

Abstract
Oculodentodigital dysplasia (ODDD) is primarily an autosomal dominant human disease caused by any one of over 60 mutations in the GJA1 gene encoding the gap junction protein Cx43. In the present study, wound healing was investigated in a G60S ODDD mutant mouse model and by using dermal fibroblasts isolated from two ODDD patients harboring the p.D3N and p.V216L mutants along with dermal fibroblasts isolated from their respective unaffected relatives. Punch biopsies revealed a delay in wound closure in the G60S mutant mice in comparison to wild type littermates and this delay appeared to be due to defects in the dermal fibroblasts. While both the p.D3N and p.V216L mutants reduced gap junctional intercellular communication in human dermal fibroblasts, immunolocalization studies revealed that Cx43 gap junctions were prevalent at the cell surface of p.D3N expressing fibroblasts but greatly reduced in p.V216L expressing fibroblasts. Mutant expressing fibroblasts were further found to have reduced proliferation and migration capabilities. Finally, in response to TGFβ1, mutant expressing fibroblasts expressed significantly less alpha smooth muscle actin suggesting they were inefficient in their ability to differentiate into myofibroblasts. Collectively, our results suggest that ODDD patients may have subclinical defects in wound healing due to impaired function of dermal fibroblasts.
Keywords: Cx43, GJA1, oculodentodigital dysplasia, ODDD, fibroblasts, gap junctions, phosphorylation
Introduction
The human GJA1 gene (BC026329; MIM# 121014) encodes the most commonly found gap junction protein found in the body, Cx43 (Solan and Lampe, 2009). The ubiquitous expression of Cx43 and primary function in establishing gap junctional intercellular communication (GJIC) has led to this connexin being one of the most widely studied members of the connexin family. GJIC plays a critical role in many physiological processes ranging from more passive roles in cell and tissue maintenance to a highly active role in synchronization of electrical coupling (Kizana, et al., 2007). Not surprisingly, dysregulation of either connexin expression or mutations in genes encoding connexins has led to a wide array of human diseases.
In 2003, Paznekas and colleagues successfully mapped a developmental disorder known as oculodentodigital dysplasia (ODDD) to mutations in the GJA1 gene (Paznekas, et al., 2003). Presently, 62 mutations have been mapped to the GJA1 gene and linked to ODDD (Paznekas, et al., 2009). ODDD is a rare, mostly autosomal dominant inherited disorder affecting the development of a variety of tissues and organs. Though ODDD patients exhibit some consistent features such as syndactyly, camptodactyly, craniofacial abnormalities, enamel loss and microdontia, other less common features include glaucoma, neurological defects (progressive spastic paraparesis), neurogenic bladder and even heart and skin diseases (Paznekas, et al., 2009). Missense mutations are by far the most widely found type of mutations, but two frame-shift mutations have also been identified (van Steensel, et al., 2005; Vreeburg, et al., 2007). Surprisingly, two autosomal recessive mutations have been reported and one of these results in the premature truncation of Cx43 after only encoding 33 amino acids, effectively resulting in a near complete Cx43 knockout (Richardson, et al., 2006). GJA1 gene mutations are found throughout the expanse of the gene but the vast majority of the mutations occur in the first two thirds of the gene. At present, over 15 specific mutations have been engineered, over-expressed and studied in reference cell systems to assess the ability of the mutant Cx43 to traffic, assemble and form functional channels (Churko, et al., 2010; McLachlan, et al., 2005). Intriguingly, all of the Cx43 mutants examined under these conditions have severely compromised intercellular channel forming abilities and exhibit moderate to severe dominant-negative effects on co-expressed endogenous Cx43. Thus, these in vitro studies would predict that patients harboring autosomal dominant Cx43 mutants are operating on far less than 50% normal Cx43 function.
A well-documented limitation of mutant overexpression studies is the difficulty in matching the expression level of the mutant and wild-type counterpart to equal the 1:1 expression level expected to be found in patients. To overcome this shortcoming, three mouse models of ODDD have been generated in an attempt to mimic the geneotype/phenotype relationship found in the human disease. The first of these models was engineered in 2005 through an ethylnitrosurea screen where a mouse, designated Gja1Jrt/+, was found to harbor a G60S missense mutation (Flenniken, et al., 2005). While this mutation has not yet been identified in the human population, the mouse phenotype was characterized to closely match the clinical symptoms exhibited by ODDD patients. This mouse model has been used extensively and has shed insight into the molecular mechanisms underlying ODDD. In more recent years, two additional mouse models have been engineered to reflect human mutations of interest. The first one was designed to generically express an I130T Cx43 mutant (Cx43I130T/+) (Kalcheva, et al., 2007). The second constitutively expressed the G138R mutant (Cx43G138R/+) (Dobrowolski, et al., 2008). Given the recent generation of these mice, they are only beginning to be studied as mouse models of ODDD. Seminal publications describing these mice indicate that while they manifest common characteristics of ODDD, there are unique features to each mouse strain that will require further study.
Although mutant Cx43 expression in reference cell models and genetic mouse models of ODDD are valuable tools to investigate the cell mechanisms associated with ODDD, they may not fully reflect the pathophysiologic aspects of ODDD exhibited by patients. In the current study, we attempt to bridge this shortcoming by isolating and culturing the first primary culture cells from ODDD patients. Here we recruited two ODDD families to obtain human skin tissue specimens for the establishment of matched fibroblast cell lines from both patients and unaffected close relatives. We further compared our findings to fibroblasts derived from the Gja1Jrt/+ mouse model to assess potential human/mouse differences. We found that fibroblasts obtained from these patients are significantly different from their genetically matched control fibroblasts in their proliferation, migration and differentiation into myofibroblasts. Here we suggest that wound healing in ODDD patients may be delayed and propose a mechanism by which fibroblasts may exacerbate disease in these patients.
Materials and Methods
Human fibroblast cultures
Fresh skin biopsy samples obtained from patients and unaffected relatives were placed in ice cold DMEM (25 mL, Cat# 11960-044, Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS, Cat# A12617DJ, Invitrogen) prior to being cut into small pieces and transported into a second dish containing DMEM with 10% FBS and 100 U/ml penicillin & 100 μg/ml streptomycin (Cat# 15140-122, Invitrogen), and incubated at 37 °C in a humidified chamber containing 5% CO2. The media was changed once a week for 3-4 weeks. Cells were subcultured once they reached confluence by treating with trypsin (0.05% trypsin with 0.25 EDTA, Cat# 25200-056 Invitrogen). For long-term storage, collected cells were resuspended in cryopreservation media (DMEM with 10% FBS, 1× antibiotic/antimycotic and 10% DMSO-filtered (Cat# D2650, Sigma-Aldrich, St. Louis, MO) and aliquoted into cryotubes. Cells were stored in liquid nitrogen. Human fibroblasts were sent to the University of Western Ontario for further analysis as approved by the Office of Research Ethics at the University of Western Ontario. All fibroblasts cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine and incubated at 37 °C in humidified air with 5% CO2. In subsequent experiments, results were compared from passage-matched fibroblasts used from passages 2 to 6.
Extraction of genomic DNA for sequencing
Genomic DNA was extracted from peripheral blood leukocytes and/or fibroblast cells of all individuals by standard methods with proteinase K (Cat# 51104, Qiagen DNA Blood kit, Qiagen, Mississauga, ON). PCR amplification of the Cx43 gene was performed by using Cx43 primers of the forward 5 ’-TGGGACAGGAAGAGTTTGCAC-3 ’ and reverse 5 ’-CACCTGGTGCACTTTCTACAGCAC-3’ and the sequencing of Cx43 gene by using primer 1: 5’-GGTGGCCTTCTTGCTGATCC-3’, primer 2: 5’-TGGGCAGGGATCTCTTTTGC-5’ and primer 3: 5’-GGTTGCCCAAACTGATGGTG.
Dye coupling assay
A preloading assay was used to quantify GJIC in primary fibroblast cultures as described previously (Goldberg, et al., 1995). Briefly, a subpopulation of primary fibroblasts was loaded with the gap junction permeable dye, calcein-AM (Cat# D1430, Molecular Probes, Eugene, OR), and the gap junction impermeable dye, DiI (Cat# D282, Molecular Probes, Eugene, OR). The preloaded cells (~ 100 cells) were seeded onto a monolayer of unlabeled primary fibroblast cells, and after 3 h, samples were observed and images recorded with a Leica DM IRE2 inverted epifluorescence microscope. The instances of calcein dye transfer from the DiI-labeled source cells were counted, and successful dye transfer was declared if there was clear evidence of dye transfer to at least one cell adjacent to DiI donor cell. Three independent trials were performed for each cell line. Standard errors were calculated, and the control and mutant cells were compared with a Student’s t-test.
Patch-clamp electrophysiology
The intercellular electrical coupling conductance between control or mutant fibroblasts was assessed by using the dual whole cell patch-clamp technique as previously described (Gong, et al., 2006). Human fibroblasts were plated on glass coverslips in cell culture dishes at low density for 24 ~48 hours. Isolated fibroblast cell pairs with side-to-side contacts were chosen for double patch-clamp recording. Data acquisition and analysis were performed via Digidata 1322A interface and pClamp9 software (Axon Instruments Inc., Union City, CA). Gap junctional conductance (Gj) was determined and presented as mean ± S.E. Online series resistance compensation at 80% was applied to improve the accuracy of measured Gj. A Student’s t-test was performed to determine statistical significance (**, p < 0.01).
Immunocytochemistry and immunohistochemistry
Cells were grown on glass coverslips and fixed with ice-cold 80% methanol/20% acetone at 4°C for 20 min and rinsed with PBS 3 times before immunolabeling. Antibodies to the gap junction protein, polyclonal Cx43 (1:500; Cat# C6219, Sigma-Aldrich, St. Louis, MO) mAb GM130, a 130 kDa Golgi matrix protein antibody (1:100; Cat# 610822, BD Transduction Laboratories, Mississauga, ON), and an intermediate filament, mAb vimentin antibody (1:500; Cat# MAB3400, Millipore, Temecula, CA), were used. Primary antibody binding was detected with appropriate Alexa-488 (anti-mouse Cat# A11017, anti-rabbit Cat# A11008) or Alexa-555–conjugated anti-rabbit (Cat# A21429) or anti-mouse (Cat# A21425) (1:500; Invitrogen, Carlsbad, CA) secondary antibodies. Nuclei were stained with Hoechst 33342 (1:1000; Cat# H3570, Molecular Probes, Eugene, OR) or DAPI (1:1000; Cat# D3571, Molecular Probes). Cells were imaged with a Zeiss LSM 510 META confocal microscope as previously described (Simek, et al., 2009).
Wound healing assay
The wound healing assay was approved by the Office of Research Ethics at the University of Western Ontario. Eleven adult G60S mutant mice and eleven WT control mice were anesthetized with isofluorane gas and the dorsal skin surface was shaved using #40 clippers. Prior to performing a punch biopsy, a three stage incision site preparation (betadine soap/alcohol/betadine scrub) was performed on the shaved surface and a sterile disposable 5 mm bore biopsy tool was used to obtain a 5 mm epidermal and dermal skin biopsy. Buprenorphine (0.05mg/kg) was administered once during anesthesia recovery, and additional doses (8-12h) were administered if the mice looked stressed. The area of wound closure was measured after day 1, 3, 6, 9, and 12 from images taken at these time points. Wound area calculations were performed using ImageJ and the resulting wound area was graphed using Graphpad Prism 4.0. A two-way ANOVA (repeated measures) from data points Day 3, 6, and 9 with a Bonferroni post-hoc test was performed to determine statistical significance (*p<0.05).
Punch biopsy migration
Circular skin punches were cultured ex vivo as previously described (Stephens, et al., 1996). Fresh 5 mm dermal punch biopsies from postnatal day 4 WT and G60S littermate mice were placed on culture wells either precoated with 10 μg/mL fibronectin (Cat# 356008, BD Bioscience, San Jose, CA), 0.3 μg/mL laminin V (Cat# CC-145 Millipore, Billerica, MA), or 2 mg/mL bovine collagen type I (Cat# 354236, BD Bioscience, San Jose, CA). Dermal punch biopsies were incubated at 37°C for one hour to facilitate adhesion of the dermal punch biopsy to the culture well and growth medium, (DMEM Cat#11960-044, 10% FBS Cat# A12617DJ, 100U/mL pen/strep, Cat#15140-122 and 2 mM glutamine, Cat# 25030149, all from Invitrogen, Carlsbad, CA) was gently added to the culture wells to surround the punch biopsy. Cultures were then incubated at 37°C and 5% CO2 for 24 hour and extra media was placed in each culture well to fully submerge the punch biopsy. After four days, the growth media was aspirated and cultures were fixed with ice-cold 80% methanol/20% acetone for 10 minutes. Cultures were then washed and stored in PBS until imaged. Eight images were taken around the circumference of the skin explants with a Zeiss 510 microscope and the total area of cellular migration from the explants was calculated. Total cellular area of migration was then graphed using Graphpad Prism 4.0. Student’s t-tests between punch biopsies derived from WT and G60S mice on the same substrate (collagen I, fibronectin, plastic or laminin V) were performed to determine statistical significance (*p < 0.05).
Human fibroblast migration
Human fibroblasts were seeded at cell density of 1×104 cells per culture well in a 12-well tissue culture dish. Once confluent, the growth media was removed and a 1000 uL pipette tip was scraped down the center of the culture well. Culture wells were then washed 2X with Opti-MEM (Cat# 11058-021, Invitrogen) and 1 mL of Opti-MEM was added to each culture well. Time-lapse imaging was performed using a Zeiss 510 microscope equipped with Axiovision software. Time-lapse monolayer closure images were acquired every 15 minutes for 48 hours. Student’s t-tests between D3 and D3N fibroblasts or between V216 and V216L fibroblasts were performed to determine statistical significance (*p < 0.05)
Fibroblast proliferation
To examine cell proliferation, 1×104 control or mutant fibroblasts were plated in 6-well dishes and allowed to grow in growth medium. At days 2, 4 and 6, cells from replicated plates were collected and counted using an automated Countess™ Cell Counter (Invitrogen). The total cell number during each time point was then graphed using Graphpad Prism 4.0. Student’s t-tests between D3 and D3N fibroblasts or between V216 and V216L fibroblasts after six days of growth were performed to determine statistical significance (*p < 0.05)
TGFβ1 treatment
D3, D3N, V216 and V216L cells were seeded at a density of 1.5×105 cells per well in a 6-well culture dish. Cultures containing each human fibroblast population were either left untreated or treated with 1 ng/mL TGFβ1 (Cat# 240-B, R&D Systems, Minneapolis, MN). After four days, cultures were lysed using a RIPA buffer for Western blot analysis.
Western blot analysis
As previously described (Penuela, et al., 2007), total proteins were isolated from human fibroblast cell cultures using a RIPA buffer. Approximately 30 μg of total protein/lane was separated by electrophoresis on 10% SDS-PAGE gels and transferred to nitrocellulose membrane for immunoblotting. A polyclonal anti-Cx43 antibody (1:5000; Cat# C6219, Sigma-Aldrich) was used to detect endogenous Cx43 species and anti-alpha smooth muscle actin (1:1000; Cat# A2547, Sigma-Aldrich). The membranes were also probed for GAPDH (1:10,000; Cat# MAB374, Chemicon, Temecula, CA) as a control for protein loading. Primary antibody labeling was detected using IRDye 800 (1:5000; Cat# 611-132-122, Rockland, Glibertsville, PA)) and Alexa Fluor® 680 (1:5000; Cat#A21076, Invitrogen) goat anti-rabbit and anti-mouse secondary antibodies and the LiCor detection system. The relative densitometry intensities were normalized to GAPDH and graphed using Graphpad Prism 4.0. Student’s t-tests between the levels of P2/Total Cx43, alpha smooth muscle actin, or fold change in alpha smooth muscle actin expression were performed to determine statistical significance. Bars denote which groups were compared and achieved significance (*p < 0.05).
Results
Clinical presentation of patients and detection of Cx43 gene mutations
We recruited two families diagnosed with ODDD; one family has multiple members with ODDD while the second family had one isolated case. All affected individuals had the typical craniofacial appearance and limb involvement characteristic of ODDD.
Patient #1
This female individual was born with a tethered tongue, heart murmur and a kidney malfunction. Her motor milestones were normal. She has a narrow nose with hypoplastic alae nasi, and microphthalmia. Her teeth are dysplastic and irregularly enameled, but are in good condition. She had fusion of digits 3-5, with the 5th digit brachydactyly and clinodactyly. Neurologic symptoms of lower extremity paresthesias and weakness are exacerbated by heat and exercise (Uthoff’s sign). Neurogenic bladder was managed with tolterodine. At age 14, her neurologic exam was notable for normal strength and gait, but lower extremity hyperreflexia and right Babinski sign, lower extremity hyperesthesia to pinprick but normal vibratory threshold.
Patient #2
This male patient had congenital umbilical hernia, and bilateral fusion of digits 4 and 5 of the hands, but no fusion of toes. He has a narrow nose with hypoplastic alae nasi, irregular and thinly enameled dentition, but no ocular involvement. Neurologic symptoms began at age 29 with spasticity. He has had episodic gastrointestinal ilius and chronic gastrointestinal hypomotility, neurogenic bladder, and also describes Uthoff’s sign. He lost ambulation at age 54; his neurologic exam was notable for hyperreflexia, spasticity, Babinski sign, and diminished vibratory threshold in the lower extremities. He has since developed lower extremity lymphedema.
DNA was extracted from each patient’s skin fibroblasts. As expected, DNA sequence analysis found that both patients had missense mutations in the GJA1 gene encoding for Cx43 (Figure 1A). Patient 1 exhibited a missense mutation in N-terminal region of Cx43 resulting in the substitution of an aspartic acid at position 3 for asparagine (p.D3N). Patient 2 was found to harbor a mutation in the region of the molecule that would represent the 4th transmembrane domain at reside 216 where valine was replaced by leucine (p.V216L). No mutation was found in the coding region of the GJA1 gene from fibroblasts of the matched family member of the D3N (the mother of the patient) or V216L patients (the daughter of the patient). Those fibroblasts were used as normal controls and are notated as D3 and V216, respectively.
Figure 1.
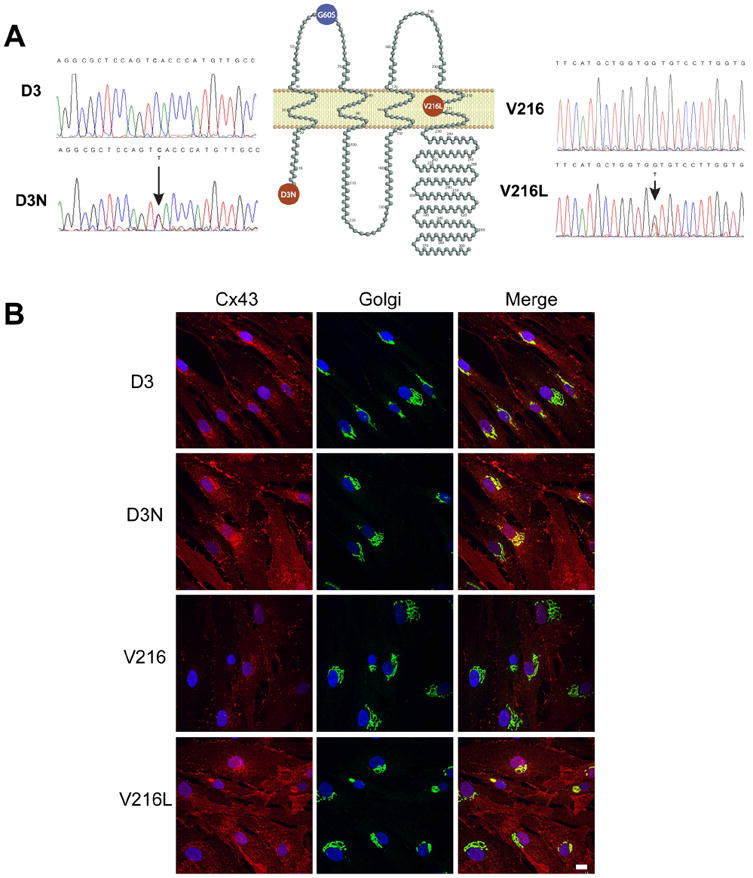
DNA sequences from two ODDD patients. (A) Genomic DNA was extracted from patient’s blood samples or skin fibroblasts. DNA sequence analysis identified the heterozygous missense mutation in the N-terminal coding region of the GJA1 gene in patient 1 where GAC (Asp, D) changed to AAC (Asn, N) (D3N). The mutation in patient 2 was found in the gene region encoding the fourth transmembrane domain of Cx43 where GTG (Val, V) changed to TTG (Leu, L)(V216L). The G60S mouse mutation is localized within the first extracellular loop domain. (B) Fibroblasts denoted as D3, D3N, V216 and V216L were double-labeled for Cx43 (red) and the resident Golgi apparatus protein GM130 (green). Overlayed images reveal that Cx43 was partially localized to the Golgi apparatus especially in cells expressing the V216L mutant. Bar=10 μm.
Characterization of Cx43 in control and mutant fibroblasts
Previous studies have reported the existence of the D3N and V216L in ODDD patients but no studies have investigated how human Cx43 mutants affect the trafficking of Cx43 and the assembly of gap junctions (Paznekas, et al., 2003; Paznekas, et al., 2009). To this end, we established the first use of primary cell culture from patients expressing the D3N and V216L mutant Cx43. Familial matched control fibroblasts (D3 and V216) were also obtained from unaffected relatives to control for the fibroblast source, cell passage and genetic variation which may have otherwise affected the interpretation of our results. Cultured cells were verified as fibroblasts due to their spindle-shaped appearance and positive staining for the intermediate filament, vimentin, confirming their mesenchymal origin (Supp. Figure S1).
To characterize the subcellular localization of Cx43 in control and mutant fibroblasts, cells were immunofluorescently double-labeled for Cx43 and a resident protein of the Golgi apparatus, GM130 (Figure 1B). In D3, D3N and V216 fibroblasts, Cx43 was localized extensively to punctate structures at sites of cell-to-cell apposition consistent with the formation of gap junction plaques. However, the prevalence of gap junction plaques was greatly reduced in the V216L fibroblasts and a large population of Cx43 co-localized with the resident Golgi protein, GM130. Collectively, these localization studies would suggest that the D3N mutant readily assembles into gap junctions while the V216L mutant is partially impaired from forming gap junctions. All fibroblasts were examined for the presence of Cx26, Cx40 and Cx45 but no specific staining was found for these connexin isoforms (data not shown).
Both human ODDD-linked mutants reduce GJIC
In order to examine GJIC in control and mutant fibroblasts, a preloading assay was performed where calcein-loaded and DiI-labeled donor cells were seeded on fibroblast cultures and the incidents of calcein spread were quantified and compared to control counterparts (Figure 2A). In all cases, the incidence of dye transfer in mutant fibroblasts were significantly reduced in comparison to their control counterparts. To further examine the degree of gap junction coupling in control and mutant human fibroblasts, the coupling conductance between cells were measured by double whole cell patch-clamp analysis (Figure 2B). Electrical conductance results confirmed that both the D3N and V216L mutants reduced the overall level of gap junction coupling compared to their control counterparts.
Figure 2.
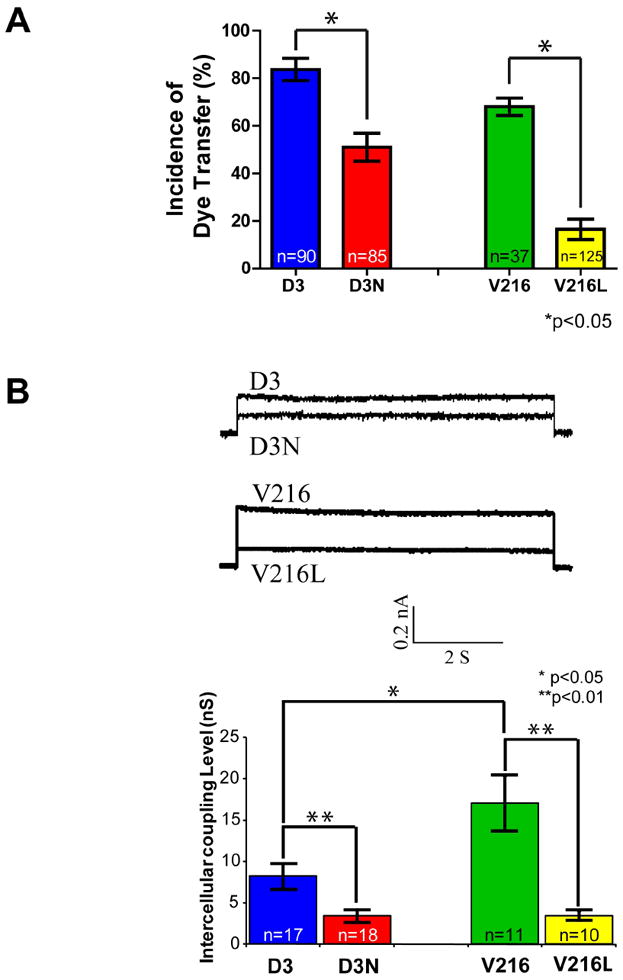
Reduced gap junctional intracellular communication in D3N and V216L fibroblasts. (A) D3, D3N, V216 and V216L fibroblasts were preloaded with calcein-AM and DiI, and seeded onto a monolayer of phenotypically-matched fibroblasts for 3 h. Calcein spread from DiI labeled cells was scored as incidences of functional dye transfer. (B) Pairs of D3, D3N, V216 and V216L fibroblasts were double whole cell patch-clamped and assessed for gap junction coupling conductance. Reduced gap junction conductance was found in D3N and V216L fibroblasts when compared to the control D3 and V216 fibroblast counterparts. *p<0.05, **p<0.01
Wound healing is delayed in mice expressing mutant Cx43
Since fibroblasts play a crucial role in wound healing (Hinz, 2007; McAnulty, 2007) and ODDD-linked Cx43 mutants reduced GJIC in dermal fibroblasts (Churko, et al., 2010), we investigated whether this decrease in GJIC could alter wound healing. To determine if wound healing is altered in an in vivo environment, skin punch biopsies were performed on wild-type (WT) and G60S (Gja1Jrt/+) mutant Cx43 mice (Figure 3) and the wound area was quantified for twelve days after the punch biopsies were performed. One to six days after the punch biopsies were performed; there was no significant difference in the size of the wound between WT and G60S mice. After nine days however, G60S mice wounds were significantly larger than those seen in the WT mice. Since myofibroblast contraction during the later stages of wound healing is vital to decrease the wound size, we sought to determine whether fibroblasts were responsible for the delay in wound healing observed in the mutant G60S mice. To achieve this, fibroblasts were assessed for their ability to proliferate, migrate and differentiate into myofibroblasts.
Figure 3.
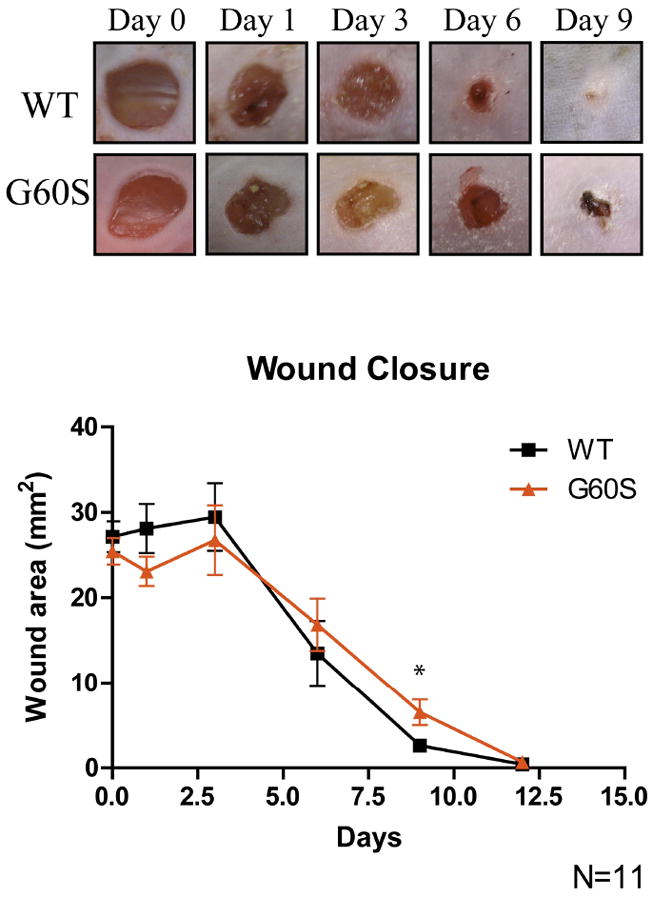
Dermal punch biopsies performed on the backs of WT and G60S mice revealed a delay in wound closure in G60S mutant mice. After nine days of healing, the wounds in the G60S mice were significantly larger than the wounds in the WT mice. *p<0.05
ODDD mutants reduce dermal fibroblast migration and proliferation
To assess whether dermal fibroblasts expressing mutant Cx43 can impact the proliferation and migration dependant events which occur during wound healing, explants derived from the WT and G60S punch biopsies were cultured on either plastic, fibronectin, laminin V or collagen type I (Figure 4). After five days in culture, a fibroblast population could easily be identified surrounding the explants. Quantification of the fibroblast population that migrated from explants derived from WT and mutant mice revealed that mutant harboring fibroblasts migrated less well on fibronectin (Figure 4B) and on collagen type I (Figure 4C).
Figure 4.
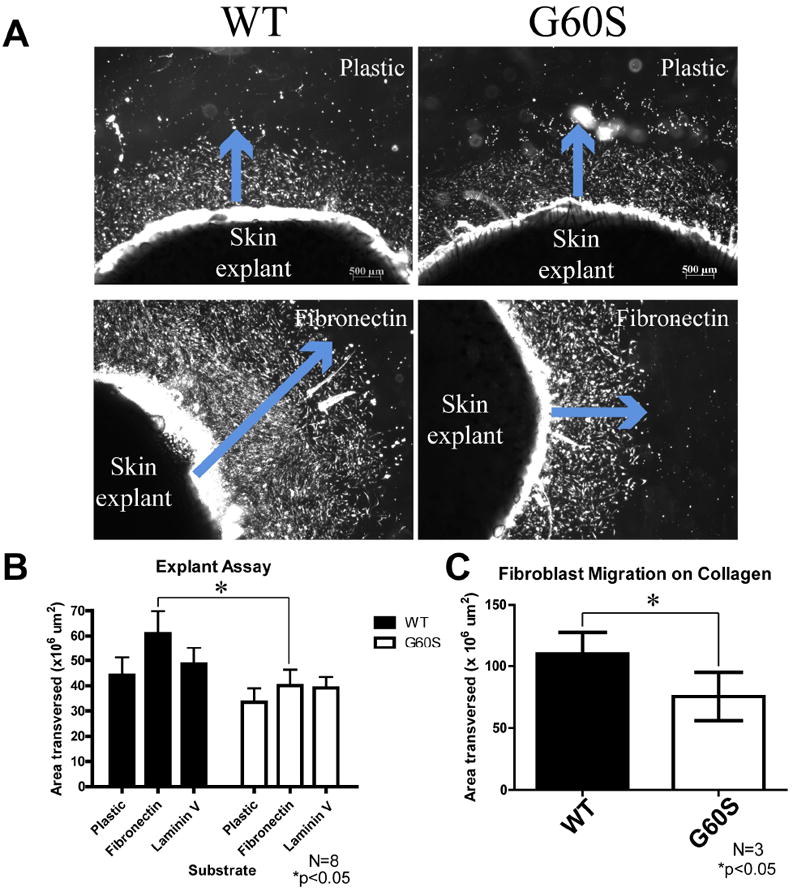
Fibroblasts from mutant mice exhibited reduced migration on fibronectin and collagen. (A) Dermal punch biopsies obtained from the backs of WT and G60S mice were plated in uncoated, fibronectin, laminin, or collagen-coated culture wells. After punch biopsies were cultured for five days, cellular emigration away from the explants could be observed (blue arrow). In these images the cells appear bright due to the nature of image acquisition. (B, C) The area of cellular emigration was quantified around the entire explant and the total cellular emigration area was quantified. There was a significant decrease in the area that fibroblasts derived from the G60S mice migrated out of the skin explants when cultured on fibronectin (B) and collagen type I (C). Bar = 500 μm.
To determine if the migration of human dermal fibroblasts was affected by the expression of mutant Cx43, time lapse imaging of scrape-wounded control and mutant fibroblasts was evaluated. When assessed over 48 hours, we observed that migration was delayed in the ODDD mutant containing fibroblast cultures (Figure 5) (see Supp. Movie S1). Surprisingly, while migration was analyzed under serum-free conditions, proliferative events were observed after 24 hours as evidenced by the presence of circular shaped cells. To quantify if the proliferation rate was different in fibroblasts expressing Cx43 mutants, cell counts were performed on human fibroblasts derived from ODDD patients and their familial matched controls (Figure 6). All cell lines were seeded at equal densities and cell counts were performed on fibroblasts grown for two, four, and six days. Both mutant fibroblasts were found to proliferate slower than their control counterparts. These results indicate that the expression of mutant Cx43 in dermal fibroblasts decreases the proliferation rate and may contribute to delays in wound healing.
Figure 5.
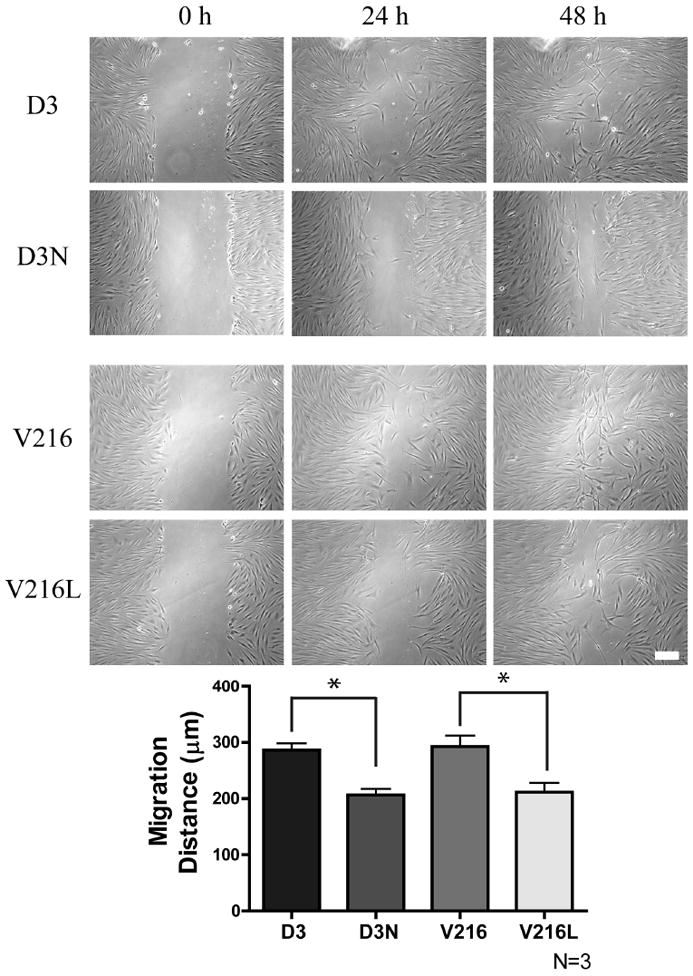
ODDD-linked human fibroblasts exhibit impaired migration. Time-lapse imaging of a scrape wound assay was performed using human fibroblasts from ODDD patients (D3N, V216L) and their matched control fibroblasts (D3, V216). After 24 hours it was evident that ODDD-linked fibroblasts migrated into the wound area slower than control fibroblasts. *p<0.05 Bar = 100μm.
Figure 6.
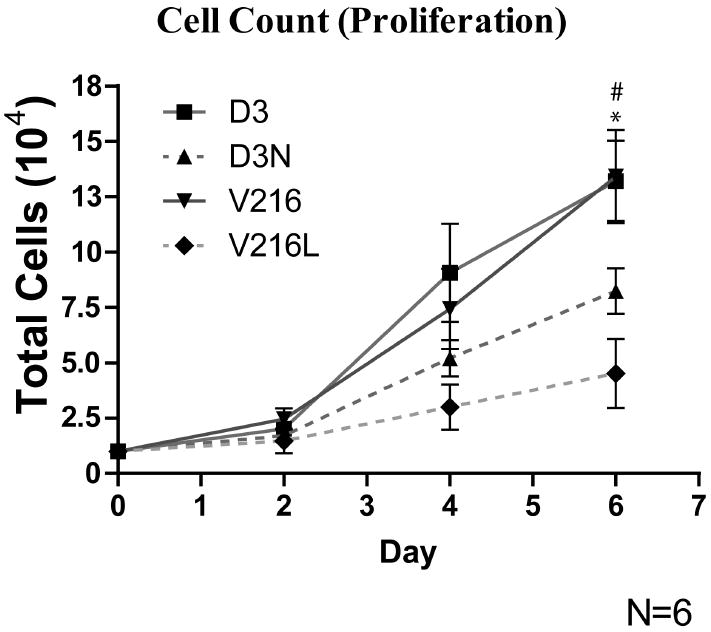
ODDD-linked human fibroblasts exhibited reduced proliferation. D3, D3N, V216 and V216L fibroblasts were plated and counted over a period of 6 days. At day 6, there were significantly fewer cells in the culture wells containing V216L and D3N fibroblasts when compared to V216 and D3 fibroblasts. # D3 vs. D3N p<0.05, * V216 vs. V216L p<0.05
ODDD mutants hinder the ability of dermal fibroblasts to differentiate into myofibroblasts
Wound closure was found to be delayed in the G60S mice nine days after a punch biopsy was performed. At this stage in wound healing, fibroblasts are exposed to TGFβ1 which facilitates the differentiation of fibroblasts into myofibroblasts. Myofibroblasts in turn express alpha smooth muscle actin which plays a role in contraction of the wounded area (Singer and Clark, 1999). To further investigate the mechanism(s) by which wound healing may be compromised, fibroblasts derived from ODDD patients, as well as their familial matched controls, were treated with TGFβ1 for four days and Western blots were performed on untreated as well as TGFβ1-treated fibroblasts to assess the expression of Cx43 and alpha smooth muscle actin. TGFβ1 treatment elevated the levels of the P2 phosphorylated species of Cx43 (P2) in D3 and V216 fibroblasts. However, in the mutant Cx43 expressing fibroblasts (D3N and V216L), this increase in the P2 species of Cx43 was not observed upon TGFβ1 treatment (Figure 7). To determine if this change in phosphorylation could impact the ability of both familial matched control and ODDD patient derived fibroblasts to differentiate into myofibroblasts, Western blots for alpha smooth muscle actin were performed with and without TGFβ1 treatment (Figure 8). Control fibroblasts (D3 and V216) expressed high levels of alpha smooth muscle actin after TGFβ1 treatment while ODDD patient-derived fibroblasts exhibited no significant increase in the expression of alpha smooth muscle actin. In addition, the fold-change in alpha smooth muscle actin expression after TGFβ1 treatment between control and ODDD fibroblasts cells was significantly different. This result demonstrates that the expression of mutant Cx43 affects the differentiation of fibroblasts into myofibroblasts. Collectively, these studies would suggest that ODDD-patients may have compromised wound healing as mutant Cx43 expressing fibroblasts display defects in their ability to proliferate, migrate, and express alpha smooth muscle actin.
Figure 7.
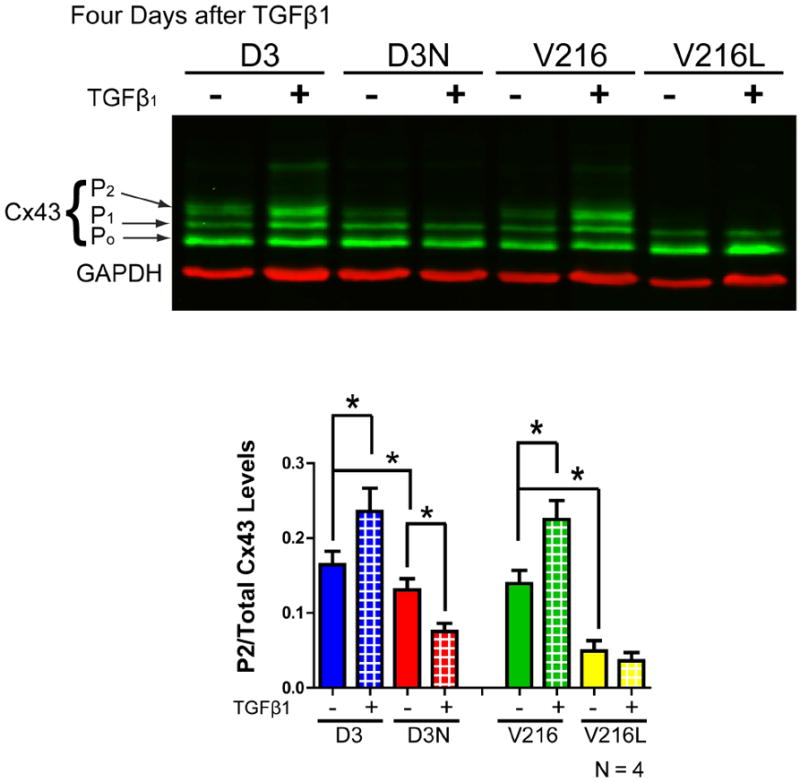
Cx43 levels (green) in untreated and TGFβ1 treated D3, D3N, V216 and V216L fibroblasts as assessed by Western blot. Three species of Cx43 were resolved reflecting various phosphorylation states where the lowest band was denoted as P0, the middle band as P1 and the highest band as P2. Four days after D3, D3N, V216, and V216L fibroblasts were treated with TGFβ1, the levels of the P2 Cx43 phosphorylated species as compared to total Cx43 levels was significantly increased in D3 and V216 fibroblasts but this increase was not observed in D3N and V216L fibroblasts. GAPDH was used as a loading control (red). *p<0.05
Figure 8.
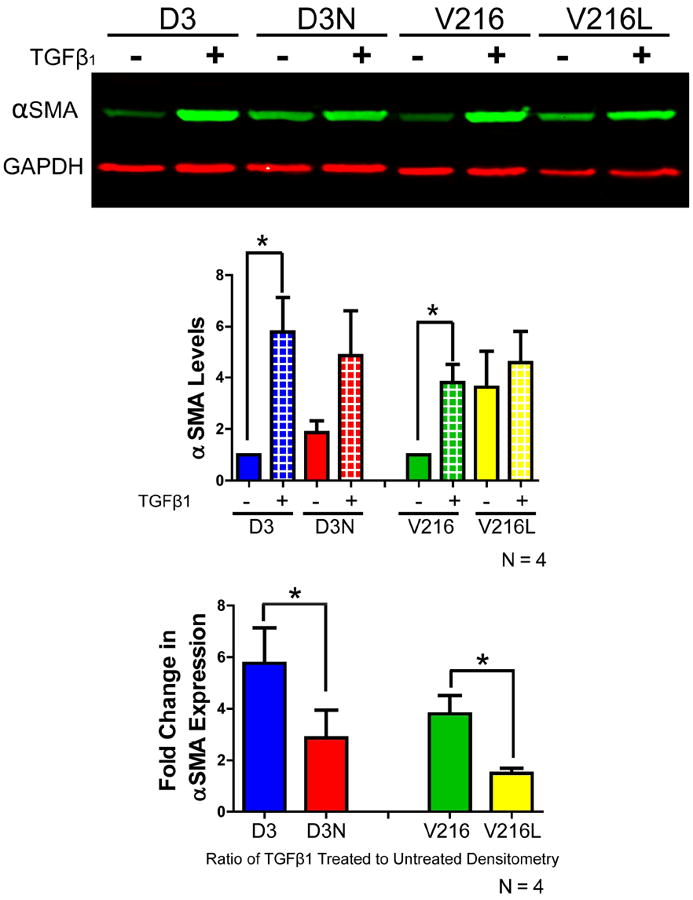
The expression of alpha smooth muscle actin was evaluated by Western blot in D3, D3N, V216 and V216L fibroblasts exposed to TGFβ1 for four days. The expression of alpha smooth muscle actin was significantly increased in D3 and V216 fibroblasts after TGFβ1 treatment. The fold change in alpha smooth muscle actin expression after TGFβ1 treatment was also significantly larger in the D3 and V216 fibroblasts when compared to the D3N and V216L fibroblasts. *p<0.05
Discussion
Since the discovery that ODDD was specifically linked to mutations in the GJA1 gene encoding Cx43 (Paznekas, et al., 2003), there has been an expansion of interest in understanding how Cx43 mutants lead to disease. Given the ubiquitous expression of Cx43 in the human body including vital organs, it was somewhat puzzling that patients harboring Cx43 mutants did not have even more disease burden. One possibility was that the wide array of missense mutations found within the GJA1 gene only mildly compromises the functional status of Cx43 in forming intercellular channels. However, this explanation is not likely correct as all the ODDD-linked mutants examined to date have moderate to severe deficiencies in forming intercellular gap junction channels when expressed in GJIC-deficient cells (McLachlan, et al., 2005; Roscoe, et al., 2005). However, these in vitro studies do not ideally mimic the human condition due primarily to the difficulty in expressing Cx43 mutants and the wild-type Cx43 counterparts at the 1:1 ratio expected to be found in ODDD patients with autosomal dominant GJA1 gene mutations. With the generation of mouse models of ODDD this genetic limitation was overcome and three ODDD-linked Cx43 mutants were also found to be dominant to endogenous Cx43 resulting in a reduction in overall Cx43 function in many tissues (Dobrowolski, et al., 2008; Flenniken, et al., 2005; Kalcheva, et al., 2007). Even in these mutant mouse studies, there was still a requirement to extrapolate the findings to the human disease condition. In the present study we report on the first efforts to design and study human models of ODDD with the anticipation that findings in these models will extend our understanding of ODDD as a disease and to further determine the functional role of Cx43.
Clinical presentation of ODDD in two patients with distinct GJA1 gene mutations
Clinically, ODDD is a complex disease as no two patients appear to have identical disease burdens. While nearly all patients have craniofacial bone deformities, fusion of digits, and thin enamel, the plethora of symptoms beyond these is highly diverse and linked to defects in any number of organs that include the heart, skin, as well as a wide array of conditions that are likely entrenched in neurogenic defects (Paznekas, et al., 2009). In the present study we obtained clinical histories and dermal fibroblasts from two patients with ODDD. Importantly, we were also fortunate to have obtained dermal fibroblasts from the parent/child of these patients which allows for all analytical tests to be matched to individuals sharing similar genetic profiles thus eliminating variance often seen in unrelated controls. Furthermore, in this study we control for any changes in Cx43 status through multiple cell passages by always using fibroblasts from control and disease conditions at the same cell passage. Collectively, we believe we established the best possible scenario to assess Cx43 and gap junctions in control and mutant-expressing human dermal fibroblasts. Moreover, we complement our approach by including in vivo wound repair studies using the G60S (GjaJrt/+) mouse model of ODDD.
Cx43 and GJIC status in control and mutant fibroblasts
Previous in vitro studies using reference cell lines strongly suggest that all over-expressed Cx43 mutants dramatically reduce GJIC and act as dominant-negatives on co-expressed Cx43 (Churko, et al., 2010; McLachlan, et al., 2005). In the case where the V216L mutant was expressed in reference cells that lack connexins, the V216L expressing cells fail to form functional intercellular channels (McLachlan, et al., 2005). However, whether these or any other ODDD-linked mutants in human cells naturally harboring the mutants can affect GJIC had not been tested. In our studies we found that human fibroblasts harboring the D3N or V216L mutant have reduced GJIC. Interestingly, the V216L mutant appears to be twice as potent at reducing GJIC compared to the D3N mutant suggesting that the impact of the autosomal dominant mutation on the coupling status in ODDD patient may be dependent on the site of the mutation. This finding is in keeping with our previous study where the G21R mutant was found to be twice as potent at reducing GJIC as a G138R mutant when ectopically expressed and examined in reference cell systems (Gong, et al., 2007). Since both the D3N and G21R mutations are found on the N-terminus of Cx43, these results may suggest that mutants within this domain are more potent than mutations found at least some other regions of Cx43. It is notable that the N-terminal domain of some connexins has been reported to be involved in transjunctional voltage gating (Purnick, et al., 2000), oligomerization of connexins into connexons (Lagree, et al., 2003), and even in plugging the gap junction pore (Maeda, et al., 2009; Oshima, et al., 2007) highlighting the functional importance of this domain.
In vitro studies using ectopically expressed Cx43 mutants have suggested that there are two clear trafficking phenotypes where Cx43 mutants are either trapped within a secretory compartment such as the endoplasmic reticulum or Golgi apparatus (e.g. fs230, fs260, G60S mutants) or where the mutant traffics to the cell surface and assembles into non-functional gap junction plaques (e.g., G21R, G138R) (Churko, et al., 2010; Gong, et al., 2006). While the D3N and V216L mutants can form gap junction plaques, a population of the V216L mutant, in particular, localized to the Golgi apparatus. Impaired trafficking of the V216L mutant may also act to inhibit the delivery of wild type Cx43 to the cell surface and partly explain how this particular mutant contributes to a disease phenotype.
Cellular consequences of reduced Cx43 function in dermal fibroblasts
Given that patient-derived dermal fibroblasts exhibit a significant reduction in GJIC and that wound healing is delayed in G60S mutant mice, we sought to determine if the function of dermal fibroblasts is compromised (Hinz, 2007; McAnulty, 2007). Interestingly, it has been reported that transient knockdown of Cx43 in the skin by siRNAs or antisense leads to enhanced wound healing (Mori, et al., 2006; Qiu, et al., 2003) but chronic down regulation of Cx43 had not previously been tested. Furthermore, the reduction of total Cx43 in a knockdown study is mechanistically distinct from the expression of a functionally dominant Cx43 mutant as exhibited in an ODDD patient as a plethora of Cx43 binding partners may remain engaged (Laird, 2010). Since Cx43 may play distinct roles at the various stages of wound repair and within cell types (e.g., lymphatic endothelial cells, keratinocytes, fibroblasts) involved in wound healing, temporal knockdown of Cx43 may be beneficial during the early stages of wound healing while potentially detrimental during a later stage. For instance, acute down regulation of Cx43 at the wound edge over a 24-48 h period leads to increased keratinocyte and fibroblast migration (Mori, et al., 2006; Wright, et al., 2009). In addition, keratinocytes were shown to play a significant role in accelerating the healing of incisional tail wounds in mice with decreased levels of Cx43 (Kretz, et al., 2003). Furthermore, transient antisense-induced knockdown of Cx43 leads to improved wound healing in the skin (Qiu, et al., 2003) and cornea (Nakano, et al., 2008) but functional recovery of Cx43 is thought to be necessary for the final stages of wound healing. Conversely, chronic expression of truncated Cx43 has been shown to be extremely detrimental as these mice die shortly after birth due to epidermal barrier defects (Maass, et al., 2004). In our mouse studies, chronic reduction in overall functional Cx43 levels delayed wound healing and suggests that some ODDD patients may have a subclinical delay in overall wound healing.
In the present study we demonstrated that ODDD fibroblasts are deficient in their ability to proliferate, migrate and differentiate when compared to their respective familial matched control fibroblasts. Collectively, these cell characteristics are intimately involved in regulating the efficacy of wound repair. Interestingly, the defect in G60S mouse fibroblast migration was only observed to occur when skin explants were cultured on fibronectin and collagen type I. It will be interesting to determine how Cx43 mutants impact the expression and localization of adhesion proteins involved in the attachment of fibroblasts to these substrates.
We have previously reported that the over-expression of several Cx43 mutants in reference cell lines reduces the phosphorylation state of total Cx43 (Churko, et al., 2010; McLachlan, et al., 2005). Importantly, the P2 phosphorylation state of Cx43 has been correlated with the presence of functional gap junctions (Solan and Lampe, 2005). In the present study, both the D3N and V216L mutants significantly decreased the levels of the P2 phosphorylated species (Churko, et al., 2010) but it is unclear if this also reflects a decrease in gap junctions. However, it is clear that fibroblasts harboring the D3N and V216L mutants are coupled to a lesser degree. Interestingly, in control fibroblasts, the P2 species of Cx43 increased after TGFβ1 treatment but this increase was not observed in fibroblasts harboring ODDD-linked mutants. TGFβ1 is a critical factor in wound repair and acts to stimulate alpha smooth muscle actin and the formation of myofibroblasts (Brenmoehl, et al., 2009; Hinz, 2007). Quantification of alpha smooth muscle actin revealed that Cx43 mutant expressing fibroblasts are deficient in regulating the differentiation of fibroblasts into myofibroblasts. Mechanistically, the delay in wound repair observed in our mutant mice is likely linked, at least in part, to a deficiency in the ability of fibroblasts to differentiate into myofibroblasts.
In summary, this is the first study documenting and employing a human model of ODDD. Here we isolated and characterized human dermal fibroblasts from two ODDD families which included both affected and unaffected family members. We also employed a punch biopsy assay to demonstrate a delay in wound course in a mouse model for ODDD. Since fibroblast contraction plays a more important role in rodent than human wound healing (Dorsett-Martin, 2004; Greenhalgh, 2005) and also given the limited number of patients studied and the lack of correlative data, we cannot generalize our findings to all ODDD patients. However, since our V216L and D3N expressing primary fibroblasts do exhibit impaired proliferation, migration, and differentiation, it is quite likely that this condition remains present but unreported or subclinical and would only be revealed in patients that have severe skin wounds where dermal fibroblasts play a more active role.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Kevin Flanigan for his role in connecting us to one of the ODDD patients used in this study. The authors would also like to thank Kevin Barr for technical animal handling assistance. This works was supported by Canadian Institutes of Health Research funding to DWL and DB and Natural Science and Engineering Research Council funding to JC.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Brenmoehl J, Miller SN, Hofmann C, Vogl D, Falk W, Scholmerich J, Rogler G. Transforming growth factor-beta 1 induces intestinal myofibroblast differentiation and modulates their migration. World J Gastroenterol. 2009;15(12):1431–42. doi: 10.3748/wjg.15.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko JM, Langlois S, Pan X, Shao Q, Laird DW. The potency of the fs260 connexin43 mutant to impair keratinocyte differentiation is distinct from other disease-linked connexin43 mutants. Biochem J. 2010;429(3):473–83. doi: 10.1042/BJ20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R, Sasse P, Schrickel JW, Watkins M, Kim JS, Rackauskas M, Troatz C, Ghanem A, Tiemann K, Degen J, et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet. 2008;17(4):539–54. doi: 10.1093/hmg/ddm329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett-Martin WA. Rat models of skin wound healing: a review. Wound Repair Regen. 2004;12(6):591–9. doi: 10.1111/j.1067-1927.2004.12601.x. [DOI] [PubMed] [Google Scholar]
- Flenniken AM, Osborne LR, Anderson N, Ciliberti N, Fleming C, Gittens JE, Gong XQ, Kelsey LB, Lounsbury C, Moreno L, et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132(19):4375–86. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- Goldberg GS, Bechberger JF, Naus CC. A pre-loading method of evaluating gap junctional communication by fluorescent dye transfer. Biotechniques. 1995;18(3):490–7. [PubMed] [Google Scholar]
- Gong XQ, Shao Q, Langlois S, Bai D, Laird DW. Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J Biol Chem. 2007;282(26):19190–202. doi: 10.1074/jbc.M609653200. [DOI] [PubMed] [Google Scholar]
- Gong XQ, Shao Q, Lounsbury CS, Bai D, Laird DW. Functional characterization of a GJA1 frameshift mutation causing oculodentodigital dysplasia and palmoplantar keratoderma. J Biol Chem. 2006;281(42):31801–11. doi: 10.1074/jbc.M605961200. [DOI] [PubMed] [Google Scholar]
- Greenhalgh DG. Models of wound healing. J Burn Care Rehabil. 2005;26(4):293–305. doi: 10.1097/01.bcr.0000169885.66639.b5. [DOI] [PubMed] [Google Scholar]
- Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–37. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- Kalcheva N, Qu J, Sandeep N, Garcia L, Zhang J, Wang Z, Lampe PD, Suadicani SO, Spray DC, Fishman GI. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc Natl Acad Sci U S A. 2007;104(51):20512–6. doi: 10.1073/pnas.0705472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizana E, Chang CY, Cingolani E, Ramirez-Correa GA, Sekar RB, Abraham MR, Ginn SL, Tung L, Alexander IE, Marban E. Gene transfer of connexin43 mutants attenuates coupling in cardiomyocytes: novel basis for modulation of cardiac conduction by gene therapy. Circ Res. 2007;100(11):1597–604. doi: 10.1161/CIRCRESAHA.106.144956. [DOI] [PubMed] [Google Scholar]
- Kretz M, Euwens C, Hombach S, Eckardt D, Teubner B, Traub O, Willecke K, Ott T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J Cell Sci. 2003;116(Pt 16):3443–52. doi: 10.1242/jcs.00638. [DOI] [PubMed] [Google Scholar]
- Lagree V, Brunschwig K, Lopez P, Gilula NB, Richard G, Falk MM. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J Cell Sci. 2003;116(Pt 15):3189–201. doi: 10.1242/jcs.00604. [DOI] [PubMed] [Google Scholar]
- Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010;20(2):92–101. doi: 10.1016/j.tcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Maass K, Ghanem A, Kim JS, Saathoff M, Urschel S, Kirfel G, Grummer R, Kretz M, Lewalter T, Tiemann K, et al. Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol Biol Cell. 2004;15(10):4597–608. doi: 10.1091/mbc.E04-04-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458(7238):597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39(4):666–71. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- McLachlan E, Manias JL, Gong XQ, Lounsbury CS, Shao Q, Bernier SM, Bai D, Laird DW. Functional characterization of oculodentodigital dysplasia-associated Cx43 mutants. Cell Commun Adhes. 2005;12(5-6):279–92. doi: 10.1080/15419060500514143. [DOI] [PubMed] [Google Scholar]
- Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci. 2006;119(Pt 24):5193–203. doi: 10.1242/jcs.03320. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Oyamada M, Dai P, Nakagami T, Kinoshita S, Takamatsu T. Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Invest Ophthalmol Vis Sci. 2008;49(1):93–104. doi: 10.1167/iovs.07-0255. [DOI] [PubMed] [Google Scholar]
- Oshima A, Tani K, Hiroaki Y, Fujiyoshi Y, Sosinsky GE. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc Natl Acad Sci U S A. 2007;104(24):10034–9. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72(2):408–18. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, Koivisto PA, Van Maldergem L, Boyadjiev SA, Bodurtha JN, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30(5):724–33. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- Penuela S, Bhalla R, Gong XQ, Cowan KN, Celetti SJ, Cowan BJ, Bai D, Shao Q, Laird DW. Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J Cell Sci. 2007;120(Pt 21):3772–83. doi: 10.1242/jcs.009514. [DOI] [PubMed] [Google Scholar]
- Purnick PE, Benjamin DC, Verselis VK, Bargiello TA, Dowd TL. Structure of the amino terminus of a gap junction protein. Arch Biochem Biophys. 2000;381(2):181–90. doi: 10.1006/abbi.2000.1989. [DOI] [PubMed] [Google Scholar]
- Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P, Green CR, Becker DL. Targeting connexin43 expression accelerates the rate of wound repair. Curr Biol. 2003;13(19):1697–703. doi: 10.1016/j.cub.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Joss S, Tomkin S, Ahmed M, Sheridan E, Dixon MJ. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. J Med Genet. 2006;43(7):e37. doi: 10.1136/jmg.2005.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe W, Veitch GI, Gong XQ, Pellegrino E, Bai D, McLachlan E, Shao Q, Kidder GM, Laird DW. Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J Biol Chem. 2005;280(12):11458–66. doi: 10.1074/jbc.M409564200. [DOI] [PubMed] [Google Scholar]
- Simek J, Churko J, Shao Q, Laird DW. Cx43 has distinct mobility within plasma-membrane domains, indicative of progressive formation of gap-junction plaques. J Cell Sci. 2009;122(Pt 4):554–62. doi: 10.1242/jcs.036970. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341(10):738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711(2):154–63. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419(2):261–72. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P, Wood EJ, Raxworthy MJ. Development of a multilayered in vitro model for studying events associated with wound healing. Wound Repair Regen. 1996;4(3):393–401. doi: 10.1046/j.1524-475X.1996.40318.x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Spruijt L, van der Burgt I, Bladergroen RS, Vermeer M, Steijlen PM, van Geel M. A 2-bp deletion in the GJA1 gene is associated with oculo-dento-digital dysplasia with palmoplantar keratoderma. Am J Med Genet A. 2005;132A(2):171–4. doi: 10.1002/ajmg.a.30412. [DOI] [PubMed] [Google Scholar]
- Vreeburg M, de Zwart-Storm EA, Schouten MI, Nellen RG, Marcus-Soekarman D, Devies M, van Geel M, van Steensel MA. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am J Med Genet A. 2007;143(4):360–3. doi: 10.1002/ajmg.a.31558. [DOI] [PubMed] [Google Scholar]
- Wright CS, van Steensel MA, Hodgins MB, Martin PE. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen. 2009;17(2):240–9. doi: 10.1111/j.1524-475X.2009.00471.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.