

Summary
In eukaryotes, lysine acetylation is a well-established posttranslational modification that has been implicated in virtually all aspects of eukaryotic physiology. Although homologs of the enzymes that catalyze protein acetylation are widely conserved and distributed amongst bacterial species, not much is known about the impact of protein acetylation on bacterial physiology. Here, we present evidence that the Gcn5-like acetyltransferase YfiQ and the sirtuin deacetylase CobB play crucial roles in the transcription regulation of the periplasmic stress-responsive promoter cpxP when cells of Escherichia coli grow in the presence of glucose, an environment that induces protein acetylation. Under this growth condition, several acetylation sites were detected on three of the RNA polymerase (RNAP) subunits: β, β’ and α. We focused on acetylations of the carboxy-terminal domain (CTD) of α because of its relative small size and its limited acetylation. We determined that K298 of α is acetylated in a glucose and YfiQ-dependent manner and that K298 is specifically required for glucose-induced cpxP transcription. Since the αCTD aids in promoter recognition by RNA polymerase, we propose its acetylation may influence bacterial physiology through effects on gene expression.
Introduction
Acetyl coenzyme A (AcCoA), the keystone molecule of central metabolism, functions as an acetyl donor. In Nε-lysine acetylation, AcCoA donates its acetyl group to lysine residues located on the surface of proteins. This posttranslational modification is reversible: Nε-lysine acetylation can be performed by several different families of acetyltransferases, including the GCN5-like acetyltransferases, while deacetylation can be catalyzed by several different families of deacetylases, including the NAD+-dependent sirtuins [reviewed by (Hu et al., 2010, Thao & Escalante-Semerena, 2011)].
Studies in eukaryotes have shown that Nε-lysine acetylation is a common posttranslational modification that can alter protein function in diverse ways [reviewed by (Kouzarides, 2000, Yang & Seto, 2008)]. In bacteria, however, the global impact of Nε-lysine acetylation is not yet well understood. To date, only two Nε-lysine-acetylated bacterial proteins have been extensively studied: acetyl-CoA synthetase (an enzyme that converts acetate to AcCoA) and CheY (a signal protein central to chemotaxis) [reviewed by (Hu et al., 2010, Thao & Escalante-Semerena, 2011)]. Recently, however, three global proteomic studies revealed the existence of a few hundred acetylated proteins associated with diverse functions in Escherichia coli and Salmonella enterica (Yu et al., 2008, Zhang et al., 2009, Wang et al., 2010). These studies indicate that the breadth of Nε-lysine acetylation in bacteria might rival that of eukaryotes.
From the global studies of Nε-lysine acetylation in bacteria, the report that RNA polymerase (RNAP) core complex could be acetylated (Yu et al., 2008, Zhang et al., 2009) was of particular interest to us. RNAP core is composed of four protein subunits: β, β’, ω, and α. The largest subunits, β and β’, respectively encoded by rpoB and rpoC, comprise the catalytic core of RNAP. ω, encoded by rpoZ, is a small subunit thought to aid in RNAP assembly (Mathew & Chatterji, 2006). α, encoded by rpoA, contains two independently folded domains connected by a flexible linker: the amino-terminal domain (αNTD) and the carboxy-terminal domain (αCTD) (Blatter et al., 1994). The αNTD promotes dimerization of the α subunits of RNAP (Busby & Ebright, 1999), a process that initiates assembly of the multi-subunit RNAP complex (Borukhov & Nudler, 2003). The αCTD helps with promoter recognition by interacting with DNA at a region upstream of the transcription initiation site known as the UP element (Ross et al., 1993) and/or by interacting with certain transcription factors (Busby & Ebright, 1999). In theory, posttranslational modification of RNAP could exert global effects on the transcription pattern and, ultimately, impose a major impact on overall bacterial physiology. To study the effect of RNAP acetylation, we used the cpxP promoter, whose transcription activation is reported to depend on RNAP and the response regulator CpxR (Wolfe et al., 2008, Danese & Silhavy, 1998).
CpxR and its cognate sensor kinase CpxA constitute the two-component signal transduction pathway CpxAR (Fig. 1). This pathway has been proposed to regulate transcription of at least 50 genes (De Wulf et al., 2002, Price & Raivio, 2009), including cpxP, which encodes a periplasmic chaperone involved in the extracytoplasmic stress response and in the regulation of the CpxAR pathway (Danese & Silhavy, 1998, Raivio et al., 1999, De Wulf et al., 2002, Price & Raivio, 2009). In response to certain extracytoplasmic signals, such as alkaline pH (Danese & Silhavy, 1998, Wolfe et al., 2008), surface attachment (Otto & Silhavy, 2002) or accumulation of misfolded periplasmic proteins, CpxA autophosphorylates a conserved histidine residue (H248) using ATP as its phosphoryl donor. Phospho-CpxA then transfers its phosphoryl group to a conserved aspartate residue (D51) of CpxR, an event that promotes CpxR-dependent transcription (Danese et al., 1995, Pogliano et al., 1997, Raivio & Silhavy, 1997).
Figure 1. The CpxAR two component system.
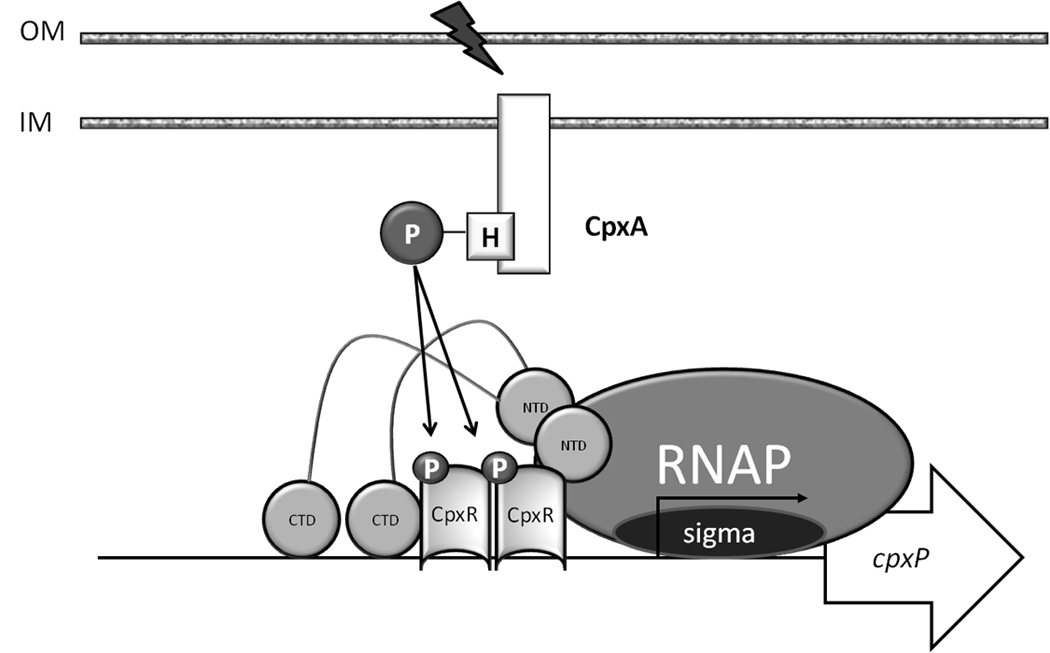
A) Activation of CpxR by the cytoplasmic membrane sensor kinase CpxA. OM, outer membrane; IM, inner membrane. Upon activation by diverse extracytoplasmic signals (thunderbolt), CpxA autophosphorylates on a conserved histidine residue (H), using ATP as its phosphoryl donor. Phospho-CpxA then acts as a phosphoryl donor to the response regulator CpxR, which autophosphorylates on the conserved aspartate residue (D51). By homology to OmpR, phosphorylation of CpxR is predicted to expose the DNA-binding domain, thus promoting the binding of CpxR to its target genes (Pogliano et al., 1997, De Wulf et al., 2002).
In the absence of its extracytoplasmic stimuli, CpxA functions as a net phosphatase, removing phosphoryl groups from phospho-CpxR (Raivio & Silhavy, 1997, Wolfe et al., 2008). When CpxA functions as a net phosphatase, CpxR can become activated in a CpxA-independent manner (Danese & Silhavy, 1998). Both WT and cpxA mutant cells growing in buffered tryptone broth (TB) in the presence of glucose, pyruvate or acetate activate the CpxR-dependent cpxP promoter, suggesting the existence of an alternate method for cpxP transcription activation (Wolfe et al., 2008).
Exposure to glucose rapidly increases the AcCoA concentration in E. coli (Chohnan et al., 1998) and promotes protein acetylation in S. enterica (Wang et al., 2010). We therefore hypothesized that CpxA-independent glucose-induced cpxP transcription involves protein acetylation. Indeed, the studies reported here support this hypothesis by implicating AcCoA, the GCN5-like acetyltransferase YfiQ, the NAD+-dependent deacetylase CobB, and an acetylated lysine (K298) located on the surface of the αCTD of RNAP. These studies also show that glucose-induced, YfiQ-dependent acetylation is not restricted to the αCTD, but also extends to multiple lysines of β and β’.
Results
cpxP transcription is induced by multiple carbon sources
We and others have previously demonstrated that cpxP transcription can be induced by adding glucose to amino acid-based media (Danese & Silhavy, 1998, Wolfe et al., 2008). This induction is independent of the sensor kinase CpxA, but requires its cognate response regulator CpxR. To better understand the mechanism by which glucose induces cpxP transcription, we first investigated whether other carbon sources could similarly induce transcription activation. To this end, we exposed a cpxA mutant strain that carries the transcriptional fusion λΦ(PcpxP’-lacZ) (strain PAD348; Table 2) to a variety of carbon sources, and measured β-galactosidase activity as a reporter of cpxP promoter function. Glucose, pyruvate, acetate and lactate, carbon sources that can be quickly metabolized and thus contribute to AcCoA synthesis, strongly induced cpxP transcription (Fig.2) in a CpxR-dependent manner (inset), while glycerol, sorbitol, succinate and citrate did not (Fig.2). Because the catabolite-repressing glucose and the non-catabolite-repressing acetate could each activate cpxP transcription, catabolite repression seemed an unlikely mechanism. Instead, we hypothesized that AcCoA could play a central role in activating cpxP transcription.
Table 2.
Strains and Plasmids used in this Study
| Strain | Relevant Characteristic | Source/Reference |
|---|---|---|
| PAD282 | F− araD139 Δ(argF-lac)U169 rpsL150(Strr) relA1 flhD5301 deoC1 λΦ(PcpxP'-lacZ) | (DiGiuseppe & Silhavy, 2003) |
| PAD348 | PAD282 cpxA::cam | DiGuiseppe & Silhavy (Princeton University) |
| PAD292 | PAD282 cpxR1::spc (spectinomycin insertion in cpxR with polar effect on cpxA) | (DiGiuseppe & Silhavy, 2003) |
| AJW3335 | PAD282 ΔaceE::kan | P1:JW0110 → PAD282 |
| AJW3143 | PAD282 yfiQ::kan | P1:JE9314 → PAD282 |
| JW0110 | ΔaceE::kan | (Baba et al., 2006) |
| JE9314 | yfiQ::kan | Escalante-Semerena (University of Wisconsin-Madison) |
| Plasmid | Relevant Characteristics | Source/Reference |
| pCA24n | Vector. Expresses chloramphenicol acetyltransferase (camR) | (Kitagawa et al., 2005) |
| pCA24n-coaA | Plasmid expressing 6xHis-CoaA under the control of an IPTG-inducible promoter | (Kitagawa et al., 2005) |
| pCA24n-cobB | Plasmid expressing 6xHis-CobB under the control of an IPTG-inducible promoter | (Kitagawa et al., 2005) |
| pTAC85 | Vector. Contain an ampicillin resistance cassette (ampR) | (Marsh, 1986) |
| pPHB3 | pTAC85 expressing phbCAB under the control of an IPTG-inducible promoter | This study |
| pREII-rpoA | Plasmid expressing wild-type rpoA or alanine substitution derivatives (ampR) | (Gaal et al., 1996) |
| pREII-Δ αCTD | Plasmid expressing ΔCTD rpoA (ampR) | Gourse (University of Wisconsin-Madison) |
Figure 2. Diverse carbon sources can induce CpxA-independent cpxP transcription.
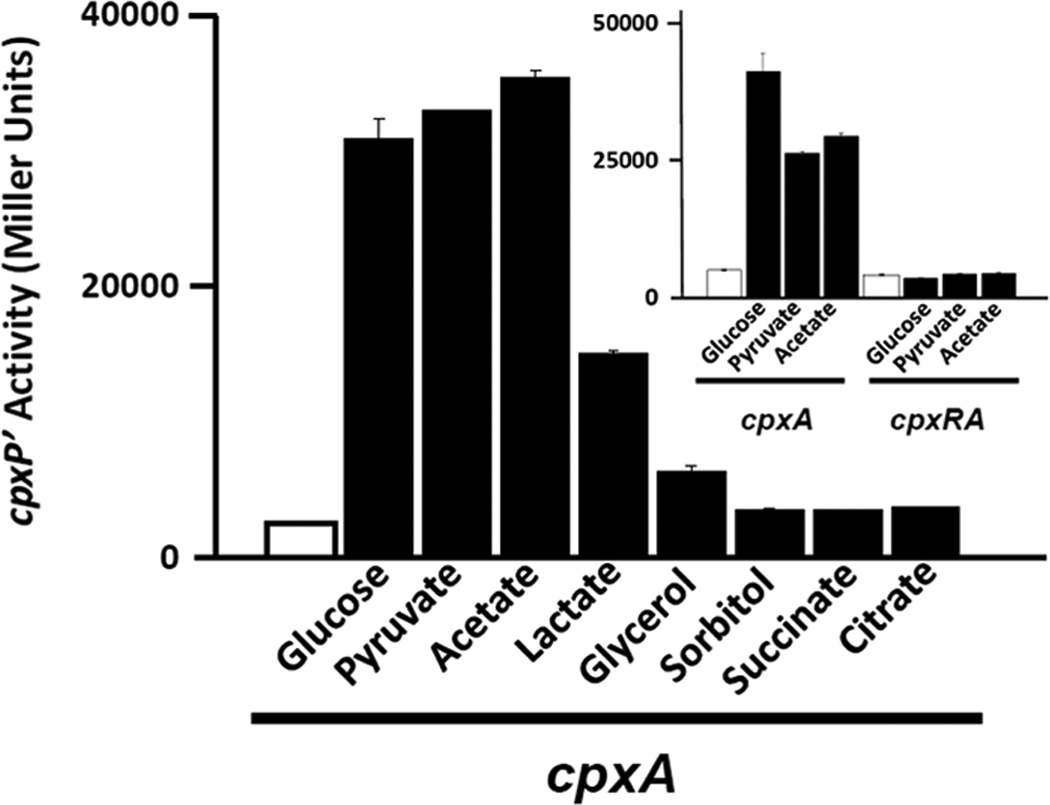
A λcpxP lysogen of the cpxA mutant (PAD348; Table 2) was grown at 37°C with shaking in buffered TB (open bars) or in the same medium supplemented with 0.4% glucose, 0.8% pyruvate, 15 mM acetate, 0.8% lactate, 0.8% glycerol, 0.4% sorbitol, 0.6% succinate or 0.4% citrate (closed bars). Cells were harvested after 7.5 hours incubation and β-galactosidase activity was measured. The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations. Inset: β-galactosidase activity of cpxA (PAD348) and cpxRA (PAD292; Table 2) mutants grown under the conditions described above.
AcCoA synthesis is required for the carbon response
To test the hypothesis that AcCoA participates in glucose-induced cpxP transcription, we disrupted the enzymatic complex required for the conversion of pyruvate to AcCoA by deleting aceE (strain AJW3335; Table 2), the gene that encodes the E1 subunit of the pyruvate dehydrogenase complex (PDH). Although alternative and less efficient routes from glucose to AcCoA exist, this aceE mutant should synthesize considerably less AcCoA than does its WT parent (PAD282; Table 2) (Wolfe, 2005). Thus, if AcCoA participates in the glucose-induced cpxP response, then this mutant should respond less robustly to exogenous glucose when compared to WT. As predicted, the aceE mutant responded less strongly to glucose (approximately 3-fold induction) than did its WT parent (approximately 6-fold induction) (Fig. 3A). The weaker response to glucose could not be explained by an overall decrease in cellular metabolism as the growth kinetics between the aceE mutant and the WT strain were similar (data not shown).
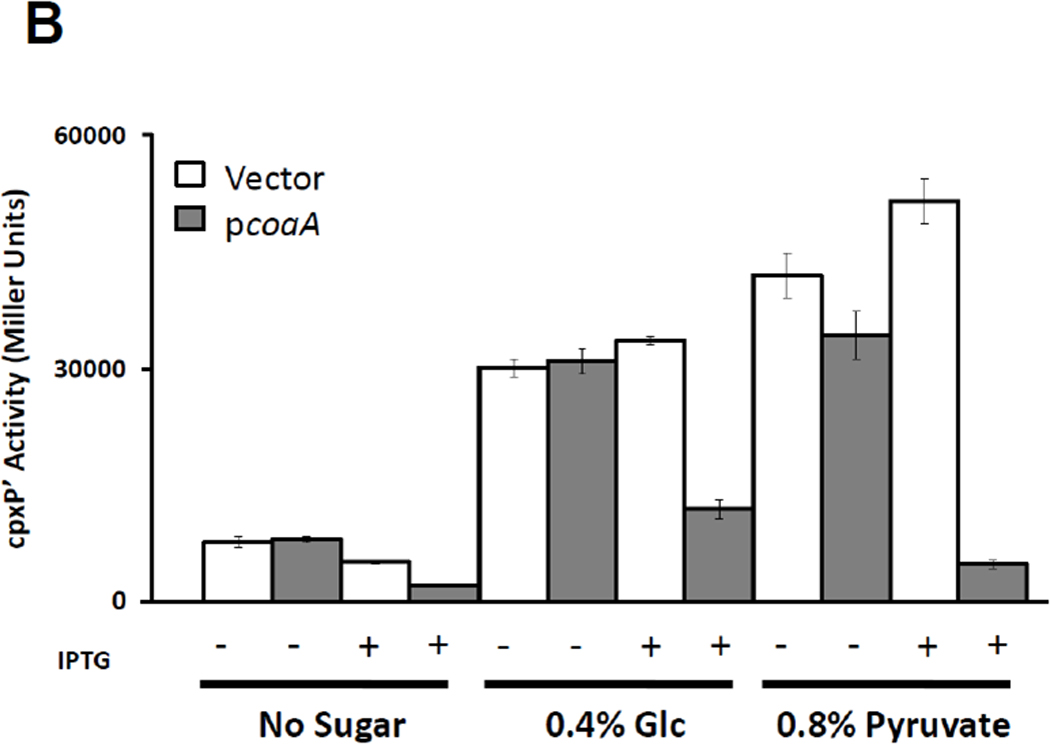
Figure 3. Glucose-induced cpxP transcription is sensitive to AcCoA and CoA manipulations.
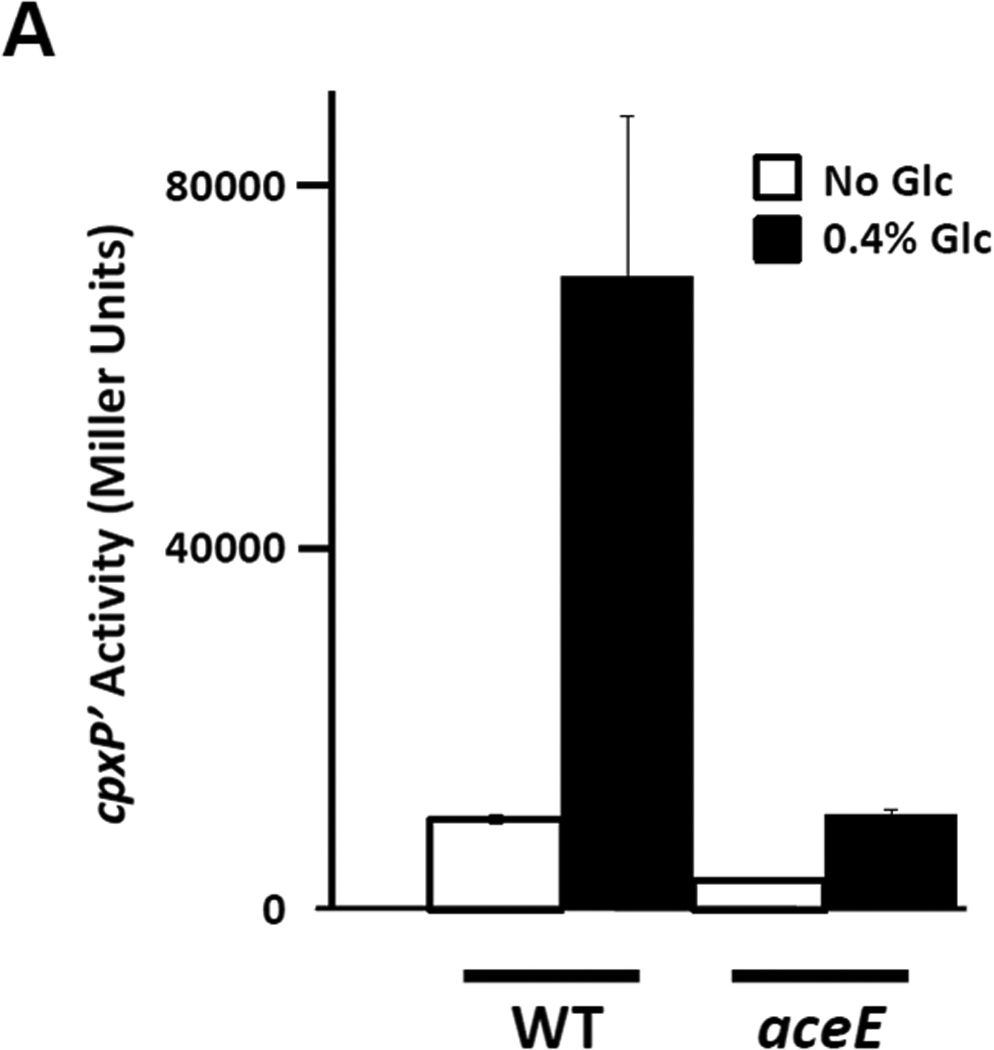
A) λcpxP lysogens of the aceE mutant (AJW3335; Table 2) and its wild-type parent (PAD282; Table 2) were grown at 37°C with shaking in buffered TB (open bars) or in the same medium supplemented with 0.4% glucose (closed bars). Cells were harvested after 7.5 hours incubation and β-galactosidase activity was measured. The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations.
B) A λcpxP lysogen of the wild-type strain (PAD282; Table 2) was transformed with the vector pCA24n (open bars) or a pCA24n derivative carrying the coaA ORF (closed bars). Transformants were grown at 37°C with shaking in buffered TB or in the same medium supplemented with 0.4% glucose or 0.8% pyruvate. IPTG was used at 50 µM to induce coaA expression. Cells were harvested after 7.5 hours incubation and β-galactosidase activity was measured. The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations.
It has been proposed that a significant regulator of protein acetylation is the AcCoA-to-CoA ratio (Albaugh et al., 2011). This proposal is supported by several experimental observations including a 6-fold decrease in this ratio when E. coli cells, grown in minimal media, are shifted from glucose to acetate as the sole carbon source (Vallari & Jackowski, 1988). If a large AcCoA-to-CoA ratio mediates the carbon response, we reasoned that decreasing that ratio would decrease the response. We used two different methods to reduce this ratio. First, we increased the CoA pool by overexpressing the native pantothenate kinase (CoaA). CoaA synthesizes the first step in CoA biosynthesis and overexpression can lead to a 3-fold increase in CoA concentration (Song & Jackowski, 1992). Into WT cells (strain PAD282), we introduced a plasmid that expresses coaA from an IPTG-inducible promoter (pCA24n-CoaA; Table 2) or its vector control (pCA24n; Table 2). Our second approach was to drain off excess acetyl groups by inducing a heterologous pathway that uses AcCoA as its substrate to synthesize the biopolymer polyhydroxybutyrate (PHB). Into WT cells (strain PAD282), we introduced a plasmid that expresses the genes phbCAB from an IPTG-inducible promoter (pPHB3; Table 2) or the vector control (pTAC85; Table 2). When induced, the PHB pathway converted AcCoA into granules of PHB, which accumulated in the cytoplasm (Steinbuchel & Schlegel, 1991, Nawrath et al., 1994) and fluoresced upon exposure to Nile Red (Ostle & Holt, 1982) (Supplemental Fig. S2A–E). Since CoaA increases the CoA pool and the PHB pathway removes acetyl groups from AcCoA, both manipulations should reduce the AcCoA-to-CoA ratio and, if our hypothesis is correct, decrease cpxP transcription upon exposure to additional carbon.
We grew the resultant transformants in TB in the absence or presence of either 0.4% glucose or 0.8% pyruvate. In the absence of these carbon sources, cpxP transcription remained low (Fig. 3B and Supplemental Fig. S2F). In their presence, however, cpxP transcription rose substantially, except when IPTG addition induced expression of either CoaA (Fig. 3B) or PHB (Supplemental Fig. S2F). Taken together, these results support the hypothesis that the AcCoA-to-CoA ratio mediates carbon–induced cpxP transcription.
cpxP transcription responds to manipulations that alter global protein acetylation status
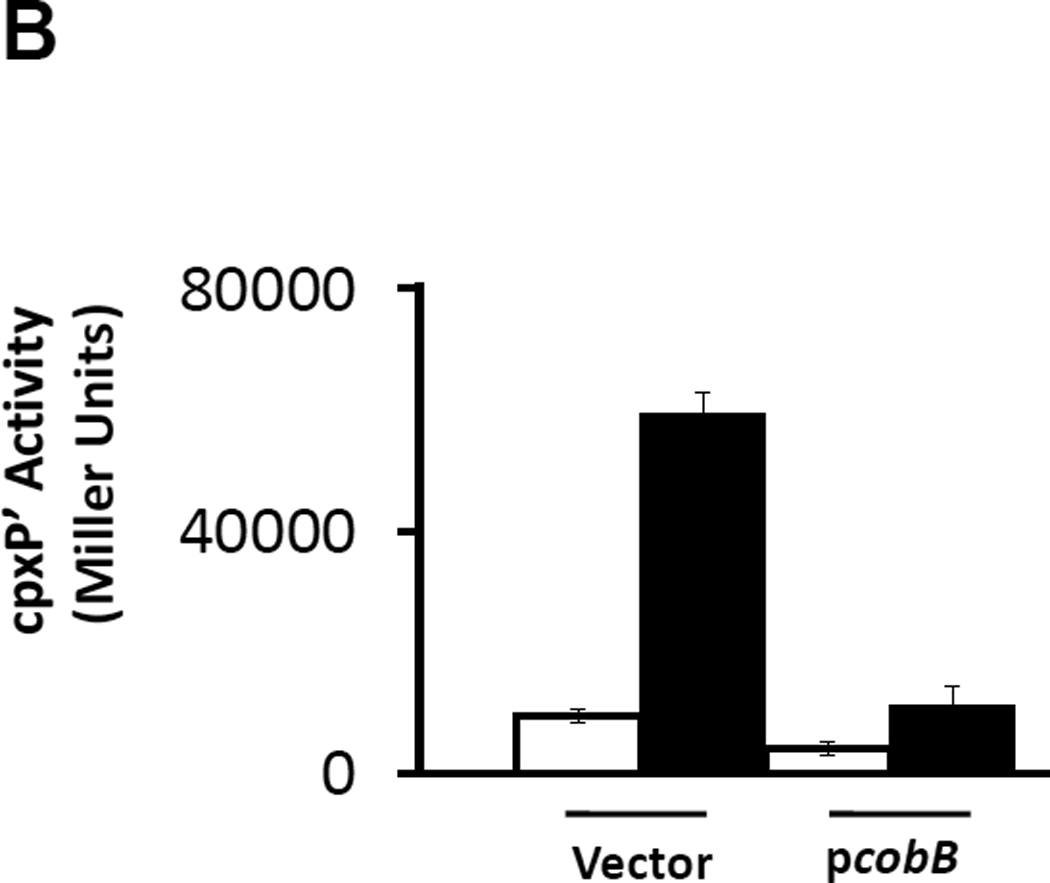
Given that acetate is directly converted to AcCoA (Wolfe, 2005), that AcCoA can function as an acetyl donor (Takahashi et al., 2006, Wellen et al., 2009), and that the addition of glucose leads to an overall increase in protein acetylation in S. enterica (Wang et al., 2010), we considered the possibility that an acetylation event could influence cpxP transcription. To test this hypothesis, we exposed the cpxA mutant strain (strain PAD348) to increasing concentrations of nicotinamide up to those previously reported for eukaryotic experiments (Chong et al., 2004, Solomon et al., 2006). Nicotinamide specifically inhibits NAD+-dependent deacetylases. Therefore, if protein acetylation influences cpxP, then we would expect to see an increase in cpxP transcription as the nicotinamide concentration increases. Conversely, WT cells (strain PAD282) that overexpressed the NAD+-dependent deacetylase CobB (pCA24n-cobB; Table 2) should respond less robustly to glucose when compared to vector control cells. Consistent with our hypothesis, cpxP transcription increased as a function of nicotinamide concentration and decreased when CobB was overexpressed. At or above 25 mM nicotinamide, cpxP transcription increased, achieving more than a 4-fold induction at 50 mM even in the absence of glucose (Fig.4A). Overexpression of CobB reduced the 6-fold glucose-dependent induction to a more modest 3-fold induction (Fig.4B).
Figure 4. Nicotinamide induces CpxA-independent cpxP transcription, whilst CobB overexpression inhibits it.
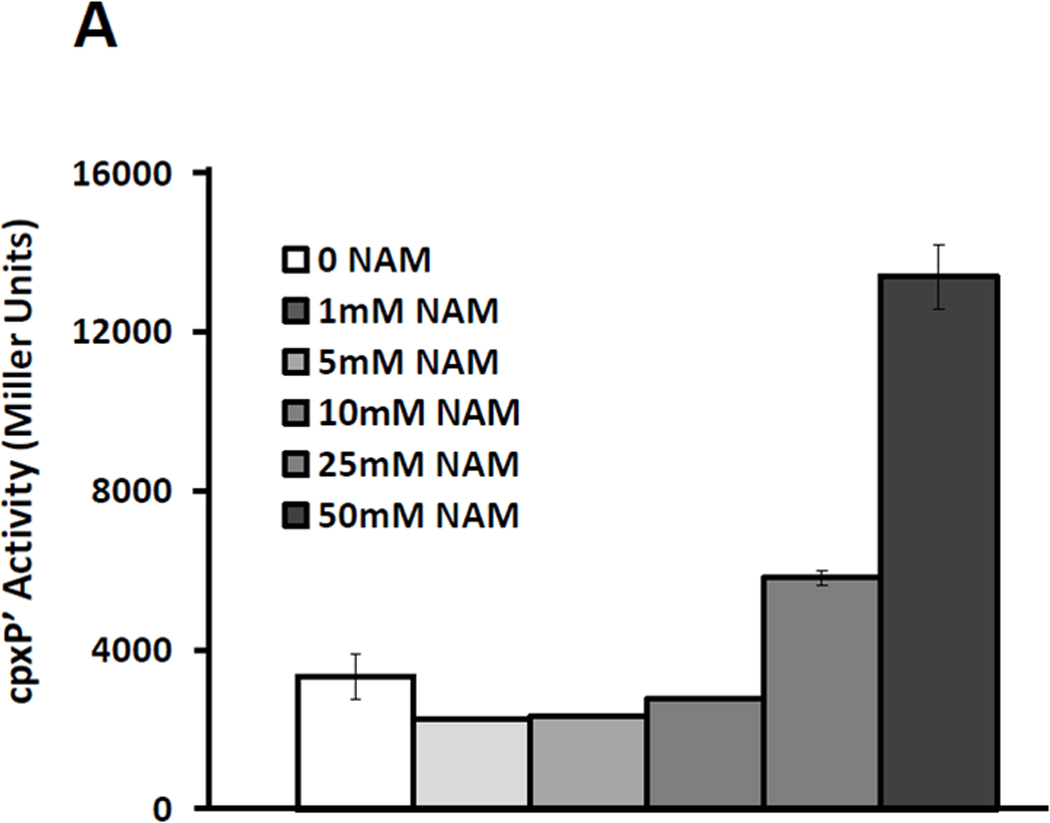
A) A λcpxP lysogen of the cpxA mutant (PAD348) was grown at 37°C with shaking in buffered TB (open bars) or the same medium supplemented with increasing concentrations of nicotinamide (NAM, closed bars). Cells were harvested after 7.5 hours incubation and β-galactosidase activity was measured. The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations.
B) A λcpxP lysogen of the wild-type strain (PAD348) was transformed with the vector pCA24n or with pCA24n derivative carrying the cobB ORF (Kitagawa et al., 2005). Transformants were grown at 37°C with shaking in buffered TB supplemented with 5 µM IPTG (open bars) or the same medium supplemented with both 0.4% glucose and 5 µM IPTG (closed bars). Cells were harvested after 7.5 hours incubation and β-galactosidase activity was measured. The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations.
To determine whether these treatments can influence the overall protein acetylation pattern (acetylome), we used a cocktail of two polyclonal anti-acetylated-lysine antibodies to perform Western immunoblot analysis on whole cell lysates (Fig 5). When WT cells (strain PAD282) were grown in the presence of either the NAD+-dependent deacetylase inhibitor nicotinamide or the extra carbon source glucose, the acetylome changed relative to cells grown without treatment. Glucose generally caused the number and/or intensity of acetylated bands to increase. In contrast, the deacetylase inhibitor nicotinamide induced more subtle changes. These subtle changes in the acetylation pattern correlated with a weaker response by the cpxP promoter to the addition of nicotinamide when compared to its response to glucose. Intriguingly, CobB overexpression in cells exposed to glucose yielded a strong effect on the acetylation pattern. This acetylome closely resembled that of cells grown in the absence of glucose or nicotinamide. Thus, the conditions that influence cpxP transcription also seem to alter the acetylome.
Figure 5. The E. coli acetylome is sensitive to conditions that affect CpxA-independent cpxP transcription.
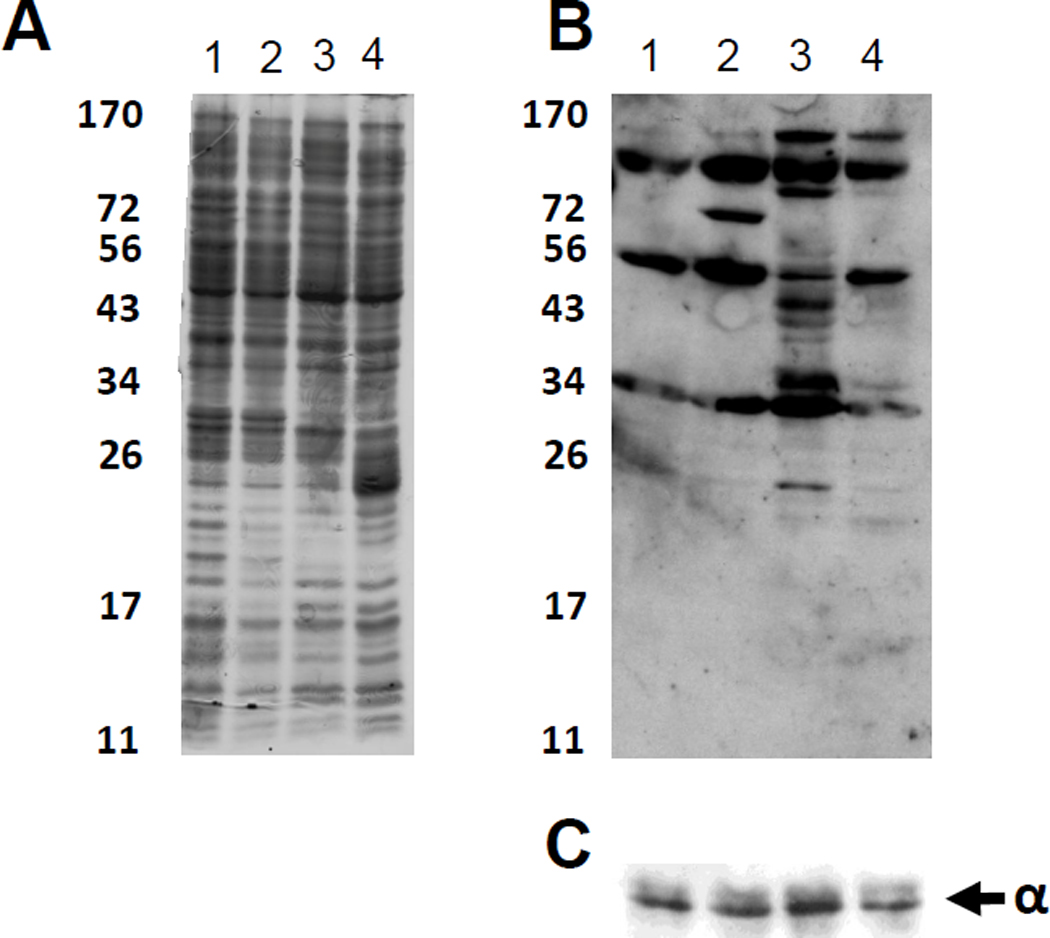
A λcpxP lysogen of the wild-type strain (PAD 282) transformed with the vector pCA24n was grown at 37°C with shaking in buffered TB supplemented with 5 µM IPTG (column 1), the same medium supplemented with 50 mM nicotinamide (column 2), or 0.4% glucose (column 3). The same strain transformed with a pCA24n derivative carrying the cobB ORF was grown in buffered TB supplemented with 5 µM IPTG and 0.4% glucose (column 4). Cells were pelleted after 7.5 hours incubation and protein was harvested as described in the Materials and Methods. In each lane, 150 µg of protein was separated by SDS-PAGE. Panels show: A) Coomassie stain, B) Western immunoblot with cocktail of anti-acetyllysine polyclonal antibodies, and C) as a loading control, a Western immunoblot using anti-α monoclonal antibody.
The carbon response requires a lysine acetyltransferase
In S. enterica, the NAD+-dependent deacetylase CobB works in conjunction with the GCN5-like acetyltransferase Pat to regulate the activity of the enzyme acetyl-CoA synthetase (Starai & Escalante-Semerena, 2004, Starai et al., 2005). The finding that CobB overexpression could inhibit cpxP transcription led us to hypothesize that glucose-induced cpxP transcription could be regulated by the E. coli Pat homolog, YfiQ. Because the acronym PatA has already been applied to the E. coli enzyme putrescine aminotransferase, we will use the term YfiQ.
To test the hypothesis that YfiQ regulates glucose-induced cpxP transcription, we compared the glucose response of a mutant that lacks YfiQ (strain AJW3143; Table 2) to that of its WT parent (strain PAD282). In contrast to its WT parent (approximately 3.5-fold induction), the yfiQ mutant responded poorly to glucose (approximately 1.5-fold induction) (Fig. 6). The requirement for YfiQ, however, did not extend to CpxA-dependent cpxP transcription, as surface-induced cpxP transcription was equivalent in both the yfiQ mutant and its WT parent (data not shown). Thus, the requirement for YfiQ is specific for the response to additional carbon. We propose that YfiQ and CobB catalyze the reversible acetylation of a protein that mediates carbon-induced cpxP transcription.
Figure 6. The carbon response requires a lysine acetyltransferase.
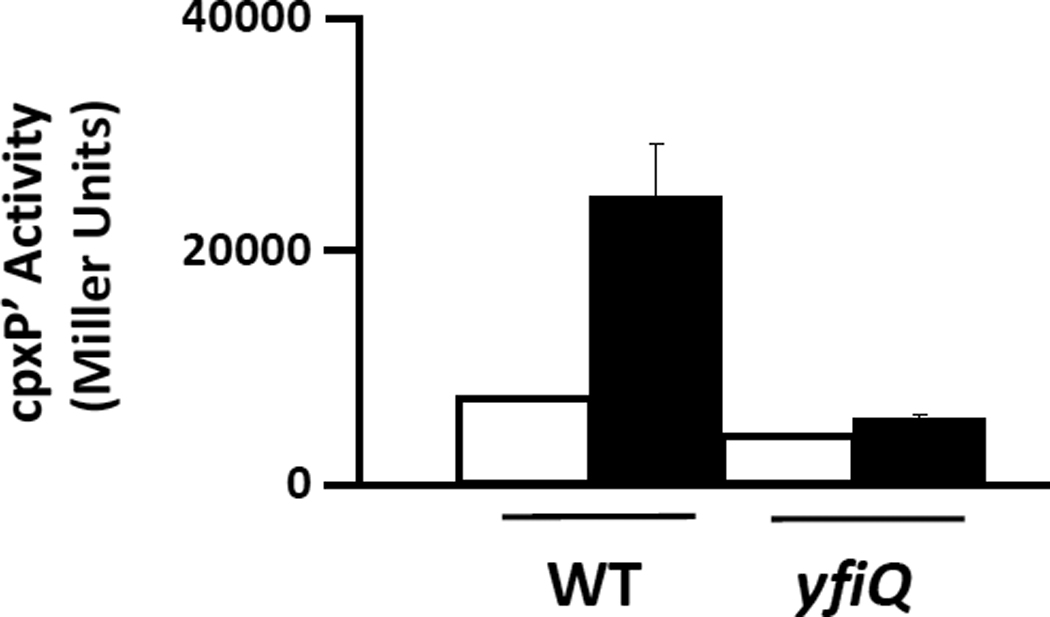
λcpxP lysogens of the yfiQ mutant (strain AJW3143) and its wild-type parent (PAD 282) were grown at 37°C with shaking in buffered TB (open bars) or the same medium supplemented with 0.4% glucose (closed bars). Cells were harvested after 7.5 hours incubation and β-galactosidase activity was measured. The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations.
YfiQ and CobB are known to catalyze the reversible acetylation of acetyl-CoA synthetase (Acs) (Starai & Escalante-Semerena, 2004, Starai et al., 2005). To test Acs involvement in glucose-induced cpxP transcription, we deleted its gene and observed no effect on that behavior (data not shown). This result was not a surprise because acs transcription is regulated by catabolite repression; thus, exposure to glucose inhibits its transcription (Kumari et al., 2000). We therefore sought the cpxP-relevant target of YfiQ and CobB elsewhere.
RNA polymerase is acetylated
The finding that protein acetylation was required for glucose-induced cpxP transcription led us to begin our search for the acetylation target(s) with CpxR and RNAP, the two proteins reported to be required for cpxP transcription (Danese & Silhavy, 1998, Wolfe et al., 2008). To identify the acetylation target(s), we grew WT cells (strain PAD282) in the presence or absence of glucose, used immunoprecipitation to purify CpxR (data not shown) or the RNAP subunits β, β’, and α (Supplemental Fig. S1), and investigated the acetylation status of these proteins by Western immunoblot analysis using anti-acetylated lysine antibodies. From cells grown in the presence of glucose, we detected acetylation of β, β’ and α, but not of CpxR. We also failed to detect acetylated β, β’ or α from cells grown in the absence of glucose (data not shown).
Using linear trap quadrupole (LTQ) Orbitrap mass spectrometry (MS), we then mapped 15 glucose-dependent acetylation sites to β (Table 1, Supplemental Table S1), 11 acetylation sites to β’ (Table 1, Supplemental Table S2), and 2 acetylation sites to α (Fig. 7, Supplemental Table S3). None of these sites were acetylated when WT cells were grown in the absence of glucose (Supplemental Tables S4 and S5). Of the 15 different acetylation sites detected from β, we detected 14 in at least two biological replicates. Of the 11 acetylation sites detected from β’, we reproducibly detected 8 (data not shown). Finally, α peptides containing acetylated K297 or acetylated K298 were detected in multiple biological replicates. We conclude that glucose induces acetylation of multiple sites on multiple subunits of RNAP, including K297 and K298 of α.
Table 1.
Lysine-acetylated β and β' peptides
| Lysine-acetylated β peptides identified from glucose-exposed wild-type cells | |||||||
|---|---|---|---|---|---|---|---|
| Sequence | Prob | SEQUEST XCorr |
SEQUEST ΔCn |
Observed Precursor m/z |
Actual Peptide Mass (MW) |
Charge | Delta PPM |
| (R)DNK236(+42)LQmELVPER(L) | 95% | 2.955 | 0.481 | 510.5923 | 1528.76 | 3 | −0.4136 |
| (R)QLEK279(+42)DDVK(L)* | 95% | 2.407 | 0.4241 | 508.7665 | 1015.52 | 2 | −0.3027 |
| (K)VTPK890(+42)GETQLTPEEK(L) | 95% | 3.296 | 0.6551 | 799.9181 | 1597.82 | 2 | 0.8714 |
| (R)AIFGEK909(+42)ASDVK(D)* | 95% | 3.424 | 0.6959 | 603.8215 | 1205.63 | 2 | −0.7829 |
| (K)ASDVK914(+42)DSSLR(V)* | 95% | 3.105 | 0.3712 | 560.2856 | 1118.56 | 2 | −0.08906 |
| (R)ALEIEEmQLK954(+42)QAK(K) | 95% | 3.883 | 0.6378 | 786.9191 | 1571.82 | 2 | 0.2837 |
| (R)AVLVAGGVEAEK988(+42)LDK(L) | 95% | 4.315 | 0.7414 | 770.9325 | 1539.85 | 2 | −0.3886 |
| (K)LDK991(+42)LPR(D)* | 95% | 1.741 | 0.3937 | 392.2397 | 782.4648 | 2 | −0.5344 |
| (R)K1035(+42)ITQGDDLAPGVLK(I) | 95% | 3.136 | 0.5483 | 748.9197 | 1495.82 | 2 | −0.07396 |
| (R)mNIGQILETHLGmAAK1122(G) | 95% | 3.105 | 0.4271 | 600.9708 | 1799.89 | 3 | −0.3436 |
| (K)INAmLK1133(+42)QQQEVAK(L) | 95% | 4.660 | 0.6422 | 779.9177 | 1557.82 | 2 | 1.362 |
| (K)QQQEVAK1140(+42)LR(E)* | 95% | 2.559 | 0.5444 | 571.3196 | 1140.62 | 2 | −0.4438 |
| (R)K1178(+42)GmPIATPVFDGAK(E) | 95% | 3.851 | 0.6688 | 745.3899 | 1488.77 | 2 | 0.1888 |
| (K)ELLK1200(+42)LGDLPTSGQIR(L) | 95% | 4.029 | 0.5342 | 841.4792 | 1680.94 | 2 | 1.418 |
| (R)STGSYSLVTQQPLGGK1262(+42) AQFGGQR(F) | 95% | 4.335 | 0.5947 | 803.744 | 2408.21 | 3 | 0.543 |
| Lysine-acetylated β’ peptides identified from glucose-exposed wild-type cells | |||||||
| Sequence | Prob | SEQUEST XCorr | SEQUEST ΔCn | Observed Precursor m/z | Actual Peptide Mass (MW) | Charge | Delta PPM |
| (K)AQTK13(+42)TEEFDAIK(I)* | 95% | 3.134 | 0.5511 | 711.8596 | 1421.70 | 2 | 0.4564 |
| (K)K40(+42)PETINYR(T) | 95% | 1.857 | 0.3906 | 531.7829 | 1061.55 | 2 | 0.5715 |
| (R)GLATTIK395(+42)AAK(K) | 95% | 2.728 | 0.654 | 508.3111 | 1014.61 | 2 | 0.1017 |
| (K)GEGmVLTGPK531(+42)EAER(L) | 95% | 3.620 | 0.5413 | 766.3754 | 1530.74 | 2 | 0.8169 |
| (K)TSLK570(+42)DTTVGR(A) | 95% | 2.799 | 0.4491 | 560.3032 | 1118.59 | 2 | −1.289 |
| (R)SGASVGIDDmVIPEK649(+42)K(H) | 95% | 3.297 | 0.5722 | 852.4306 | 1702.85 | 2 | 0.9404 |
| (R)AAAESSIQVK953(+42)NK(G) | 95% | 3.437 | 0.5779 | 644.3492 | 1286.68 | 2 | 0.6021 |
| (K)GSIK959(+42)LSNVK(S) | 95% | 2.737 | 0.5114 | 494.2947 | 986.5748 | 2 | −1.35 |
| (K)LSNVK964(+42)SVVNSSGK(L) | 95% | 3.015 | 0.6372 | 680.8746 | 1359.73 | 2 | −0.8405 |
| (R)NTELK983(+42)LIDEFGR(T) | 95% | 3.479 | 0.5128 | 492.9273 | 1475.76 | 3 | −1.452 |
| (R)LQGVK1247(+42)INDK(H) | 95% | 1.808 | 0.2969 | 528.8063 | 1055.60 | 2 | 0.2463 |
Immunoprecipitated β and β’ proteins were separated by SDS-PAGE and β and β’ bands were digested with trypsin, as described (Chi et al., 2010). The resulting peptides were analyzed in a LTQ OrbitrapXL™ mass spectrometer, as described in the Methods section and analyzed using the Scaffold software. The table shows the Xcorr, ΔCn scores, charges, both the observed precursor m/z and actual peptide mass, and the mass deviation (delta ppm) of all 15 K-acetylated peptides detected for β and 11 K-acetylated peptides detected for β’ in the glucose-exposed samples. In these tables, the CID MS/MS spectra of the acetylated β and β’ peptides are shown in Tables S1 and S2, respectively. The complete b and y ion series are cognizable and labelled in red and blue, respectively. Acetylated peptides detected in at least two biological replicates are underlined.
An asterisk indicates acetylated peptides identified in samples collected from glucose-exposed yfiQ mutants (Supplemental Tables S6 and S7). None of these peptides was detected in the absence of glucose (Supplemental Tables S4, S5, S9 and S10).
Figure 7. The α subunit of RNAP is acetylated on K297 and K298.
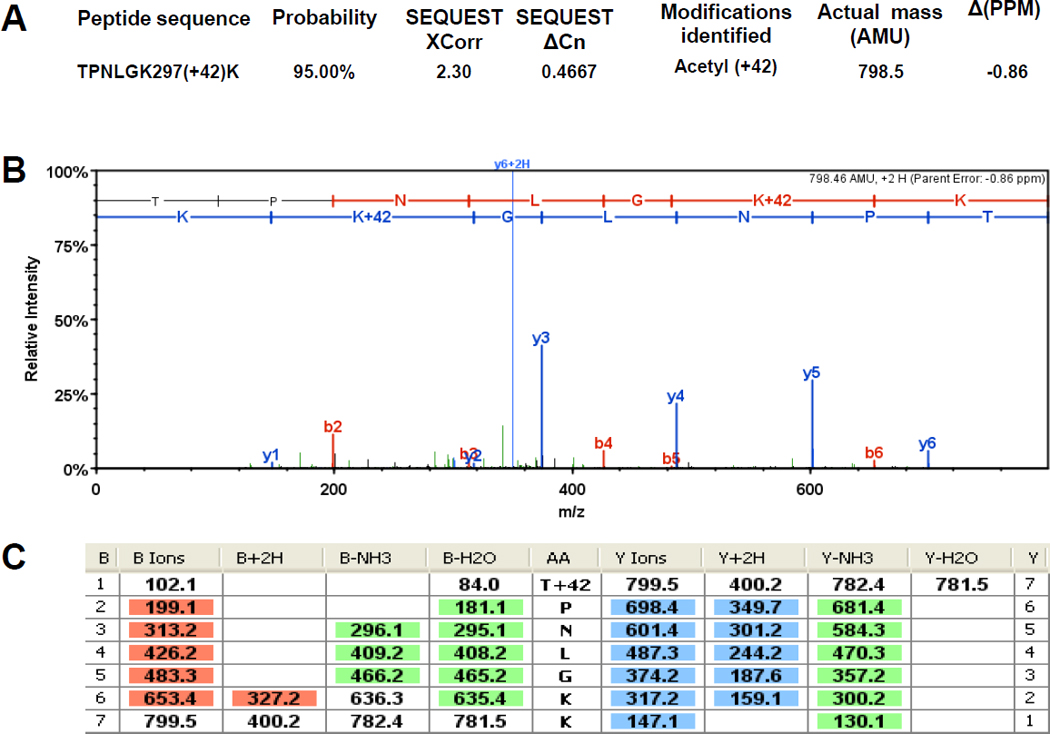
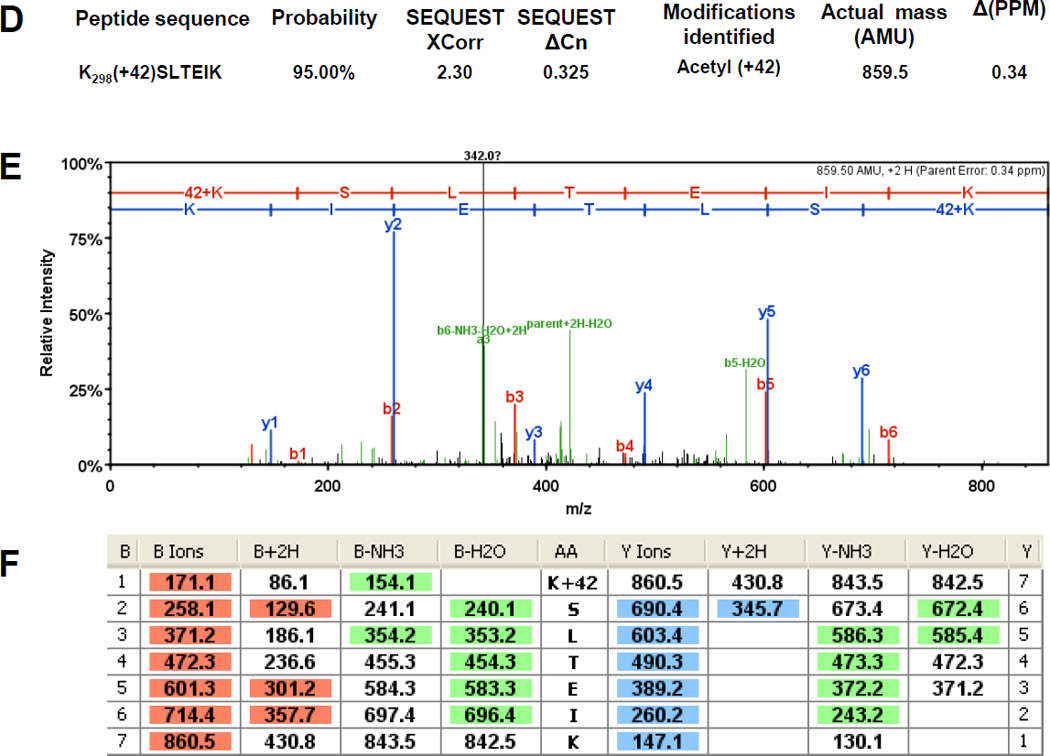
Immunoprecipitated subunits of RNAP were separated by SDS-PAGE. The α bands were excised and tryptically digested as described previously (Chi et al., 2010). The resulting peptides were analyzed in a LTQ OrbitrapXL™ mass spectrometer as described in the Materials and Methods. The double charged acetyllysine-modified peptides TPNLGK297(+42)K and K298(+42)SLTEIK were detected as mass peaks of m/z=400.237 and m/z=430.7582, respectively, in the digested α sample. A and D) The Xcorr and ΔCn scores, the actual peptide masses and mass deviations of the acetylated peptides. B and E) The corresponding CID MS/MS spectra. C and F) Complete b and y fragment ion series for these peptides.
YfiQ is required for acetylation of multiple residues of RNAP
To determine whether the protein acetyltransferase YfiQ was responsible for any of the glucose-induced acetylations of RNAP, we grew the yfiQ mutant (strain AJW3143) in the absence or presence of glucose, isolated the RNAP subunits by immunoprecipitation and subjected them to MS analysis. Ten of the fifteen glucose-dependent β acetylations, ten of the eleven glucose-dependent β’ acetylations, and one of the two glucose-dependent α acetylations (K298) required YfiQ (Table 1 and Supplemental Tables S6–8). As with WT, we detected no acetylated peptides from yfiQ mutant cells grown in the absence of glucose (Supplemental Tables S9–11). Thus, we conclude that YfiQ is required for most of the glucose-induced acetylations of RNAP.
The carbon response requires K298 of the αCTD
That acetylation of several lysines on RNAP is both glucose and YfiQ-dependent led us to hypothesize that at least one of these residues could be required for glucose-induced cpxP transcription. To test this hypothesis, we focused our attention on α. We chose α due to its smaller size relative to β and β’ and to our finding that acetylation of only one residue (K298) was both glucose and YfiQ-dependent. Since α is essential for cellular viability, we used a merodiploid system previously used for studies of α (Gaal et al., 1996) to test whether K298 or any of the other four lysines on the αCTD are required for glucose-induced cpxP transcription. We introduced plasmids that carry either the WT rpoA allele or lysine-to-alanine mutant derivatives into the cpxA mutant (strain PAD348), which carries the WT rpoA gene at its native location on the chromosome. We grew the resultant transformants in the absence or presence of glucose and compared the β-galactosidase activity of cells expressing each lysine-to-alanine substitution to that of cells expressing WT α. Of the five mutants tested, only K298A exerted a defect on glucose-induced cpxP transcription (Fig. 8A).
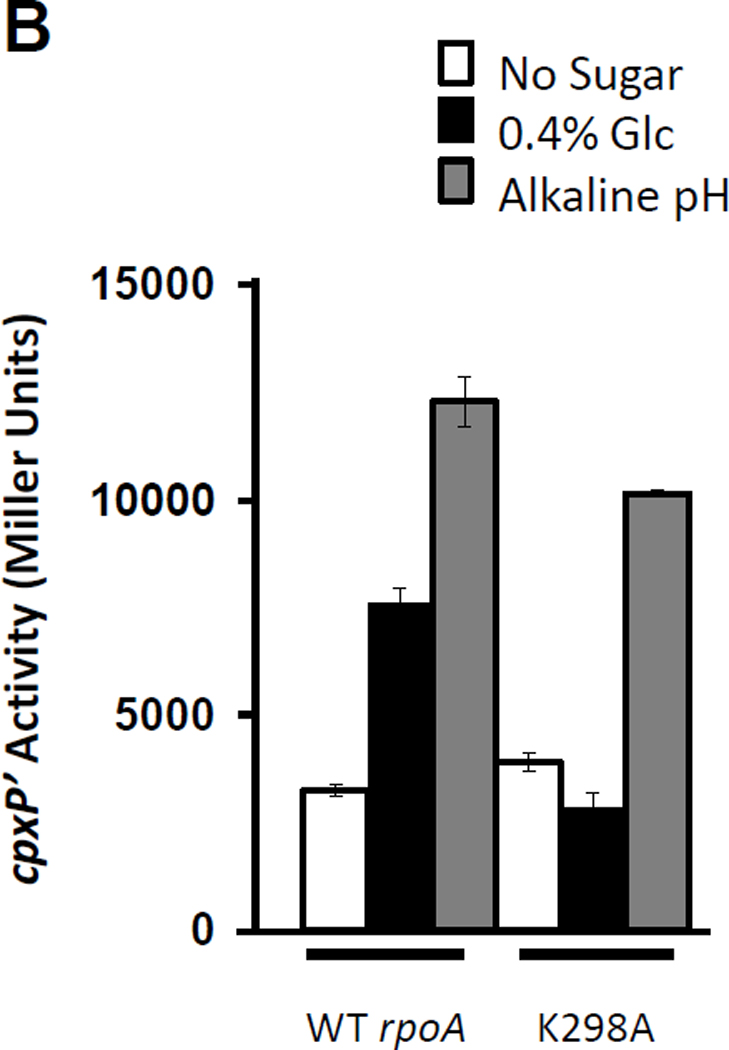
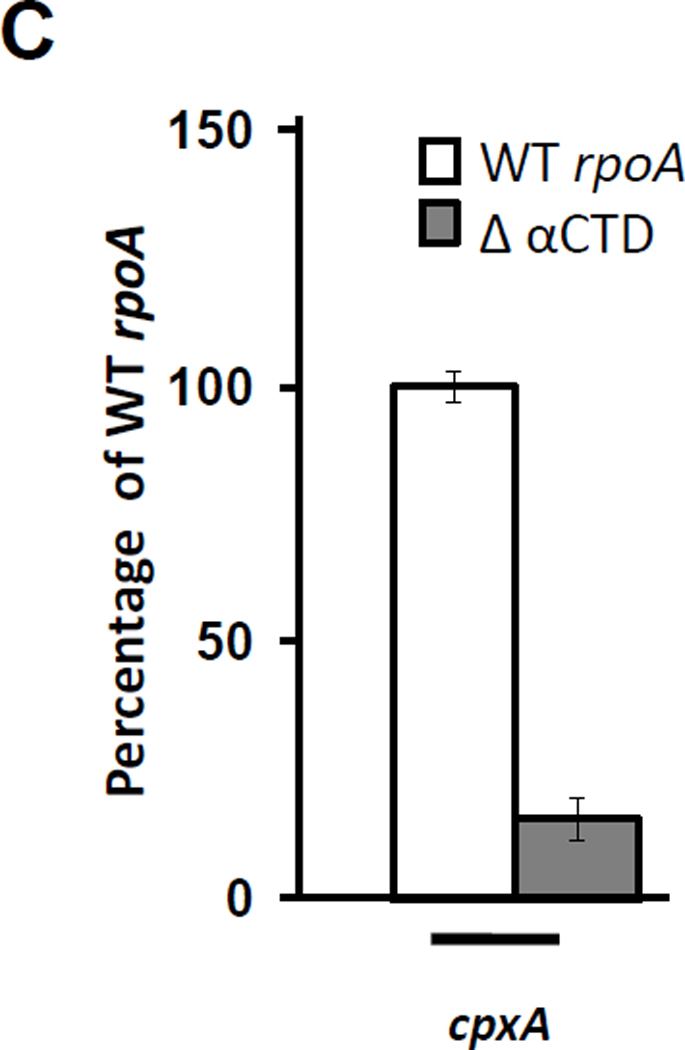
Figure 8. Glucose-induced cpxP transcription requires K298 of αCTD.
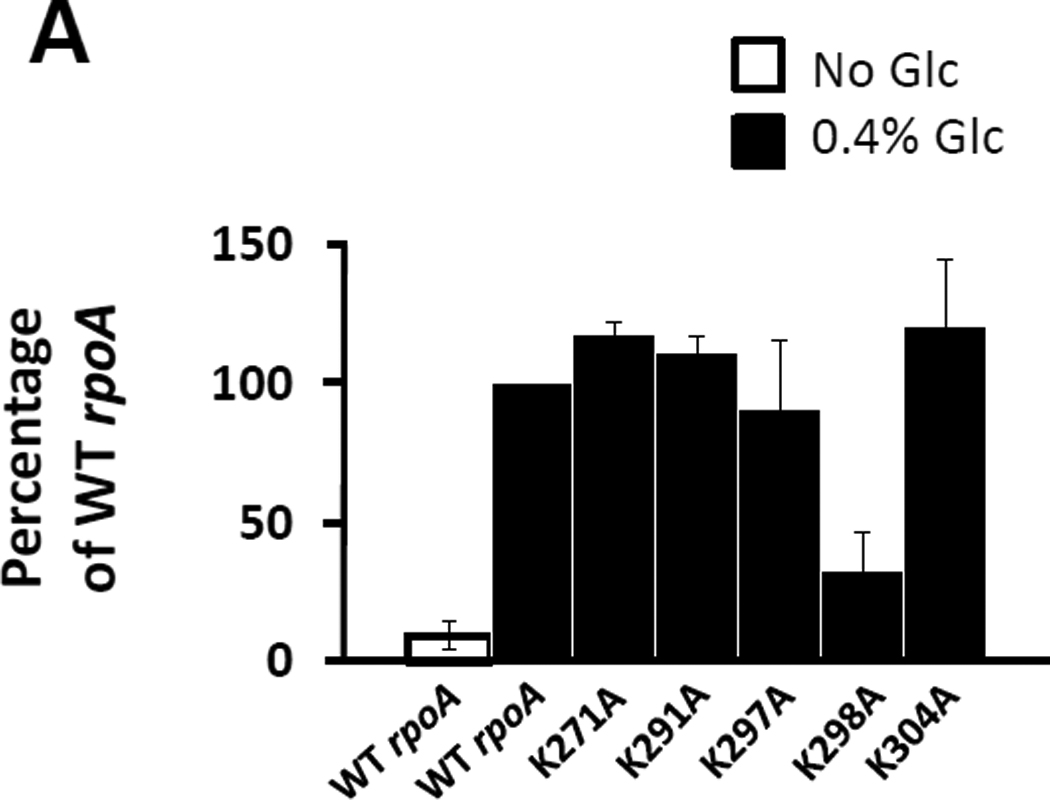
A) β-galactosidase activity of a λcpxP lysogen of the cpxA mutant (strain PAD348) transformed with plasmid pREII carrying the wild-type allele of rpoA, or lysine-to-alanine mutant derivatives of residues 271, 291, 297, 298 and 304. The resultant transformants were grown at 37°C with shaking in buffered TB (open bars) or the same medium supplemented with 0.4% glucose (closed bars). The wild-type response to glucose was set at 100% and the response observed by each of the mutant derivatives was expressed as a percentage of the activity exhibited by wild-type when exposed to 0.4% glucose. The bars indicate the mean of 5 independent experiments carried out with three independent cultures, and the error bars indicate the standard deviations.
B) β-galactosidase activity of λcpxP lysogens of the cpxA (PAD348) transformed with plasmid pREII carrying either the wild-type rpoA allele or the K298A mutant allele. Transformants were grown at 37°C with shaking in TB buffered at pH 7.0 (open bars), the same medium supplemented with 0.4% glucose (closed bars), or TB buffered at pH 7.8 (gray bars). The bars indicate the means of triplicate independent cultures, and the error bars indicate the standard deviations.
C) β-galactosidase activity of λcpxP lysogens of the cpxA mutant (strain PAD348) transformed with plasmid pREII carrying the WT allele of rpoA (white bar), or an rpoA mutant allele lacking the CTD (gray bar). Transformants were grown at 37°C with shaking in buffered TB medium supplemented with 0.4% glucose. cpxP transcription of the wild-type response to glucose was set at 100% and the response observed by the Δ αCTD mutant derivative was expressed as a percentage of the activity exhibited by the wild-type when exposed to 0.4 % glucose. The bars indicate the mean of 5 independent experiments carried out with three independent cultures, and the error bars indicate the standard deviations.
To ensure that the K298A mutant retained function, we tested if a CpxA-dependent stimulus (i.e., exposure to alkaline pH) could induce cpxP transcription in cells WT for CpxA (strain PAD282). Indeed, cells expressing the K298A mutant activated cpxP transcription when grown in TB buffered at pH 7.8, but not in TB buffered at pH 7.0 and supplemented with glucose. In contrast, cells expressing WT rpoA activated cpxP transcription under both conditions (Fig. 8B).
We further tested whether the loss of glucose-induced cpxP transcription observed with the K298A mutant was due to a dominant-negative effect by measuring β-galactosidase activity of cells that carried a mutant α lacking its CTD. Into the cpxA mutant (PAD348), we introduced plasmids that carry either the WT rpoA allele or a mutant derivative with the CTD and the linker region deleted. We grew resultant transformants in the absence or presence of glucose and measured β-galactosidase activity. Like the K298A mutant, the αCTD deletion mutant exhibited a defect in glucose-induced cpxP transcription (Fig. 8C). We interpret these results to mean that K298A allele encodes a functional form of αCTD and that K298 is required for glucose-induced cpxP transcription, but not for CpxA-induced cpxP transcription.
Discussion
We propose that Nε-lysine acetylation can modulate bacterial transcription (Fig. 9). We base this proposal on the following observations: (1) exposure to glucose or other carbon sources predicted to increase the AcCoA-to-CoA ratio can induce cpxP transcription in a sensor kinase-independent manner, (2) genetic manipulations predicted to decrease that ratio can inhibit induction, (3) exposure to the NAD+-dependent deacetylase inhibitor nicotinamide induces cpxP transcription in the absence of glucose or other AcCoA-producing carbon sources, (4) overexpression of the NAD+-dependent deacetylase CobB inhibits glucose-induced cpxP transcription, and (5) glucose-induced cpxP transcription specifically requires the GCN5-like acetyltransferase YfiQ and a lysine residue (K298) located on the surface of the αCTD that, under our growth conditions, is acetylated in a glucose- and YfiQ-dependent manner.
Figure 9. Transcription activation of cpxP by protein acetylation: a model.
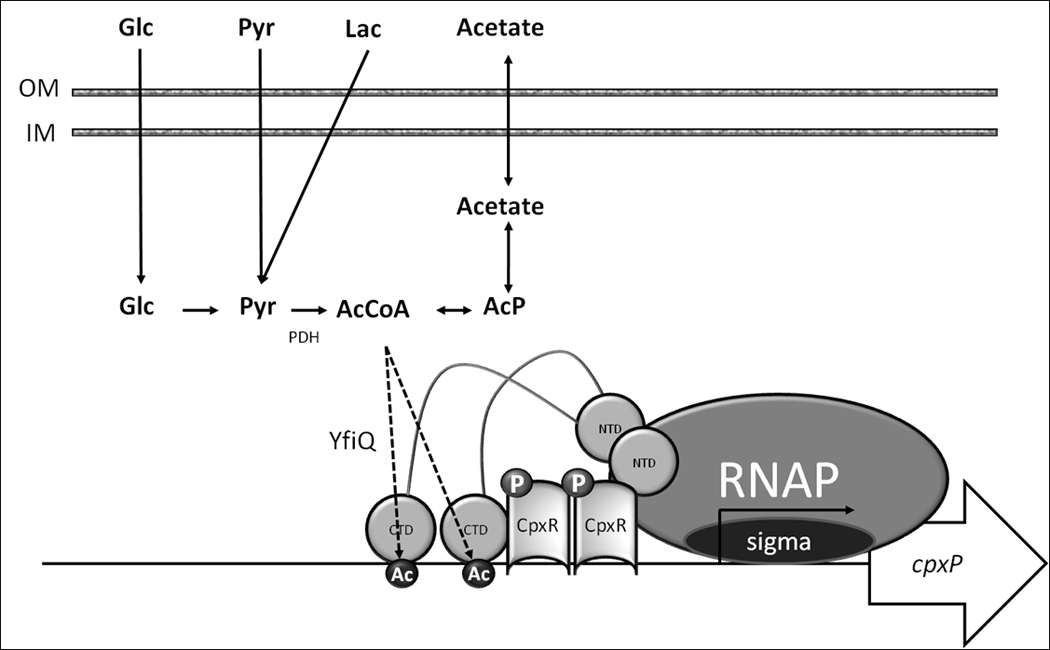
Glucose (Glc) and lactate (Lac) are converted to pyruvate (Pyr), which is converted to AcCoA by the enzyme complex pyruvate dehydrogenase (PDH). AcCoA is interconverted with acetate by the Pta-Acka pathway, with acetyl phosphate (AcP) as an intermediate (Wolfe, 2005). AcCoA serves as an acetyl-donor to the acetyltransferase YfiQ, which is required for acetylation of several sites on RNAP, including K298 on α, which is required for CpxA-independent activation of cpxP transcription.
A role for acCoA
While one could initially consider the involvement of catabolite repression in the glucose-induced cpxP transcription, the observation that both catabolite-repressing and non-catabolite-repressing carbon sources (e.g. glucose and acetate, respectively) can trigger CpxA-independent cpxP transcription (Fig. 2) argues against it. Instead, this finding focuses attention on the alternative hypothesis that transcription-activating carbon sources contribute to the total intracellular AcCoA pool, perhaps causing an AcCoA-to-CoA imbalance that the cell perceives as a stress. That AcCoA plays a central role in glucose-induced cpxP transcription is supported by the involvement of the PDH complex, as its inactivation diminishes the response to glucose (Fig. 3A). The possibility that cpxP transcription might respond to an AcCoA-to-CoA imbalance is supported by the loss of either glucose- or pyruvate-induced cpxP transcription when pantothenate kinase (CoaA) is overexpressed (Fig. 3B). The 3-fold increase in the CoA pool brought about by this genetic manipulation (Song & Jackowski, 1992) is presumably enough to alleviate the increased demand exerted by glucose, pyruvate, lactate or acetate metabolism on the normally limited CoA pool (Wolfe, 2005). That cpxP transcription responds to an AcCoA-to-CoA imbalance is further supported by the observation that drainage of acetyl groups by PHB pathway activity reduces glucose-induced cpxP transcription (Supplemental Fig. S2). Taken together, these observations provide evidence to support the proposal that the AcCoA-to-CoA ratio is a significant contributor to glucose-induced cpxP transcription, perhaps by contributing to the regulation of protein acetylation as previously proposed (Albaugh et al., 2011).
A role for Nε-acetylation
Like exposure to glucose, pyruvate or acetate (Fig. 2), exposure to nicotinamide induced cpxP transcription (Fig. 4A). In contrast, deletion of the GCN5-like acetyltransferase YfiQ (Fig. 6) or overexpression of CobB, the only NAD+-dependent deacetylase encoded by the E. coli genome, prevented glucose-induced cpxP transcription (Fig. 4B). Taken together, these observations support the hypothesis that acetylation of some protein triggers activation of cpxP transcription.
Exposure to glucose also altered the acetylation status of multiple proteins (Fig. 5). This was primarily, but not exclusively, observed as an increase in the number and/or intensity of acetylated bands as detected by Western immunoblot analysis, an observation that agrees with a recent report of protein acetylation in S. enterica (Wang et al., 2010). Unlike glucose, the CobB inhibitor nicotinamide caused relatively few changes in the total acetylation profile, as previously reported (Zhang et al., 2009). Thus, nicotinamide and therefore CobB inhibition likely exert a more limited effect on the E. coli acetylome than does glucose. Several factors could contribute to this observation: (1) the deacetylase CobB might have a limited number of acetylated targets, whereas the additional AcCoA could be used as an acetyl donor by multiple acetyltransferases; (2) CobB might not be fully expressed and active when the samples are collected for analysis; and (3) presently unidentified deacetylases could exist (L. I. Hu and A. J. Wolfe, unpublished data). Under any of these circumstances, inhibition of the deacetylase CobB would not be expected to exert a large impact on the acetylome. It should be noted, however, that CobB overexpression antagonized glucose-induced acetylation. This behavior is consistent with one of three possibilities: (1) CobB could directly reverse glucose-induced acetylations, (2) CobB could activate another deacetylase(s) that reverses those acetylations, or (3) CobB could inhibit the acetyltransferase(s) responsible for those acetylations.
Despite exerting quite different effects upon the acetylome, glucose and nicotinamide both activated cpxP transcription. This argues that the key acetylated lysine likely resides on a protein not obviously detected by Western immunoblot analysis of cell lysates.
A role for an acetylated lysine
Following enrichment by immunoprecipitation, mass spectrometry analysis revealed acetylation of several surface-exposed lysines of the β, β’, and α subunits of RNAP isolated from WT cells exposed to glucose (Fig. 7, Table 1, Supplemental Tables S1–S3). In contrast, we obtained no evidence that CpxR becomes acetylated (data not shown). Because α was acetylated on only two lysines, because YfiQ controlled acetylation of K298 but not of K297, and because the αCTD is instrumental in several aspects of transcription initiation, we further investigated the effect of αCTD acetylation on cpxP promoter. Using a merodiploid system, we asked whether K298 played a substantial role in glucose-induced cpxP transcription and found that it did. Expression of the K298A mutant inhibited transcription, whereas similar expression of the other α K-to-A mutants (including K297A) had no significant effect on glucose-induced cpxP transcription (Fig 8A). Furthermore, the requirement for K298 was specific: extracytoplasmic stimuli that activate CpxA kinase activity and thus rapid phosphorylation of CpxR did not require K298 (Fig 8B).
The findings presented in this manuscript substantially add to data presented by previous reports that RNAP subunits can become acetylated (Yu et al., 2008, Zhang et al., 2009). We detected the previously reported acetylation sites on β (K1122 and K1200) and β’ (K983) (Table 1), but not on α (K291). Our data, however, increase the number of identified acetylated RNAP lysines from 3 to almost 30. It also establishes at least one growth condition under which these acetylation events likely occur and implicate the acetyltransferase YfiQ in acetylation of the majority of those sites, including K1200 of β. Thus, it appears that RNAP can become acetylated on multiple lysine residues on different subunits and that the acetylation pattern of RNAP can differ depending on the growth condition. In this case, one could imagine that distinct acetylation events could differentially impact RNAP activities, including its ability to bind to promoters and to interact with transcription factors. Indeed, we possess evidence that relatively slight alterations to either the external or genetic environments can shift the RNAP acetylation profile and that this shift is accompanied by a change in output from activation to inhibition of cpxP transcription (B. P. Lima et al., in preparation).
Model
We envision a simple model that accounts for all our observations (Fig. 9). When a cell grows in buffered TB, the lack of a strong extracytoplasmic stimulus permits CpxA to function as a net phosphatase, removing phosphoryl groups from phospho-CpxR. If that cell becomes exposed to a carbon source (e.g. glucose, pyruvate, lactate or acetate) whose metabolism results in an AcCoA-to-CoA imbalance, the acetyltransferase YfiQ catalyzes the acetylation of several lysines on the surface of RNAP, including K298 of α. If metabolism of that carbon source (e.g. glucose or lactate) requires an NAD+-dependent enzyme (e.g. glyceraldehyde dehydrogenase or lactate dehydrogenase, respectively), then a competition might ensue with the NAD+-dependent deacetylase CobB for the limited supply of NAD+. This competition would result in reduced deacetylation of CobB targets. The outcome of increased acetylation and/or decreased deacetylation would be activation of cpxP transcription.
Several important questions remain unanswered. What is the function of CpxR in the cpxP response to glucose? In addition to AcCoA and NAD+ availability, how does the cell regulate YfiQ and CobB expression and/or activity? Do other protein acetyltransferases and deacetylases exist? Importantly to this report, does YfiQ directly acetylate K298 and does K298 acetylation induce cpxP transcription? While our genetic and MS evidence supports the existence of a YfiQ/K298 relationship, it might not be direct. For example, YfiQ could work indirectly, regulating the synthesis of a signal and/or the activity of another acetyltransferase and/or deacetylase. Efforts to obtain direct biochemical evidence by reconstituting RNAP acetylation in vitro are proceeding with caution, as we do not know the requirements beyond RNAP, CpxR, YfiQ, AcCoA and the cpxP promoter. For example, properly targeted acetylation may require assembly of a nucleoprotein complex that could include nucleoid histone-like proteins, a scaffold protein analogous to the eukaryotic bromodomain to correctly dock the acetyltransferase or deacetylase, and/or a second covalent modification to dictate whether acetylation of a particular lysine can occur as observed for p53 and many other eukaryotic acetylated proteins (Kouzarides, 2000, Yang & Seto, 2008).
We also do not know the percentage of α that is acetylated on K298, a value that can only be obtained by quantitative mass spectrometry methods. However, we have a few clues that suggest that the majority of α isolated from glucose-exposed WT cells is acetylated. These clues come from the fact that trypsin, the protease used to obtain the MS-analyzed peptides, cleaves after lysines but not after acetyllysines. From glucose-naïve WT cells, we detected the non-acetylated peptides (291–297) TPNLGK and (299–310) SLTEIKDVLASR, suggesting that K297 and K298 were not acetylated. In contrast, from glucose-exposed WT cells, we detected the related but acetylated peptides (291–298) TPNLGK297(+42)K and (298–304) K298(+42)SLTEIK (Fig. 7), suggesting that K297 and K298 could be acetylated. Strikingly, the inability to detect the non-acetylated peptides TPNLGK and SLTEIKDVLASR in glucose-exposed samples (Supplemental Table S3) suggests that much of the α isolated from glucose-exposed WT cells is acetylated on K297 and K298.
If our proposed model is true, then how could acetylation of K298 affect cpxP transcription? Analysis of the crystal structure of CRP and αCTD bound to a synthetic DNA fragment containing two CRP binding sites flanked by αCTD binding sites suggests that K298 of α makes direct interaction with DNA adjacent to the CRP binding site (Benoff et al., 2002). Thus, K298 likely plays a role in closed complex formation, the first step in transcription initiation. So how could acetylation of K298 work? While we do not know how acetylation of K298 contributes to cpxP transcription, it is possible that non-acetylated αCTD binds to a site that is not conducive to CpxR-dependent activation. If so, the acetylation of K298 could re-position the αCTD, permitting it to make productive contact with CpxR. Alternatively, CpxR and RNAP could form an overly stable complex that binds tightly to the cpxP promoter. If so, then acetylation of K298 might loosen this interaction, facilitating promoter clearance. Whichever is true, it should be noted that although K298 is required for glucose-induced cpxP transcription, it may not be sufficient; other acetylated lysines on the surface of the other RNAP subunits might also participate. A more direct test of this model requires in vitro transcription, in which CpxR and properly acetylated RNAP are used drive transcription from the cpxP promoter.
Other questions remain: What are the general rules for activation? How general is the effect of K298 acetylation? Does protein acetylation influence transcription from other CpxR-dependent promoters? Does it influence CpxR-independent promoters? We possess evidence that the response to glucose and other carbon sources is context-dependent, i.e. some CpxR-dependent promoters respond to carbon source exposure and some do not (A. Cosgrove, B. P. Lima and A. J. Wolfe, unpublished data). This context-dependence suggests that the mechanism(s) by which acetylation influences transcription is not simple, and may involve factors such as promoter architecture and nucleoprotein complex composition.
Concluding Remarks
Great strides have been made to understand the role of protein acetylation in eukaryotes. In contrast, our knowledge of protein acetylation in bacteria remains in its infancy. Our current knowledge is restricted to three lists of acetylated bacterial proteins, the structures of several bacterial GNATs, some functions of a single acetyltransferase (YfiQ/Pat), a single deacetylase (the NAD+-dependent sirtuin CobB), and the effects of acetylation on the functions of two acetylated proteins that function in central metabolism and chemotaxis [reviewed in (Hu et al., 2010)]. This manuscript extends that knowledge by (1) providing support for the hypothesis that the AcCoA-to-CoA ratio is a major determinant in controlling protein acetylation (Albaugh et al., 2011), (2) showing that several RNAP subunits can become acetylated on multiple lysines, (3) determining that the GCN5-like acetyltransferase YfiQ plays a major role in RNAP acetylation, and (4) demonstrating that the protein acetylation machinery of E. coli affects transcription of at least one promoter, potentially by its ability to modify RNAP, the molecular machine that performs transcription of all bacterial genes.
Materials and Methods
Bacterial strains, plasmids, and bacteriophage
All bacterial strains used in this study are listed in Table 2. Derivatives were constructed by generalized transduction with P1kc, as described previously (Silhavy et al., 1984). The transcriptional fusion Φ(PcpxP-lacZ), carried by λPcpxP, and described previously (Danese & Silhavy, 1998), was a generous gift from Thomas Silhavy (Princeton University, Princeton, NJ). Construction of monolysogens was performed and verified as described previously (Simons et al., 1987, Powell et al., 1994).
Culture conditions
For strain construction, cells were grown in LB containing 1% (wt/vol) tryptone, 0.5% (wt/vol) yeast extract, and 0.5% (wt/vol) sodium chloride; LB plates also contained 1.5% agar. For promoter activity assays, cells were grown in buffered TB which consisted of 1% (wt/vol) tryptone buffered at pH 7.0 with potassium phosphate (100 mM). Cell growth was monitored spectrophotometrically (DU640; Beckman Instruments, Fullerton, CA) by determining the optical density at 600 nm (OD600). Kanamycin (40 µg/ml), chloramphenicol (25 µg/ml), and ampicillin (100 µg/ml) were added as needed. When necessary, isopropyl β-D-1-thiogalatopyranoside (IPTG) was added to the media at the indicated concentration to induce gene expression from a plasmid vector. Nile red (Sigma Aldrich), at a final concentration of 25 µg/ml, was added to LB plates to detect in vivo PHB accumulation as previously described (Spiekermann et al., 1999).
Plasmid construction
To construct plasmid pPHB1, alleles phbCAB+ were amplified from Ralstonia solanacearum strain GMI1000 (a kind gift from Caitilyn Alen, UW-Madison) using forward primer 5’—ATC GAA CCC GGG ATG GCA TCG CGT CAC AA—3’ and reverse primer 5’—AGC ATG AGT GCC CCG GGT CAG CCC ATG T—3’. Bases underlined indicate the SmaI restriction site engineered into each primer. The resulting ~4-kb fragment was A-tailed and gel-purified using the QIAquick® gel extraction kit (Qiagen). This product was ligated into the multiple cloning site of plasmid pGEM-T-Easy (Promega). The resulting plasmid was ~7-kb and was named pPHB1.
To construct plasmid pPHB3, the Rs phbCAB+ genes were cut out of pPHB1 using SmaI. The resulting ~4-kb fragment was gel-extracted and ligated into plasmid pTAC-85 (Marsh, 1986) cut with NcoI where the single-stranded DNA overhangs were blunted with mung bean nuclease and dephosphorylated using shrimp alkaline phosphatase. The resulting plasmid was ~9.1-kb and was named pPHB3.
Promoter activity assays
To monitor promoter activity from Φ(PcpxP-lacZ), cells were grown aerobically at 37°C with agitation at 250 rpm in buffered TB, unless otherwise noted. At regular intervals, 50 µl aliquots were harvested and added to 50 µl of All-in-One β-galactosidase reagent (Pierce Biochemical). β-Galactosidase activity was determined quantitatively using a microtiter format, as described previously (Beatty et al., 2003). Promoter activity was plotted versus OD600. Although promoter activity was determined over the entire growth curve, only the peak activity is shown. This peak activity invariably occurred as the cultures entered stationary phase (Wolfe et al., 2008). Each experiment included three independent measurements and was performed at least twice.
Western immunoblot analysis
To identify the protein acetylation profile, 3 ml cell cultures were grown at 37°C with agitation at 250 rpm in buffered TB for 7.5 hours (i.e., entry into stationary phase), harvested and pelleted by centrifugation. The pellets were resuspended with 500 µl TE buffer solution containing 10 mM Tris HCl pH8.0 and 1 mM EDTA and subjected to sonication for protein extraction. Samples were separated by electrophoresis on a SDS-PAGE 12% polyacrylamide gel, transferred to PVDF membrane, and subjected to Western immunoblot analysis. To determine the acetylation state of proteins, a cocktail of two polyclonal anti-acetyllysine antibodies was used (Cell Signaling Technology®) at a 1:200 dilution and (ImmuneChem®) at a 1:500 dilution. To visualize the α subunit of RNAP, a monoclonal antibody specific for α (NeoClone Biotechnology) was used at a 1:2000 dilution. Incubation was carried out overnight at 4°C with shaking. Secondary goat anti-rabbit (Cell Signaling Technology®) immunoglobulin G antibody at a 1:1000 dilution, or goat anti-mouse (Millipore) immunoglobulin G antibody at 1:5000 dilution both conjugated to horseradish peroxidase were used for 1 hour at room temperature with shaking. Enhanced chemiluminescence Western immunoblotting reagents (Cell Signaling Technology®) were used for visualization, according to manufacturer’s instructions. Membrane stripping was done with OneMinute® Western Blot Stripping Buffer (GM Biosciences), according to manufacturer’s instructions.
Immunoprecipitation
For immunoprecipitation, 50 ml buffered TB cultures were collected after 7.5 hours of incubation at 37°C, pelleted by centrifugation, resuspended in 5 ml TE buffer and lysed by sonication. 1 ml of lysate was used for immunoprecipitation of RNAP with RNAP β mouse monoclonal antibodies (NeoClone Biotechnology) or immunoprecipitation of CpxR with MBP-CpxR rabbit polyclonal antibody (Raivio & Silhavy, 1997) and rotated overnight at 4°C. Protein G-Sepharose® (Sigma) was used to pull down antibodies from cell lysate. Pulled down beads were washed three times with TE buffer and once with wash buffer containing 100 mM NaCl and 50 mM Tris-HCl at pH 7.2. Loading buffer was added directly to the beads and samples were heated to 95°C for 5 minutes. Samples were separated by SDS-PAGE and stained with NOVEX® Colloidal Blue Stain (Invitrogen), according to manufacturer’s instructions.
Orbitrap-mass spectrometry and protein identification
The α, β and β’ bands were excised and subjected to tryptic digestion, as described previously (Chi et al., 2010). Tryptic peptides were separated and measured online by ESI-mass spectrometry using a nanoACQUITY UPLC™ system (Waters, Milford, MA) coupled to an LTQ Orbitrap™ XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA). A trap column (Symmetry® C18, 5 µm, 180µm inner diameter × 20mm, Waters) was used for desalting. Elution was performed onto an analytical column (BEH130 C18, 1,7 µm, 100 µm inner diameter × 100mm, Waters) by a binary gradient of buffers A (0.1% (v/v) acetic acid) and B (99.9 % (v/v) acetonitrile, 0.1% (v/v) acetic acid) over a period of 80 min with a flow rate of 400 nl/min. The Orbitrap XL was operated in data-dependent MS/MS mode using the lockmass option for real time recalibration.
Proteins were identified by searching all MS/MS spectra in “dta” format against an E. coli database (extracted from the Uniprot-KB database: http://www.uniprot.org/uniprot/?query=Escherichia+coli+K12&sort=score) using Sorcerer™-SEQUEST® (Sequest v. 2.7 rev. 11, Thermo Electron including Scaffold_3_00_05, Proteome Software Inc., Portland, OR). The Sequest search was carried out considering the following parameters: a parent ion mass tolerance - 10 ppm, fragment ion mass tolerances of 1.00 Da. Up to two tryptic miscleavages were allowed. Methionine oxidation (+15.99492 Da), cysteine carbamidomethylation (+57.021465 Da) and lysine acetylation (+42.010571 Da) were set as variable modifications. Proteins were identified by at least two peptides applying a stringent SEQUEST filter. Sequest identifications required at least ΔCn scores of greater than 0.10 and XCorr scores of greater than 1.9, 2.2, 3.3 and 3.8 for singly, doubly, triply and quadruply charged peptides. Acetylated peptides that passed these filter criteria were examined manually and accepted only when b− or y− ions confirmed the acetylation site.
Supplementary Material
Acknowledgments
We thank Thomas Silhavy, Patricia DiGiuseppe-Champion, Rick Gourse, and Jorge Escalante-Semerena for their generous donations of strains, plasmids, and/or antibodies. We thank Niyati Parikh for her help with the carbon source experiments and Noriko Shibata and Linda Fox (Loyola University SSOM Core Imaging Facility) for their help with the transmission electron micrograph. We also thank Jorge Escalante-Semerena, Sandy Thao, John Kirby and members of the Wolfe and Visick laboratories for discussions and/or critical reading of the manuscript. This work was supported by NIH grant GM066130 and Loyola University Chicago Potts Foundation award LU11200 awarded to A.J.W, by a grant of the Deutsche Forschungsgemeinschaft AN746/2-1 to H.A., and by NIH grant GM62203 to J. C. Escalante-Semerena, which supported S.R.B.
References
- Albaugh BN, Arnold KM, Denu JM. KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. Chembiochem. 2011;12:290–298. doi: 10.1002/cbic.201000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty CM, Browning DF, Busby SJ, Wolfe AJ. Cyclic AMP receptor protein-dependent activation of the Escherichia coli acsP2 promoter by a synergistic class III mechanism. J Bacteriol. 2003;185:5148–5157. doi: 10.1128/JB.185.17.5148-5157.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoff B, Yang H, Lawson CL, Parkinson G, Liu J, Blatter E, Ebright YW, Berman HM, Ebright RH. Structural basis of transcription activation: the CAP-alpha CTD-DNA complex. Science. 2002;297:1562–1566. doi: 10.1126/science.1076376. [DOI] [PubMed] [Google Scholar]
- Blatter EE, Ross W, Tang H, Gourse RL, Ebright RH. Domain organization of RNA polymerase alpha subunit: C-terminal 85 amino acids constitute a domain capable of dimerization and DNA binding. Cell. 1994;78:889–896. doi: 10.1016/s0092-8674(94)90682-3. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Nudler E. RNA polymerase holoenzyme: structure, function and biological implications. Curr Opin Microbiol. 2003;6:93–100. doi: 10.1016/s1369-5274(03)00036-5. [DOI] [PubMed] [Google Scholar]
- Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- Chi BK, Albrecht D, Gronau K, Becher D, Hecker M, Antelmann H. The redox-sensing regulator YodB senses quinones and diamide via a thiol-disulfide switch in Bacillus subtilis. Proteomics. 2010;10:3155–3164. doi: 10.1002/pmic.201000230. [DOI] [PubMed] [Google Scholar]
- Chohnan S, Izawa H, Nishihara H, Takamura Y. Changes in size of intracellular pools of coenzyme A and its thioesters in Escherichia coli K-12 cells to various carbon sources and stresses. Biosci Biotechnol Biochem. 1998;62:1122–1128. doi: 10.1271/bbb.62.1122. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004;24:728–743. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- Danese PN, Silhavy TJ. CpxP, a stress-combative member of the Cpx regulon. J Bacteriol. 1998;180:831–839. doi: 10.1128/jb.180.4.831-839.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 1995;9:387–398. doi: 10.1101/gad.9.4.387. [DOI] [PubMed] [Google Scholar]
- De Wulf P, McGuire AM, Liu X, Lin EC. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J Biol Chem. 2002;277:26652–26661. doi: 10.1074/jbc.M203487200. [DOI] [PubMed] [Google Scholar]
- DiGiuseppe PA, Silhavy TJ. Signal detection and target gene induction by the CpxRA two-component system. J Bacteriol. 2003;185:2432–2440. doi: 10.1128/JB.185.8.2432-2440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaal T, Ross W, Blatter EE, Tang H, Jia X, Krishnan VV, Assa-Munt N, Ebright RH, Gourse RL. DNA-binding determinants of the alpha subunit of RNA polymerase: novel DNA-binding domain architecture. Genes Dev. 1996;10:16–26. doi: 10.1101/gad.10.1.16. [DOI] [PubMed] [Google Scholar]
- Hu LI, Lima BP, Wolfe AJ. Bacterial protein acetylation: the dawning of a new age. Mol Microbiol. 2010;77:15–21. doi: 10.1111/j.1365-2958.2010.07204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S, Beatty CM, Browning DF, Busby SJ, Simel EJ, Hovel-Miner G, Wolfe AJ. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J Bacteriol. 2000;182:4173–4179. doi: 10.1128/jb.182.15.4173-4179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh P. Ptac-85, an E. coli vector for expression of non-fusion proteins. Nucleic Acids Res. 1986;14:3603. doi: 10.1093/nar/14.8.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Chatterji D. The evolving story of the omega subunit of bacterial RNA polymerase. Trends Microbiol. 2006;14:450–455. doi: 10.1016/j.tim.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Nawrath C, Poirier Y, Somerville C. Targeting of the polyhydroxybutyrate biosynthetic pathway to the plastids of Arabidopsis thaliana results in high levels of polymer accumulation. Proc Natl Acad Sci U S A. 1994;91:12760–12764. doi: 10.1073/pnas.91.26.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostle AG, Holt JG. Nile blue A as a fluorescent stain for poly-beta-hydroxybutyrate. Appl Environ Microbiol. 1982;44:238–241. doi: 10.1128/aem.44.1.238-241.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto K, Silhavy TJ. Surface sensing and adhesion of Escherichia coli controlled by the Cpx-signaling pathway. Proc Natl Acad Sci U S A. 2002;99:2287–2292. doi: 10.1073/pnas.042521699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogliano J, Lynch AS, Belin D, Lin EC, Beckwith J. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev. 1997;11:1169–1182. doi: 10.1101/gad.11.9.1169. [DOI] [PubMed] [Google Scholar]
- Powell BS, Rivas MP, Court DL, Nakamura Y, Turnbough CL., Jr Rapid confirmation of single copy lambda prophage integration by PCR. Nucleic Acids Res. 1994;22:5765–5766. doi: 10.1093/nar/22.25.5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Raivio TL. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J Bacteriol. 2009;191:1798–1815. doi: 10.1128/JB.00798-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivio TL, Popkin DL, Silhavy TJ. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J Bacteriol. 1999;181:5263–5272. doi: 10.1128/jb.181.17.5263-5272.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivio TL, Silhavy TJ. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J Bacteriol. 1997;179:7724–7733. doi: 10.1128/jb.179.24.7724-7733.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, Gourse RL. A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science. 1993;262:1407–1413. doi: 10.1126/science.8248780. [DOI] [PubMed] [Google Scholar]
- Silhavy TJ, Berman ML, Enquist LW. Experiments with gene fusions. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1984. [Google Scholar]
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, Huber LJ. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Jackowski S. Cloning, sequencing, and expression of the pantothenate kinase (coaA) gene of Escherichia coli. J Bacteriol. 1992;174:6411–6417. doi: 10.1128/jb.174.20.6411-6417.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiekermann P, Rehm BH, Kalscheuer R, Baumeister D, Steinbuchel A. A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch Microbiol. 1999;171:73–80. doi: 10.1007/s002030050681. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Gardner JG, Escalante-Semerena JC. Residue Leu-641 of Acetyl-CoA synthetase is critical for the acetylation of residue Lys-609 by the Protein acetyltransferase enzyme of Salmonella enterica. J Biol Chem. 2005;280:26200–26205. doi: 10.1074/jbc.M504863200. [DOI] [PubMed] [Google Scholar]
- Steinbuchel A, Schlegel HG. Physiology and molecular genetics of poly(beta-hydroxy-alkanoic acid) synthesis in Alcaligenes eutrophus. Mol Microbiol. 1991;5:535–542. doi: 10.1111/j.1365-2958.1991.tb00725.x. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Thao S, Escalante-Semerena JC. Control of protein function by reversible Nvarepsilon-lysine acetylation in bacteria. Curr Opin Microbiol. 2011;14:200–204. doi: 10.1016/j.mib.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallari DS, Jackowski S. Biosynthesis and degradation both contribute to the regulation of coenzyme A content in Escherichia coli. J Bacteriol. 1988;170:3961–3966. doi: 10.1128/jb.170.9.3961-3966.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AJ, Parikh N, Lima BP, Zemaitaitis B. Signal integration by the two-component signal transduction response regulator CpxR. J Bacteriol. 2008;190:2314–2322. doi: 10.1128/JB.01906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. The diversity of lysine-acetylated proteins in Escherichia coli. J Microbiol Biotechnol. 2008;18:1529–1536. [PubMed] [Google Scholar]
- Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.