effect of glucocorticoids at the transcriptional level (30a, 38, 64). Corticosteroid pharmacodynamics include: 1) the binding of the corticosteroid to its cytosolic receptor; 2) the subsequent formation of the corticosteroid-receptor complex; and 3) the translocation of the cytosolic complex to the nucleus that alters the transcriptional machinery, activating or repressing numerous genes. This is modeled by equations governing the inflammation-induced production of cortisol along with the endogenous circadian pattern in cortisol secretion (Eq. 4m), transcription and translation of cytosolic glucocorticoid receptor (Eq. 4, n and o), and the intracellular dynamics as the signal is transduced from the cytoplasm (Eq. 4p) to the nucleus (Eq. 4q).
| (4m) |
| (4n) |
| (4o) |
| (4p) |
| (4q) |
Proinflammatory cytokines interact with neural-based pathways that modulate the progression of the immune response. As a result of the activation of neuroendocrine axis, anti-inflammatory hormones are secreted and recognized by immune cells. In the case of catecholamines, the secretion of catecholamines from SNS and adrenal medulla attenuates the proinflammatory manifestations of human endotoxemia, as evidenced by reduced TNF levels (78). The anti-inflammatory influence is mediated by intracellular cAMP signaling potentiating anti-inflammatory (IL-10) signaling (77, 78). Epinephrine is modeled as being secreted in response to stimulation by the proinflammatory response (19) and ultimately leads to increased anti-inflammatory signaling, as shown in Eq. 4, r–u. Cortisol produced in the adrenal cortex interacts with the adrenal medulla, ultimately stimulating epinephrine production (84). As experimental data shows that plasma epinephrine levels lag cortisol levels (17, 44), cortisol is modeled as stimulating epinephrine production, thereby producing a slightly delayed circadian peak in the baseline epinephrine profile.
| (4r) |
| (4s) |
| (4t) |
| (4u) |
These elements in Eq. 4, and the corresponding parameter values in Table 2, comprise a semimechanistic model of human endotoxemia, including physiologic diurnal rhythms (20, 21, 23, 70).
Table 1.
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| HR | 1 | kpar,HF | 1 |
| kosc | 0.05 | fHF | 0.275 |
| kpar,LF | 0.5 | kcirc | 0.04 |
| fLF | 0.105 |
Frequencies for the high frequency (HF) and low frequency (LF) bands are set to the mean value of the standard limits of those bands. The other parameters are set manually. In practice, the parameters in the integral pulse frequency modulation model would need to be tuned to an individual subject due to significant person-to-person variability in cardiac dynamics, such as the mean heart rate (HR) and the amplitude of circadian rhythms.
Table 2.
Model parameter values, as set in our previous publications
| Parameter | Value | Parameter | Value | Parameter | Value | Parameter | Value | Parameter | Value | Parameter | Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Kin,RM1 | 0.406 | KA,E | 0.534 | K2,REPI | 5.465 | K4 | 2.240 | KP,NFkBn | 29.741 | Kin,Tpar | 45.181 |
| Kin,RM2 | 0.032 | Kout,A | 0.810 | K3,EPIR | 5.546 | Kin,mRNA,R | 0.091 | KP,E | 9.051 | KTpar,Tsym | 9.756 |
| Kout,RM | 0.421 | KA,FRN | 0.401 | τ | 0.053 | KmRNA,R,P | 1.740 | Kout,P | 0.333 | Kout,Tpar | 4.201 |
| Kin,F1 | 0.992 | Kin,Fen | 0.843 | n | 5.509 | Kout,mRNA,R | 0.251 | Kin,E | 0.080 | Ksyn_Rm | 2.900 |
| KEPI,FRN | 0.090 | KFen,P | 0.256 | Kin,HRV | 1.185 | KNFkB,1 | 16.294 | KE,P | 2.210 | IC50_Rm | 26.200 |
| KP,M | 0.973 | Kout,F | 1.058 | Kout,HRV | 1.045 | KNFkB,2 | 1.186 | Kout,E | 0.257 | Kdeg | 0.112 |
| KA,M | 1.000 | Kin,EPI | 5.921 | Klps,1 | 4.500 | Kin,IkBa | 0.463 | Ka1 | 3.654 | Ksyn_R | 1.199 |
| TF1 | 12.082 | KEPI,P | 0.231 | Klps,2 | 6.790 | KIkBa,1 | 13.273 | Ka2 | 0.055 | rf | 0.490 |
| TF2 | 16.530 | Kout,EPI | 7.286 | Ksyn | 0.020 | Kout,IkBa | 0.463 | Ka3 | 2.927 | Kre | 0.570 |
| TM1 | 1.732 | K0REPI | 11.011 | K2 | 0.040 | KI,1 | 1.400 | KC | 11.286 | Kon | 0.003 |
| TM2 | 20.149 | K1,REPI | 3.006 | K1 | 3.000 | KI,2 | 0.870 | KTsym,Tpar | 7.764 | Kdgr_R | 0.057 |
| Kin,A | 0.461 | KREPI,EPI | 0.845 | K3 | 5.000 | Kin,P | 0.033 | Ka4 | 3.435 | KT | 0.630 |
| KA,cAMP | 0.145 |
GRANTS
J. D. Scheff, P. D. Mavroudis, and I. P. Androulakis acknowledge support from National Institute of General Medical Sciences (NIGMS) Grant GM-082974. J. D. Scheff, S. E. Calvano, and S. F. Lowry are supported, in part, from NIGHMS Grant GM-34695.
DISCLOSURES
No conflicts of interest (financial or otherwise) are declared by the author(s).
REFERENCES
- 1. Almon RR, DuBois DC, Brandenburg EH, Shi W, Zhang S, Straubinger RM, Jusko WJ. Pharmacodynamics and pharmacogenomics of diverse receptor-mediated effects of methylprednisolone in rats using microarray analysis. J Pharmacokinet Pharmacodyn 29: 103–129, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Almon RR, Dubois DC, Jin JY, Jusko WJ. Pharmacogenomic responses of rat liver to methylprednisolone: an approach to mining a rich microarray time series. Aaps J 7: E156–E194, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Almon RR, DuBois DC, Jusko WJ. A microarray analysis of the temporal response of liver to methylprednisolone: a comparative analysis of two dosing regimens. Endocrinology 148: 2209–2225, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Almon RR, Lai W, DuBois DC, Jusko WJ. Corticosteroid-regulated genes in rat kidney: mining time series array data. Am J Physiol Endocrinol Metab 289: E870–E882, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alvarez SM, Katsamanis Karavidas M, Coyle SM, Lu SE, Macor M, Oikawa LO, Lehrer PM, Calvano SE, Lowry SF. Low-dose steroid alters in vivo endotoxin-induced systemic inflammation but does not influence autonomic dysfunction. J Endotoxin Res 13: 358–368, 2007 [DOI] [PubMed] [Google Scholar]
- 6. Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem 15: 1697–1705, 2008 [DOI] [PubMed] [Google Scholar]
- 7. Bayly EJ. Spectral analysis of pulse frequency modulation in the nervous systems. IEEE Trans Biomed Eng 15: 257–265, 1968 [DOI] [PubMed] [Google Scholar]
- 8. Bayston KF, Cohen J. Bacterial endotoxin and current concepts in the diagnosis and treatment of endotoxaemia. J Med Microbiol 31: 73–83, 1990 [DOI] [PubMed] [Google Scholar]
- 9. Brennan M, Palaniswami M, Kamen P. Do existing measures of Poincare plot geometry reflect nonlinear features of heart rate variability? IEEE Trans Biomed Eng 48: 1342–1347, 2001 [DOI] [PubMed] [Google Scholar]
- 10. Brennan M, Palaniswami M, Kamen P. Poincare plot interpretation using a physiological model of HRV based on a network of oscillators. Am J Physiol Heart Circ Physiol 283: H1873–H1886, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Buchman TG. The digital patient: predicting physiologic dynamics with mathematical models. Crit Care Med 37: 1167–1168, 2009 [DOI] [PubMed] [Google Scholar]
- 12. Buchman TG. Physiologic stability and physiologic state. J Trauma 41: 599–605, 1996 [DOI] [PubMed] [Google Scholar]
- 13. Burger AJ, Charlamb M, Sherman HB. Circadian patterns of heart rate variability in normals, chronic stable angina and diabetes mellitus. Int J Cardiol 71: 41–48, 1999 [DOI] [PubMed] [Google Scholar]
- 14. Burgess HJ, Trinder J, Kim Y, Luke D. Sleep and circadian influences on cardiac autonomic nervous system activity. Am J Physiol Heart Circ Physiol 273: H1761–H1768, 1997 [DOI] [PubMed] [Google Scholar]
- 15. Buttenschoen K, Kornmann M, Berger D, Leder G, Beger HG, Vasilescu C. Endotoxemia and endotoxin tolerance in patients with ARDS. Langenbecks Arch Surg 393: 473–478, 2008 [DOI] [PubMed] [Google Scholar]
- 16. Chiu HW, Kao T. A mathematical model for autonomic control of heart rate variation. IEEE Eng Med Biol Mag 20: 69–76, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Dimitrov S, Benedict C, Heutling D, Westermann J, Born J, Lange T. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood 113: 5134–5143, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DuBois DC, Xu ZX, McKay L, Almon RR, Pyszcznski N, Jusko WJ. Differential dynamics of receptor down-regulation and tyrosine aminotransferase induction following glucocorticoid treatment. J Steroid Biochem Mol Biol 54: 237–243, 1995 [DOI] [PubMed] [Google Scholar]
- 19. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52: 595–638, 2000 [PubMed] [Google Scholar]
- 20. Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. In silico simulation of corticosteroids effect on an NFkB-dependent physicochemical model of systemic inflammation. PLoS One 4: e4706, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. Modeling endotoxin-induced systemic inflammation using an indirect response approach. Math Biosci 217: 27–42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. Multiscale model for the assessment of autonomic dysfunction in human endotoxemia. Physiol Genomics 42: 5–19, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. A physiological model for autonomic heart rate regulation in human endotoxemia. Shock 35: 229–239, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. Translational potential of systems-based models of inflammation. Clin Transl Sci 2: 85–89, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Freeman BD, Natanson C. Anti-inflammatory therapies in sepsis and septic shock. Expert Opin Investig Drugs 9: 1651–1663, 2000 [DOI] [PubMed] [Google Scholar]
- 26. Gherghel D, Hosking SL, Orgul S. Autonomic nervous system, circadian rhythms, and primary open-angle glaucoma. Surv Ophthalmol 49: 491–508, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Godin PJ, Buchman TG. Uncoupling of biological oscillators: a complementary hypothesis concerning the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med 24: 1107–1116, 1996 [DOI] [PubMed] [Google Scholar]
- 28. Godin PJ, Fleisher LA, Eidsath A, Vandivier RW, Preas HL, Banks SM, Buchman TG, Suffredini AF. Experimental human endotoxemia increases cardiac regularity: results from a prospective, randomized, crossover trial. Crit Care Med 24: 1117–1124, 1996 [DOI] [PubMed] [Google Scholar]
- 29. Haimovich B, Calvano J, Haimovich AD, Calvano SE, Coyle SM, Lowry SF. In vivo endotoxin synchronizes and suppresses clock gene expression in human peripheral blood leukocytes. Crit Care Med 38: 751–758, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haimovich B, Reddell MT, Calvano JE, Calvano SE, Macor MM, Coyle SM, Lowry SF. A novel model of common Toll-like receptor 4- and injury-induced transcriptional themes in human leukocytes. Crit Care 14: R177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30a. Hazra A, Pyszczynski N, DuBois DC, Almon RA, Jusko WJ. Modeling receptor/gene-mediated effects of corticosteroids on hepatic tyrosine aminotransferase dynamics in rats: dual regulation by endogenous and exogenous corticosteroids. J Pharmacokin Pharmacodyn 34: 643–667, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hrushesky WJM, Langevin T, Kim YJ, Wood PA. Circadian dynamics of tumor-necrosis-factor-alpha (cachectin) lethality. J Exp Med 180: 1059–1065, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hrushesky WJM, Wood PA. Circadian time structure of septic shock: Timing is everything. J Infect Dis 175: 1283–1284, 1997 [PubMed] [Google Scholar]
- 33. Huikuri HV, Niemela MJ, Ojala S, Rantala A, Ikaheimo MJ, Airaksinen KE. Circadian rhythms of frequency domain measures of heart rate variability in healthy subjects and patients with coronary artery disease. Effects of arousal and upright posture. Circulation 90: 121–126, 1994 [DOI] [PubMed] [Google Scholar]
- 34. Ihekwaba AE, Broomhead DS, Grimley RL, Benson N, Kell DB. Sensitivity analysis of parameters controlling oscillatory signalling in the NF-kappaB pathway: the roles of IKK and IkappaBalpha. Syst Biol (Stevenage) 1: 93–103, 2004 [DOI] [PubMed] [Google Scholar]
- 35. Jan BU, Coyle SM, Macor MA, Reddell M, Calvano SE, Lowry SF. Relationship of basal heart rate variability to in vivo cytokine responses after endotoxin exposure. Shock 33: 363–368, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jan BU, Coyle SM, Oikawa LO, Lu SE, Calvano SE, Lehrer PM, Lowry SF. Influence of acute epinephrine infusion on endotoxin-induced parameters of heart rate variability: a randomized controlled trial. Ann Surg 249: 750–756, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Javorka M, Zila I, Balharek T, Javorka K. Heart rate recovery after exercise: relations to heart rate variability and complexity. Braz J Med Biol Res 35: 991–1000, 2002 [DOI] [PubMed] [Google Scholar]
- 38. Jin JY, Almon RR, DuBois DC, Jusko WJ. Modeling of corticosteroid pharmacogenomics in rat liver using gene microarrays. J Pharmacol Exp Ther 307: 93–109, 2003 [DOI] [PubMed] [Google Scholar]
- 39. Jusko WJ. Receptor-mediated pharmacodynamics of corticosteroids. Prog Clin Biol Res 387: 261–270, 1994 [PubMed] [Google Scholar]
- 41. Karemaker JM. Autonomic integration: the physiological basis of cardiovascular variability. J Physiol 517: 316, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kleiger RE, Miller JP, Bigger JT, Jr, Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol 59: 256–262, 1987 [DOI] [PubMed] [Google Scholar]
- 43. Korpelainen JT, Sotaniemi KA, Huikuri HV, Myllyla VV. Circadian rhythm of heart rate variability is reversibly abolished in ischemic stroke. Stroke 28: 2150–2154, 1997 [DOI] [PubMed] [Google Scholar]
- 44. Kronfol Z, Nair M, Zhang Q, Hill EE, Brown MB. Circadian immune measures in healthy volunteers: relationship to hypothalamic-pituitary-adrenal axis hormones and sympathetic neurotransmitters. Psychosom Med 59: 42–50, 1997 [DOI] [PubMed] [Google Scholar]
- 45. Lahiri MK, Kannankeril PJ, Goldberger JJ. Assessment of autonomic function in cardiovascular disease: physiological basis and prognostic implications. J Am Coll Cardiol 51: 1725–1733, 2008 [DOI] [PubMed] [Google Scholar]
- 46. Lake DE, Richman JS, Griffin MP, Moorman JR. Sample entropy analysis of neonatal heart rate variability. Am J Physiol Regul Integr Comp Physiol 283: R789–R797, 2002 [DOI] [PubMed] [Google Scholar]
- 47. Lombardi F, Malliani A, Pagani M, Cerutti S. Heart rate variability and its sympatho-vagal modulation. Cardiovasc Res 32: 208–216, 1996 [DOI] [PubMed] [Google Scholar]
- 48. Lowry SF. Human endotoxemia: a model for mechanistic insight and therapeutic targeting. Shock 24, Suppl 1: 94–100, 2005 [DOI] [PubMed] [Google Scholar]
- 49. Lowry SF. The stressed host response to infection: the disruptive signals and rhythms of systemic inflammation. Surg Clin North Am 89: 311–326 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lowry SF, Calvano SE. Challenges for modeling and interpreting the complex biology of severe injury and inflammation. J Leukoc Biol 83: 553–557, 2008 [DOI] [PubMed] [Google Scholar]
- 51. Miyata T, Torisu M. Plasma endotoxin levels and functions of peripheral granulocytes in surgical patients with respiratory distress syndrome. Jpn J Surg 16: 412–417, 1986 [DOI] [PubMed] [Google Scholar]
- 52. Morris JA, Jr, Norris PR, Waitman LR, Ozdas A, Guillamondegui OD, Jenkins JM. Adrenal insufficiency, heart rate variability, and complex biologic systems: a study of 1,871 critically ill trauma patients. J Am Coll Surg 204: 885–892, 2007 [DOI] [PubMed] [Google Scholar]
- 53. Nakagawa M, Iwao T, Ishida S, Yonemochi H, Fujino T, Saikawa T, Ito M. Circadian rhythm of the signal averaged electrocardiogram and its relation to heart rate variability in healthy subjects. Heart 79: 493–496, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakao M, Norimatsu M, Mizutani Y, Yamamoto M. Spectral distortion properties of the integral pulse frequency modulation model. IEEE Trans Biomed Eng 44: 419–426, 1997 [DOI] [PubMed] [Google Scholar]
- 55. Niklasson U, Wiklund U, Bjerle P, Olofsson BO. Heart-rate variation: what are we measuring? Clin Physiol 13: 71–79, 1993 [DOI] [PubMed] [Google Scholar]
- 56. Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest 117: 1162–1172, 2000 [DOI] [PubMed] [Google Scholar]
- 57. Opal SM, Scannon PJ, Vincent JL, White M, Carroll SF, Palardy JE, Parejo NA, Pribble JP, Lemke JH. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis 180: 1584–1589, 1999 [DOI] [PubMed] [Google Scholar]
- 58. Pagani M. Heart rate variability and autonomic diabetic neuropathy. Diabetes Nutr Metab 13: 341–346, 2000 [PubMed] [Google Scholar]
- 59. Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med 328: 1471–1477, 1993 [DOI] [PubMed] [Google Scholar]
- 60. Peng CK, Havlin S, Stanley HE, Goldberger AL. Quantification of scaling exponents and crossover phenomena in nonstationary heartbeat time series. Chaos 5: 82–87, 1995 [DOI] [PubMed] [Google Scholar]
- 61. Petrovsky N, Harrison LC. The chronobiology of human cytokine production. Int Rev Immunol 16: 635–649, 1998 [DOI] [PubMed] [Google Scholar]
- 62. Pincus SM. Greater signal regularity may indicate increased system isolation. Math Biosci 122: 161–181, 1994 [DOI] [PubMed] [Google Scholar]
- 63. Ponikowski P, Anker SD, Chua TP, Szelemej R, Piepoli M, Adamopoulos S, Webb-Peploe K, Harrington D, Banasiak W, Wrabec K, Coats AJ. Depressed heart rate variability as an independent predictor of death in chronic congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 79: 1645–1650, 1997 [DOI] [PubMed] [Google Scholar]
- 64. Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn 29: 1–24, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rassias AJ, Holzberger PT, Givan AL, Fahrner SL, Yeager MP. Decreased physiologic variability as a generalized response to human endotoxemia. Crit Care Med 33: 512–519, 2005 [DOI] [PubMed] [Google Scholar]
- 66. Richman JS, Moorman JR. Physiological time-series analysis using approximate entropy and sample entropy. Am J Physiol Heart Circ Physiol 278: H2039–H2049, 2000 [DOI] [PubMed] [Google Scholar]
- 67. Saito Y, Shimizu T, Takahashi Y, Mishima K, Takahashi K, Ogawa Y, Kogawa S, Hishikawa Y. Effect of bright light exposure on muscle sympathetic nerve activity in human. Neurosci Lett 219: 135–137, 1996 [DOI] [PubMed] [Google Scholar]
- 68. Sayk F, Vietheer A, Schaaf B, Wellhoener P, Weitz G, Lehnert H, Dodt C. Endotoxemia causes central downregulation of sympathetic vasomotor tone in healthy humans. Am J Physiol Regul Integr Comp Physiol 295: R891–R898, 2008 [DOI] [PubMed] [Google Scholar]
- 69. Schaller MD, Waeber B, Nussberger J, Brunner HR. Angiotensin II, vasopressin, and sympathetic activity in conscious rats with endotoxemia. Am J Physiol Heart Circ Physiol 249: H1086–H1092, 1985 [DOI] [PubMed] [Google Scholar]
- 70. Scheff JD, Calvano SE, Lowry SF, Androulakis IP. Modeling the influence of circadian rhythms on the acute inflammatory response. J Theor Biol 264: 1068–1076, 2010 [DOI] [PubMed] [Google Scholar]
- 71. Schmidt H, Müller-Werdan U, Werdan K. Autonomic dysfunction: A relevant component in multiple organ dysfunction syndrome. In: Yearbook of Intensive Care and Emergency Medicine, edited by Vincent JL. Berlin: Springer, 2007, p. 455–467 [Google Scholar]
- 72. Seely AJ, Christou NV. Multiple organ dysfunction syndrome: exploring the paradigm of complex nonlinear systems. Crit Care Med 28: 2193–2200, 2000 [DOI] [PubMed] [Google Scholar]
- 73. Shanker BA, Coyle SM, Reddell MT, Choi CW, Calvano J, Macor MA, Calvano SE, Lowry SF. Modeling the human injury response. J Am Coll Surg 211: S53–S54, 2010 [Google Scholar]
- 74. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6: 318–328, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sun YN, DuBois DC, Almon RR, Jusko WJ. Fourth-generation model for corticosteroid pharmacodynamics: a model for methylprednisolone effects on receptor/gene-mediated glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm 26: 289–317, 1998 [DOI] [PubMed] [Google Scholar]
- 76. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology Heart rate variability: standards of measurement, physiological interpretation and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Circulation 93: 1043–1065, 1996 [PubMed] [Google Scholar]
- 77. van der Poll T. Effects of catecholamines on the inflammatory response. Sepsis 4: 159–167, 2001 [Google Scholar]
- 78. van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J Clin Invest 97: 713–719, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vodovotz Y. Translational systems biology of inflammation and healing. Wound Repair Regen 18: 3–7, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vodovotz Y, Clermont G, Chow C, An G. Mathematical models of the acute inflammatory response. Curr Opin Crit Care 10: 383–390, 2004 [DOI] [PubMed] [Google Scholar]
- 81. Vodovotz Y, Constantine G, Rubin J, Csete M, Voit EO, An G. Mechanistic simulations of inflammation: current state and future prospects. Math Biosci 217: 1–10, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vodovotz Y, Csete M, Bartels J, Chang S, An G. Translational systems biology of inflammation. PLoS Comput Biol 4: e1000014, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Warner HR, Cox A. A mathematical model of heart rate control by sympathetic and vagus efferent information. J Appl Physiol 17: 349–355, 1962 [DOI] [PubMed] [Google Scholar]
- 84. Wurtman RJ, Pohorecky LA, Baliga BS. Adrenocortical control of the biosynthesis of epinephrine and proteins in the adrenal medulla. Pharmacol Rev 24: 411–426, 1972 [PubMed] [Google Scholar]
- 85. Xu ZX, Sun YN, DuBois DC, Almon RR, Jusko WJ. Third-generation model for corticosteroid pharmacodynamics: roles of glucocorticoid receptor mRNA and tyrosine aminotransferase mRNA in rat liver. J Pharmacokinet Biopharm 23: 163–181, 1995 [DOI] [PubMed] [Google Scholar]