

Abstract
Objective
The serum of most neuromyelitis optica (NMO) patients contains autoantibodies (NMO-IgGs) directed against the aquaporin-4 (AQP4) water channel located on astrocyte foot processes in the perivessel and subpial areas of the brain. Our objectives were to determine the source of central nervous system (CNS) NMO-IgGs and their role in disease pathogenesis.
Methods
Fluorescence activated cell sorting and single-cell reverse transcriptase PCR were used to identify overrepresented plasma cell immunoglobulin (Ig) sequences in the cerebrospinal fluid (CSF) of an NMO patient after a first clinical attack. Monoclonal recombinant antibodies (rAbs) were generated from the paired heavy and light chain sequences and tested for target specificity and Fc effector function. The effect of CSF rAbs on CNS immunopathology was investigated by delivering single rAbs to rats with experimental autoimmune encephalomyelitis (EAE).
Results
Repertoire analysis revealed a dynamic, clonally expanded plasma cell population with features of an antigen-targeted response. Using multiple independent assays, 6 of 11 rAbs generated from CSF plasma cell clones specifically bound to AQP4. AQP4-specific rAbs recognized conformational epitopes and mediated both AQP4-directed antibody-dependent cellular cytotoxicity and complement-mediated lysis. When administered to rats with EAE, an AQP4-specific NMO CSF rAb induced NMO immunopathology: perivascular astrocyte depletion, myelinolysis and complement and Ig deposition.
Interpretation
Molecular characterization of the CSF plasma cell repertoire in an early NMO patient demonstrates that AQP4-specfic Ig is synthesized intrathecally at disease onset and directly contributes to CNS pathology. AQP4 is now the first confirmed antigenic target in human demyelinating disease.
Introduction
Neuromyelitis optica (NMO) is a severe demyelinating disorder that primarily affects the optic nerve and spinal cord resulting in vision loss and paralysis.1 Serum autoantibodies (NMO-IgGs) directed against the aquaporin-4 (AQP4) water channel has been shown to be a disease-specific marker of NMO pathology.2, 3 Three observations suggest that anti-AQP4 IgG plays a role in the pathophysiology of NMO. First, a loss of AQP4 expression on astrocytes is observed early in NMO lesions;4 second, NMO-IgG titers at the nadir of exacerbations correlate with the length of longitudinally-extensive spinal cord lesions;5 third, serum AQP4 levels correlate with clinical disease activity6and fourth, Ig deposition in NMO lesions occurs in vasculocentric areas of high AQP4 expression.4 Nevertheless, not all NMO patients have serum NMO-IgG.3, 7 Thus, it remains unclear whether NMO-IgG contributes directly to disease pathogenesis or is a serologic marker of a broader autoimmune response.
To address this question, we examined the intrathecal humoral immune response of an NMO patient after a first clinical attack using single cell reverse transcriptase PCR (RT-PCR). Bivalent human IgG1 monoclonal recombinant antibodies (rAbs) were reconstructed from the paired heavy- and light-chain sequences of plasma cell clones and examined for AQP4 reactivity, antibody-mediated effector function, and pathogenicity in the experimental autoimmune encephalomyelitis (EAE) model.
Patients and Methods
Patient
CSF was obtained from an NMO-IgG seropositive patient 8 weeks after the onset of unilateral monosymptomatic optic neuritis as part of the standard clinical evaluation. Informed consent was obtained prior to participation in this study. The CSF revealed 20 white blood cells (96% mononuclear), total protein 61 mg/dl, glucose 51 mg/dl, IgG index 0.49, IgG synthesis 0.0 and no oligoclonal bands. Her subsequent clinical history is remarkable for additional exacerbations of optic neuritis and transverse myelitis meeting the revised criteria for NMO.8
CSF Cell Labeling and FACS
CSF cell collection, fluorescent-labeling, and cell sorting were performed as described.9 Briefly, CSF is placed on ice immediately after collection, and the cells are pelleted and resuspended in a small volume of residual CSF. A combination of fluorescently-tagged murine Abs specific for the human cell surface markers CD19-AP, CD138-PE, CD14-APC-Cy7, and CD3-FITC (Caltag Laboratories, Burlingame, CA) is added to the CSF cell suspension, incubated at room temperature, and then diluted with sterile phosphate-buffered saline (PBS). Cells are sorted on a MoFlo cytometer (Cytomations, Fort Collins, CO). Cells are first selected in the size range of lymphocytes and plasmacytes by forward and side light scattering. CD138+ plasma cells are then identified and sorted into single wells of a 96-well PCR plate containing 20 µl of 1X RT buffer.
cDNA Synthesis and Amplification of VH and VL Chain Sequences
cDNA synthesis, nested PCR amplification, and purification of PCR products were performed as described.10 Purified PCR products were sequenced at the University of Colorado Cancer Center DNA Sequencing Core. Sequences were analyzed and edited with 4Peaks software (Mek&Tosj.com) and then aligned human immunoglobulin germline sequences using IMGT/V-QUEST (http://www.imgt.org/IMGT_vquest/share/textes/).
Construction, Expression and Purification of rAbs
RAbs were produced using a dual vector transient transfection system. VH and VL PCR products were cloned into the expression vectors pIgG1Flag11 and pCEP4, respectively, as described.12 Final constructs were sequenced verified. Constructs were cotransfected into HEK293 cells (Invitrogen, Carlsbad, CA, R620-07) using Lipofectamine 2000 (Invitrogen). After transfection, the cells were grown for 6–7 days in DMEM medium + 10% fetal bovine serum (FBS), the supernatant harvested, fresh medium added, and cells are propagated for 6–7 days. The cell culture supernatant was subsequently removed and combined with the previous collection. The cell culture supernatant was centrifuged to pellet cells and debris. Cell-free supernatants were incubated overnight with protein A-sepharose (Sigma-Aldrich, St. Louis, MO) at 4°C. The overnight slurry was transferred to columns and the rAbs were eluted in 0.1M glycine/1M NaCl (pH 3.0) and immediately adjusted to pH 7.5 by the addition of 0.1M Tris-HCl, pH 8.0. Recombinant IgG was subsequently exchanged and concentrated in storage buffer (PBS + 0.1% IgG/protease-free BSA) using Ultracel YM-30 microconcentrators (Millipore). Antibody integrity was confirmed by nondenaturing gel electrophoresis, and the IgG concentration was determined by a human IgG capture ELISA.
Cloning and Expression of Human AQP4
Forward (5’-ACTAGTGCAATGAGAGCTG CACTCTGGCTG-3’) and reverse (5’-CCGCGGGTCTGCTTTCAGTGCGATCTTCTAG-3’) primers with SpeI and SacII restriction sites at their 5’ and 3’ ends, respectively were used to amplify full length M1 AQP4 cDNA from reverse-transcribed total human brain RNA (BD Biosciences). The PCR amplified product was cloned into the plasmid pLenti6/V5 (Invitrogen) using the SpeI and SacII restriction sites. To generate gene-containing virus particles, the pLenti6/V5-AQP4 construct and packaging mix were combined to transfect a 293 FT cell line using Lipofectamine 2000 (Invitrogen). Virus containing supernatant was subsequently used to transduce the human LN18 glioblastoma cell line to create LN18AQP4.13 The LN18 glioblastoma cell line was transduced with an empty pLenti6/V5 vector to create the control cell line LN18CTR. These stably transduced cell lines were maintained under identical selectable pressure and conditions throughout the experiments.
Cell-Based Flow Cytometry Assay of AQP4 Antibody Binding
The following bioassay was used to quantify antibody reactivity of patient sera, CSF, and rAbs to native AQP4. Serum, CSF, or purified rAb was diluted in RPMI 1640 growth medium and 20 µl added to a 96-well plate, each well containing 30,000 LN18AQP4 or LN18CTR cells diluted in 20 µl of RPMI 1640. The final concentration of IgG in serum and CSF ranged from 14.6 µg/ml to 28.6 ng/ml, and the final concentration of rAb ranged from 5.0 µg/ml to 2.4 ng/ml. The plates were incubated on ice on an orbital shaker for 20 minutes. Cells were then washed twice with FACS buffer (1% FBS in PBS). 20 µl of diluted (1:100 in washing buffer) Alexa Fluor 488-labeled goat anti-human IgG secondary antibody (Invitrogen) was added to each well. After incubation on ice for 20 minutes, cells were washed twice and resuspended in 140 µl of FACS buffer. Cell surface staining was then analyzed on a FACS cell analyzer (CyAn ADP, Beckman Coulter, Fullerton, CA) using Summit software (Beckman Coulter). The antibody titer (ΔMFI) was determined by subtracting the median fluorescence intensity (MFI) obtained with the LN18CTR cell line from the MFI obtained with the LN18AQP4 cell line. Before analysis, the total IgG concentration in sera and CSF were measured by nephelometry (BN ProSpec) and diluted to 5 mg/ml.
AQP4 Indirect Immunofluorescence Microscopy
Immunofluorescence microscopy on frozen mouse cerebellum was performed as described.3 NMO CSF rAb (1 µg/ml), NMO patient sera (1:60 dilution), or NMO patient CSF (1:6 dilution) was applied to the treated tissue section and incubated at 4°C overnight in a humidified chamber. The secondary antibody, Alexa-488 goat-anti-human IgG (Molecular Probes, Portland, OR) was applied for 1 hour at room temperature, washed, and viewed on a Nikon E800 microscope equipped with epifluorescence.
Human Fetal Astrocyte (HFA) Flow Cytometry
HFA flow cytometry was performed as described.14 Briefly, HFAs were detached with warm PBS + 2mM EDTA, washed with FACS buffer, and then incubated for 30 minutes with either patient or control sera or recombinant NMO antibodies. After two washes with FACS buffer, PE-conjugated anti-human IgG (Sigma-Aldrich) and APC-conjugated anti-HLA-ABC (BD biosciences) were added for 30 minutes. Finally, HFAs were washed twice and fixed with 1% formaldehyde for later flow cytometry analysis.
Flow Cytometric Analysis of AQP4-Specific Complement Cytotoxicity
To eliminate any transfer of human complement into the cytotoxicity assay, whole IgG from patient serum and CSF was purified using Sepharose-G columns (GE Bio-sciences, Piscataway, NJ) according to the manufacturer’s protocol. Patient serum IgG (125 µg/ml), CSF IgG (125 µg/ml), or CSF rAbs (2.5 µg/ml) were added in duplicate to wells in a U-Shape 96-well microtiter plate (Greiner Bio-One, Monroe, NC) containing 50,000 LN18CTR or LN18AQP4 cells in 40 µl of RPMI medium. Cells were incubated on ice for 25 minutes on an orbital shaker. Cells were washed twice with PBS containing 1% FBS and once with RPMI medium. Cells were then transferred to a 96-well cell culture plate (F-bottom, Greiner Bio-One). 7.5 µl of fresh human, anti-AQP4 seronegative serum was added in a final volume of 150 µl RPMI to each well. After incubation at 37°C in a humidified CO2 incubator for 12 hours, the supernatant was discarded, and the remaining cells were detached with 0.05% trypsin-EDTA and transferred into flow cytometry tubes. Cell number and viability was determined by flow cytometry (CyAn ADP, Beckman Coulter).
In Vitro Antibody-Dependent Cell-Mediated Cytotoxicity Assay (ADCC)
To eliminate serum complement from the ADCC assay, whole IgG from anti-AQP4 antibody positive and negative sera was purified using Sepharose-G columns (GE Bio-sciences, Piscataway, NJ) according to the manufacturer’s protocol. Successful isolation of IgG was shown using the LN18AQP4 cell-based flow cytometry assay described above. CD56+ human NK cells were isolated from PBMCs of healthy donors separated by density gradient centrifugation (Biocoll Separating Solution, Biochrom). 107 PBMCs were incubated with 20 µl of CD56 MACS beads (Miltenyi Biotec) and 80 µl of MACS buffer (2.5g BSA and 2ml EDTA in 500µl 1× PBS) on ice on an orbital shaker for 15 minutes. After incubation, they were washed with and diluted in 2 ml of MACS buffer. CD56+ cells were then separated using MACS (AutoMACS, Miltenyi Biotec). A purity of >95% NK cells was obtained.
5 µg/ml of rAb diluted in 40 µl of RPMI growth medium was added in duplicates to a U-Shape 96-well plate (Greiner Bio-One), each well containing 30,000 LN18CTR or LN18AQP4 cells in 40 µl of RPMI medium, and incubated on ice for 25 minutes on an orbital shaker. Cells were then washed twice with FACS buffer and once with RPMI medium and transferred into a 96-well cell culture plate (F-Bottom, Greiner Bio-One). 60,000 CD56+ NK cells resuspended in 40 µl of RPMI medium were added to each well. Medium was added to yield a final volume of 150 µl. After incubation at 37°C in a humidified CO2 incubator for 10 hours, the supernatant was collected, remaining cells were detached using 70 µl of 0.05% trypsin-EDTA and transferred into FACS tubes. Cell number and viability was detected using FACS based cell counting (CyAn ADP, Beckman Coulter).
Human NK Cell Isolation and CD107a Mobilization Assay
Human NK cells were isolated and the CD107a mobilization assay performed as described14. Briefly, HFAs were incubated either with media, 10% patient or control serum or recombinant NMO antibodies at 40 µg/ml for one hour at 37°C. HFAs were then washed twice with fresh media and NK cells were added at a 1:1 ratio in addition to 1uL monensin and 1uL of PE-conjugated CD107a (BD biosciences) antibody. After 5 hours, cells were harvested and analyzed on a FACSCalibur flow cytometer.
NMO CSF rAb Transfusion in the Rat MBP EAE Model
2.5 mg of NMO CSF rAbs-168, -51, -43, and -10 (n=3 each), an anti-MOG monoclonal antibody (mAb 8-18C5) (n=2), a control rAb (rAb 2B4) against measles virus nucleocapsid protein (n=3), or PBS vehicle (n=3) was transferred into the retrobulbar venous plexus of female Lewis rats previously immunized with guinea pig MBP72– 85 (gpMBP-peptide) emulsified in complete Freund’s adjuvant containing 5 mg/ml inactivated Mycobacterium tuberculosis H37 Ra. The rAb was administered at a clinical disease score of 1.0 to 1.5. Thirty hours after injection, the animals were euthanized and the brains, optic nerves, and spinal cords were dissected, and processed for cryosectioning and paraffin embedding.
Histology and Immunohistochemistry
CNS tissue was evaluated for inflammation, demyelination, astrocyte destruction, and myelin vacuolization by hematoxylin/eosin staining and immunohistochemistry for myelin basic protein (MBP, Dako) and glial fibrillary acidic protein (GFAP; Dako). Polyclonal antibodies against C9 (kindly provided by B.P. Morgan, Cardiff), human IgG (Dako), and CD68 (ED1, Serotec) were used to detect complement, human IgG, and macrophage infiltration, respectively. Transected axons were visualized by immunohistochemistry for amyloid precursor protein (APP, Chemicon) as described.13
Quantification of Astrocyte loss in EAE Animals
The extent of astrocyte loss was assessed in animals with gpMBP-peptide induced EAE transferred with rAb-2B4; NMO CSF rAbs-168, -51, -43, and -10; PBS; and mAb 8-18C5 using GFAP immunocytochemistry. Histological sections were immunostained with anti-GFAP antibodies (Dako) and assessed by light microscopy. GFAP-immunostained sections were scanned using a BX51 Olympus light microscope equipped with a DP71 digital camera. At least 15 spinal cord cross sections were examined per animal, corresponding to an area of at least 55 mm2. Areas of complete astrocyte loss were measured using Analysis™ software. The total area of astrocyte loss between groups was evaluated by ANOVA.
Results
The Humoral Immune Response in NMO CSF is Antigen-Driven
Cerebrospinal fluid (CSF) was obtained from an AQP4 seropositive 48 year-old woman 8 weeks after an attack of monosymptomatic optic neuritis. Although CSF was negative for oligoclonal bands and elevated Ig synthesis, there was a pronounced CSF cell pleocytosis (see Patient in Patients and Methods). Flow cytometry revealed a prominent CSF humoral immune response: 3.7% of the CSF lymphocyte population were CD19+CD138− B cells and 0.9% were CD138+ plasma cells. Like CSF from multiple sclerosis (MS) patients,15, 16 most of the CD138+ CSF plasma cells (70.5%) were CD19+CD138+ plasma blasts. Single CD138+ plasma cells were isolated, and the heavy- (VH) and light-chain (VL) variable region sequences were amplified from single cells by RT-PCR (Supplemental Table). The CD138+ VH repertoire showed numerous clonal populations, widespread intraclonal diversity, and the preferential use of VH2 family germline sequences. CD138+ plasma cell heavy chains were extensively mutated with an average germline homology of 93.7% (range 80.1–99.2%). The VH family germline distribution of the CSF CD138+ plasma cell repertoire was significantly skewed from the expected germline distribution.17. VH2 family germline sequences dominated the plasma cell VH repertoire (Fig 1a) and accounted for 37% of the total sequences (expected germline frequency - 5.6%,17 p< 0.0001); VH4 family sequences were also significantly elevated (Fig 1a) and comprised 32% of the total repertoire (expected germline frequency - 21.6%,17 p<0.01). The VH2 and VH4 germline bias was unchanged when only unique sequences were tabulated.
Fig 1.
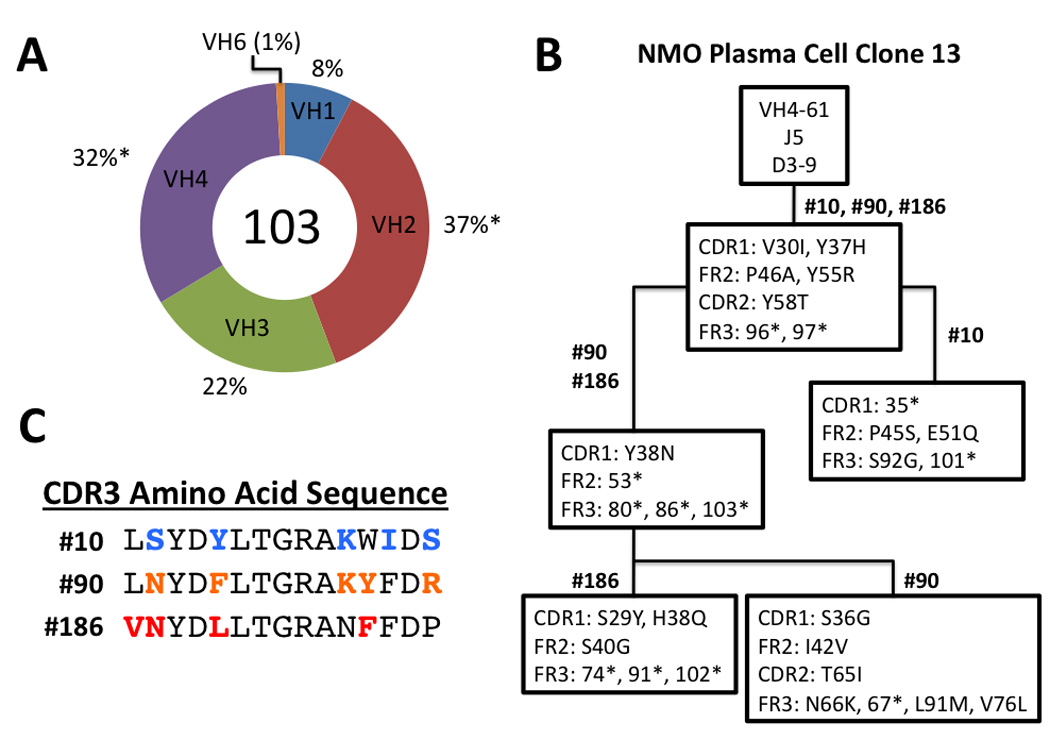
The CD138+ NMO CSF plasma cell repertoire shows VH2 family germline bias and extensive intraclonal diversity. (A) The percentage of VH usage in 103 CD138+ CSF plasma cells is indicated. *Statistically significant difference (binomial distribution, P <0.01). (B) Lineage analysis of the three plasma cell VH sequences comprising plasma cell clone 13 demonstrates intraclonal diversity. The topmost box contains the germline V, D, and J segments used in the clonal heavy chain rearrangement. The numbers above each box indicate the plasma cells (Supplemental Table) that contain the mutations listed inside. The location of each mutation within the FR or CDR region of VH sequence is noted on the left. Replacement mutations are demarcated by the initial amino acid, amino acid position, and the new amino acid resulting from the mutation (e.g., Y37H). Silent mutations are demarcated by the amino acid position followed by an asterisk (e.g., 96*). (C) CDR3 amino acid sequence of each plasma cell VH in clone 13. Bolded, colored amino acids denote somatic hypermutations resulting in amino acid substitutions within the germline V(D)J rearrangement.
Most of the CD138+ plasma cell VH repertoire was contained within clonal populations: plasma cells expressing heavy-chain sequences with identical V(D)J rearrangements. 53% of the total CD138+ plasma cell VH sequences (Table 1) were found within 20 plasma cell clones. Most of these clonal populations showed marked intraclonal diversity with individual VH sequences displaying distinct somatic mutations within both the framework (FR) and complementarity-determining regions (CDRs). For example, sequence analysis of plasma cell clone 13 revealed a common set of somatic mutations punctuated by a large number of sequence-specific replacement mutations (Fig 1b). This high degree of intraclonal diversity was particularly manifest in CDR3 where widespread replacement mutations readily distinguished between individual members of the clonal population (Fig 1c and Table 1). Overall, the CSF plasma cell repertoire in early NMO displayed features of a highly dynamic, compartmental, antigen-driven humoral immune response.
Table 1.
CD138+ CSF Plasma Cell Clone VH Sequences
| Clone No. |
CDR3 | Rep Seq No. |
Sequence Variants in Clone |
VH Family |
Germline | JH |
|---|---|---|---|---|---|---|
| 1 | RLVVVPHATAGGWFDP | 20 | 3/3 | VH2 | 2–5 | J5 |
| 2 | RGGRLQNWFDP | 51 | 9/10 | VH2 | 2–5 | J5 |
| 3 | GGPNIVLMVYATPRRVSGNGFDI | 52 | 2/2 | VH3 | 3–11 | J3 |
| 4 | HTLLEGGGLRRFGASPLYAFDI | 43 | 0/2 | VH4 | 4–39 | J3 |
| 5 | RQGGSGGAKYFDF | 35 | 2/2 | VH2 | 2–5 | J4 |
| 6 | RHILDAFDI | 146 | 3/3 | VH2 | 2–5 | J3 |
| 7 | DETQRTSYIISRYGFDT | 1 | 2/2 | VH4 | 4–39 | J5 |
| 8 | DEAQKTPYSSRSYGGDNWFDP | 46 | 3/3 | VH4 | 4–39 | J3 |
| 9 | AEGRGWSAFYYYYMEV | 53 | 2/3 | VH4 | 4–59 | J6 |
| 10 | GDYVFDY | 58 | 2/2 | VH3 | 3–23 | J4 |
| 11 | HRRVAVGGYSYYQYMDV | 138 | 2/3 | VH3 | 3–73 | J6 |
| 12 | YRRVAVAGYTYYYYMDV | 31 | 5/5 | VH3 | 3–73 | J6 |
| 13 | LSYDYLTGRAKWIDS | 10 | 3/3 | VH4 | 4–61 | J5 |
| 14 | GPLGGGYYLPLDS | 165 | 2/2 | VH4 | 4–61 | J4 |
| 15 | DRSAFRGGFGLFFDAFNI | 71 | 0/2 | VH4 | 4-4 | J3 |
| 16 | LHYFYASGSPSYMDV | 173 | 2/2 | VH4 | 4–61 | J6 |
| 17 | RLIPSSVFDY | 180 | 2/2 | VH2 | 2–5 | J4 |
| 18 | LRRDGYNRYYFDY | 188 | 2/2 | VH3 | 3–7 | J4 |
| 19 | IPLRYYDLWRGSFDI | 157 | 0/2 | VH1 | 1-f | J3 |
| 20 | DGASGSYHHFDH | 94 | 2/2 | VH3 | 3–43 | J4 |
Each row contains the clone number (clone no.), heavy-chain CDR3 amino acid sequence (CDR3), representative sequence number (rep seq no.), number of CDR3 sequence variants in the clonal population, most homologous human V region family (VH Family), germline segment (Germline), and joining segment (JH). CDR3 amino acids subject to replacement mutation within the demarcated clonal population are denoted in bold.
Most Recombinant Antibodies Derived from NMO CSF Plasma Cells Target Aquaporin-4
Bivalent IgG1 recombinant monoclonal antibodies (rAbs) were generated from paired heavy- and light-chains recovered from 11 (10 clonal and 1 non-clonal) individual CSF plasma cells (Table 2). Because a light chain variable region sequence was not amplified from plasma cell #90, two rAbs (rAb-90 and rAb-90b) were synthesized using the heavy-chain from plasma cell #90 (clone 13, Table 1) and light-chains from plasma cells #10 and #186, respectively. Since plasma cells within the same clone usually share identical recombination events in the light chain locus, we generated two rAbs using somatically mutated light chain sequences from two fellow plasma cell clones in an attempt to reproduce the specificity of the natural plasma cell #90 Ab. Although the heavy chain constant region expressed by plasma cell #53 (clone 9, Table 1; Supplemental Table) was IgG2, rab-53 was generated using an IgG1 constant region. The other plasma cells in clone 9 (see plasma cells #27 and #167 in Supplemental Table) employed IgG1 heavy chains.
Table 2.
NMO Serum, CSF and CSF Plasma Cell-Derived Recombinant Antibodies
| rAb | VH CDR3 | VL CDR3 | LN18AQP4 Binding |
HFA Binding |
Mouse IIF Microscopy |
Complement Activation |
ADCC | CD107a Mobilization |
NMO Pathology |
|---|---|---|---|---|---|---|---|---|---|
| rAb-51 | RGGRLQNWFDP | QQANSFPYT | − | − | − | ND | ND | − | − |
| rAb-43 | HTLLEGGGLRRFGASPLYAFDI | QLRSNWPGIT | ++ | ++ | − | + | ++ | − | − |
| rAb-53b | AEGRGWSAFYYYYMEV | QQYGSSPWTF | ++ | ++ | + | + | +++ | + | ND |
| rAb-58 | GDYVFDY | QHYNSYPYTF | +++ | ++ | + | + | +++ | + | ND |
| rAb-69 | YRRVTVAGYSYYYYMDV | QQSHSLPRTF | − | − | − | ND | ND | − | ND |
| rAb-31 | YRRVAVAGYTYYYYMDV | QQSHSLPRTF | ++ | +++ | + | + | + | − | ND |
| rAb-10 | LSYDYLTGRAKWIDS | QQYGSTPLTF | +++ | ++ | + | + | +++ | + | + |
| rAb-90 | LNYDFLTGRAKYFDR | QQYGSTPLTF | − | − | − | − | − | − | ND |
| rAb-90b | LNYDFLTGRAKYFDR | QQYGSSPLTF | − | − | − | ND | ND | − | ND |
| rAb-186 | VNYDLLTGRANFFDP | QQYGSSPLTF | + | + | + | + | ++ | + | ND |
| rAb-168a | RRQNPIMISSGGVIANAFDI | QQSHSLPRTF | − | − | − | ND | ND | − | − |
| Serum | NA | NA | +++ | +++ | + | + | ++ | + | ND |
| CSF | NA | NA | +++ | +++ | + | + | ++ | + | ND |
Recombinant antibodies rAb-90 and rAb-90b (clone 13) were synthesized using the light chains from rAb-10 and rAb-186, respectively.
Serum – patient serum; CSF – patient cerebrospinal fluid
Non-clonal CD138+ VH sequence;
original heavy chain sequence was IgG2.
LN18AQP4 Binding (median fluorescence intensity, [rAb] = 0.25 µg/ml): +, 100–399; ++, 400–799; +++, ≥ 800.
HFA Binding (percentage binding, [rAb] = 45 µg/ml): +, 25–50%; ++, 51–75%; +++, ≥ 75%
Mouse IF Microscopy (indirect immunofluorescence microscopy): +, positive; −,negative.
Complement Activation (specific lysis of LN18AQP4 cell line): +, positive; −, negative.
ADCC (percentage cell survival LN18AQP4 cell line): ND, not done; −, negative; +, 21–30%; ++, 11–20; +++, 0–10%.
CD107 Mobilization Assay: +, positive; −, negative
NMO Pathology: +, positive; −, negative; ND, not done.
Abbreviations: rAb – recombinant antibody; VH CDR3 - variable region heavy-chain complementarity-determining region 3; VL CDR3 - variable region light-chain complementarity-determining region 3; LN18AQP4 – AQP4-transfected LN18 glioblastoma cell line; HFA – human fetal astrocyte; IIF – indirect immunofluorescence; ADCC – antibody-dependent cell-mediated cytotoxicity.
The patient’s serum, CSF, and rAbs were tested for binding to AQP4 by quantitative flow cytometry and indirect immunofluorescence. Patient serum and CSF, but not control serum, bound to the surface of glioblastoma cells (LN18AQP4) transfected with human AQP4 (Fig 2a), human fetal astrocytes14 (HFAs) (Fig 2b), and mouse cerebellar sections3 (Figs 3a–c,f). Six of 11 NMO CSF rAbs bound with high affinity to LN18AQP4 cells (Figs 2a,c) and HFAs (Figs 2b,d). The titration curves of the AQP4-specific rAbs, serum, and CSF on the LN18AQP4 cell line were congruent (Fig 2c), indicating that the CSF plasma cell rAbs had similar binding affinity to serum and CSF NMO-IgG. The binding of CSF rAbs to HFAs and AQP4-transfected LN18AQP4 cells was quite similar (Figs 2c,d). While 5 of 6 AQP4-specific rAbs produced the characteristic staining of CNS microvessels, pia, and subpia on mouse cerebellar sections (Fig 3d), rAb-43 did not stain mouse cerebellar sections (Fig 3e), indicating that this plasma cell rAb recognized a species-specific epitope on the human AQP4 protein. AQP4-specific rAbs recognized conformational epitopes and did not bind AQP4 on western blotting (data not shown).
Fig 2.

Most of the NMO CSF rAbs were directed against AQP4. (A) Histograms demonstrate staining of LN18CTR control (red line) and LN18AQP4 (green line) cells with serum, CSF, or rAb from our NMO patient. Antibody binding to AQP4 was detected using an anti-human IgG secondary antibody and quantitated by flow cytometry. The difference between the median fluorescence intensity observed with LN18CTR and LN18AQP4 (ΔMFI) corresponds to the amount of IgG bound to AQP4 on the surface of cells. (B) Dot plots show combined staining of MHC class I human fetal astrocytes (HFAs) with control serum, NMO patient serum, NMO patient CSF, or NMO CSF rAb. Serum, CSF, or rAb binding to HFAs was detected with PE-conjugated anti-human IgG and quantified by flow cytometry. Control serum was routinely diluted to 1:50. Patient CSF was diluted to the identical IgG concentration of the matched patient serum (300 µg/ml). (C) Change in median fluorescence intensity (ΔMFI) is plotted against IgG concentration for the serum, CSF, and AQP4-specific CSF rAbs of our NMO patient. (D) The percentage HFA binding is plotted for each NMO CSF rAb. 6 of 11 rAbs bound HFAs (mean = 69 ± 17%; range, 40 to 94.6%).
Fig 3.

CSF NMO rAbs bind murine cerebellar tissue. Control NMO serum, NMO patient serum, NMO patient CSF and CSF rAb-10 display the same pattern of binding to murine cerebellum by indirect immunofluorescence microscopy: linear staining of pia (arrows) and white matter microvessels (arrowheads). (A) Negative control MS patient serum (1:60 dilution). (B) Positive control NMO serum (1:60 dilution). (C) Our NMO patient’s serum (1:60 dilution). (D) CSF rAb-10 (50 µg/ml). (E) CSF rAb-43 (50 µg/ml). (F) Our NMO patient’s CSF (1:6 dilution). Scale bars indicate 50µm.
Using change in median fluorescence intensity or percentage HFA binding as quantitative measures of serum and CSF NMO-IgG titers, we calculated the NMO-IgG antibody synthesis index18 using both the LN18AQP4 and HFA flow cytometric assays. The NMO-IgG antibody synthesis index was significantly elevated (2.2 and 2.0) when calculated using either assay. Overall, both CSF repertoire analysis and CSF IgG protein synthesis confirmed the intrathecal production of AQP4 IgG at the time of the initial demyelinating event in this patient.
Binding of NMO CSF rAbs to AQP4 Mediate IgG1 Fc Effector Functions
The histopathology of actively demyelinating NMO lesions demonstrates perivascular Ig deposition, complement deposition, T and B cells, and prominent eosinophilic and granulocytic perivascular infiltrates.19 The composition of these lesions suggests that NMO IgG mediates tissue injury through both complement-mediated cell lysis and antibody-dependent cell-mediated cytotoxicity (ADCC). Serum and CSF IgG from our NMO patient were evaluated for their ability to lyse LN18AQP4 glial cells in the presence of serum complement or human NK cells. Similar to the observations of Hinson et al.20, we observed a selective reduction in the viability of LN18AQP4 glial cells exposed to serum complement and NMO patient serum IgG or CSF IgG (Fig 4a) but not in glial cells transfected with an empty vector (LN18CTR) (p < 0.001, paired t-test). In the presence of human NK cells, NMO patient serum IgG and CSF IgG directed selective ADCC of LN18AQP4 but not LN18CTR target cells (Fig 4b) (p < 0.001, paired t-test). There was no difference in the ability of serum and CSF IgG to direct complement-mediated lysis or ADCC of LN18AQP4 target cells (p=NS, t-test).
Fig 4.

Serum and CSF IgG from our NMO patient directs complement-mediated lysis and antibody-dependent cell-mediated cytotoxicity (ADCC) of target cells expressing AQP4. (A) AQP4-specific complement-mediated killing of LN18AQP4 and LN18CTR cells by CSF rAbs was quantitated using FACS analysis. The experiment was performed in duplicate, and the percentage cell survival was normalized to the negative control sample (No IgG or complement). Mean and standard deviation are shown. IgG purified from the serum (NMO Patient Serum IgG + complement) or CSF (NMO Patient CSF IgG + complement) significantly reduced the viability of LN18AQP4 target cells (p < 0.001, paired t-test). Serum and CSF IgG purified from an MS patient resulted in no significant change in LN18AQP4 viability (p = 0.34, paired t-test). (B) ADCC of LN18AQP4 and LN18CTR cells in the presence of human NK cells and NMO patient serum or CSF IgG was evaluated by FACS analysis. The number of viable cells in culture was quantitated and normalized to the number of surviving cells in the negative control (No IgG or NK cells) sample. In the presence of human NK cells, NMO patient serum (NMO Patient Serum IgG + NK Cells) and CSF IgG (NMO Patient CSF IgG + NK Cells) resulted in a specific loss of more than 80% of LN18AQP4 target cells (p < 0.001, paired t-test). ADCC of LN18AQP4 target cells with NMO patient serum and CSF IgG was significantly greater than that resulting from AQP4-negative MS serum (MS Serum IgG + NK Cells) and CSF IgG (MS CSF IgG + NK Cells) (p < 0.001, t-test).
To examine the antibody effector function of our NMO CSF rAbs, AQP4-specific rAbs were tested for their ability to mediate complement activation, ADCC and NK cell degranulation in vitro. In the presence of serum complement, AQP4-specific CSF rAbs resulted in a marked and specific reduction in cell viability in the LN18AQP4 cell line but not in LN18CTR cells (Fig 5a) (p < 0.001, paired t-test). Control Ig and AQP4-negative CSF rAbs did not affect the viability of either cell line. Similarly, in an in vitro ADCC assay, AQP4-specific CSF rAbs specifically reduced the viability of LN18AQP4 but not LN18CTR target cells after prolonged incubation with human NK cells (Fig 5b) (p < 0.001, paired t-test). In contrast, purified Ig from control MS serum, AQP4-negative CSF rAb-90, NK cells alone, or purified Ig from our NMO patient’s serum alone did not affect target cell survival (Fig 5b). Compared to the other AQP4-specific CSF rAbs, rAbs-43, -186, and -31 showed decreased ADCC of LN18AQP4 target cells (Fig 5b). The NK cell-mediated killing of LN18AQP4 cells preincubated with CSF rAb was dose-dependent. Serial dilution of the AQP4-specific rAb-10 demonstrated a clear lytic dose response from 5 µg/ml (5.9% survival) to 0.05 µg/ml (74% survival) (Fig 5c).
Fig 5.

AQP4-specific CSF rAbs are functional IgG1 antibodies. (A) AQP4-specific complement-mediated killing of LN18AQP4 and LN18CTR cells by CSF rAbs was quantitated using FACS analysis. The experiment was performed in duplicate, and the percentage cell survival was normalized to the negative control (No Serum) sample. The loss of LN18AQP4 cell viability was greater than 75% in the presence of AQP4-specific rAb (rAbs-10, -31, -43, -53, -58, and -186) and complement. Non-specific reduction in cell viability rarely exceeded 20% in the absence of serum (No Serum) or the presence of nonspecific serum (MS Ig) or CSF rAb (rAb-90). Mean and standard deviation are shown. (B) The induction of antibody-dependent cell-mediated cytotoxicity (ADCC) of LN18AQP4 and LN18CTR cells in the presence of human NK cells and CSF rAbs was evaluated by FACS analysis. After 12 hrs of cocultivation, the number of viable LN18 cells in culture was quantitated and normalized to the number of surviving cells in the negative control (AQP4 only) sample that contained NMO patient serum in the absence of human NK cells. AQP4-specific rAbs-10, -53, and -58 resulted in a specific loss of more than 94% of the target LN18AQP4 cells; AQP4-specific rAbs-43, -186, and -31 caused a slightly lower amount of LN18AQP4-specific target cell death. No impact on LN18AQP4 cell survival was observed in the absence of rAb (NK only), the absence of NK cells (rAb-10 only), the presence of nonspecific serum (MS Ig) or CSF rAb (rAb-90). The experiment was performed in duplicates; mean and standard deviation are shown. (C) Serial dilution of CSF rAb-10 demonstrates that NK-mediated lysis of LN18AQP4 cells is dose dependent. AQP4-specific rAb-10 (5 µg/ml, 0,5 µg/ml and 0,05 µg/ml) was preincubated with either LN18AQP4 or LN18CTR target cells and the level of ADCC in the presence of human NK cells was quantitated by FACS analysis. Cell survival was determined after 10 hr cocultivation and normalized to the negative control (LN18CTR) cell line. (D) NK cell surface mobilization of CD107a was quantified by FACS analysis after coincubation of HFAs with NMO patient serum, NMO patient CSF, or AQP4-specific NMO CSF rAb. NMO patient serum, NMO patient CSF, and AQP4-specific CSF rAbs-10, --53, -58, and -186 resulted in comparable elevations in CD107a surface expression on human NK cells. CSF rAbs-43 and -31 triggered only modest CD107a surface mobilization compared to a measles virus nucleocapsid-specific control rAb (rAb-2B4). The experiment was performed in duplicates; mean and standard deviation are shown.
Surface mobilization of CD107a by NK cells is strongly correlated with NK-mediated cytotoxic activity.14 To test whether AQP4-specific NMO CSF rAbs could trigger ADCC of HFAs, we quantified expression of the degranulation marker CD107a on the surface of human NK cells cocultivated with HFAs in the presence or absence of NMO CSF rAbs. As reported by Vincent et al.14, there was a significant increase in the percentage of NK cells expressing surface CD107a after contact with HFAs preincubated with patient serum or CSF (Fig 5d). Preincubation of HFAs with 4 of 6 AQP4-specific NMO CSF rAbs resulted in a similar increase in cell surface expression of CD107a (Fig 5d). There was a strong inverse correlation of CD107a surface induction and LN18AQP4 cell viability in the in vitro ADCC assay (p = 0.04, r = −0.69). Similar to their attenuated performance in the ADCC assay (Fig 5b), rAb-43 and rAb-31 showed reduced induction of CD107a surface expression on NK cells cocultivated with HFAs relative to other AQP4-specific rAbs (Fig 5d). Interestingly, these rAbs displayed lower performance in ADCC measures despite their robust affinity for AQP4 (Fig 2c) and their ability to induce complement-mediated lysis of LN18AQP4 cells (Figs 5a). Since each CSF rAb contains the identical heavy chain IgG1 constant region, it is likely that the target epitope of AQP4-specific rAbs influences antibody effector function.
NMO CSF rAbs Alter CNS Immunopathology in Experimental Autoimmune Encephalomyelitis (EAE)
The rat myelin basic protein (MBP) EAE model has been used to document the deleterious effect of antimyelin antibodies in acute CNS demyelination.13 Four NMO CSF rAbs (rAb-10, rAb-168, rAb-51, rAb-43), a monoclonal antibody against myelin oligodendrocyte glycoprotein (mAb 8-18C5), and rAb-2B4, a control rAb against measles virus nucleocapsid, were transfused by retrobulbar venous plexus injection into rats previously immunized with guinea pig MBP72–85. Thirty hours after antibody transfusion, animals were euthanized and the brains, optic nerves, and spinal cords assessed for immunopathology. Intravenous transfer of the measles virus-specific rAb-2B4 did not alter the CNS immunopathology observed in any of the EAE animals (Fig 6). Intravenous transfer of the AQP4-specific rAb-10, however, resulted in novel pathologic changes prototypic for NMO such as perivascular astrocyte depletion and complement deposition (Fig 6). These pathologic changes were consistently noted in each of the three treated animals. We used GFAP immunocytochemistry to quantify the area of astrocyte loss in 15 representative spinal cord sections in each experimental animal. No loss of GFAP immunoreactivity was observed in rats transferred with rAb-2B4, rAb-168, rAb-51, rAb-43, mAb 8-18C5 or PBS. However, between 3 and 7% of total spinal cord area were devoid of astrocytes in rats transferred with rAb-10 (p < 0.001, ANOVA).
Fig 6.

AQP4-specific CSF rAb-10 induced prototypic NMO pathology in guinea pig MBP72–85-induced EAE rats. The rows are arranged by NMO CSF rAb, and the columns are ordered by immunohistochemical stain. In rats injected with rAb-10, glial fibrillary acidic protein (GFAP) immunohistochemistry revealed prominent perivascular loss of astrocyte cell bodies and processes. Staining for complement C9 (C9) and human Ig (hIg) demonstrated significant deposits of perivascular complement protein and human Ig deposition beyond the zone of perivascular astrocyte depletion. Intravenous transfer of negative control measles virus-specific rAb-2B4 or human AQP4-specific rAb-43 produced no additional immunopathology. Myelin basic (MBP) immunohistochemistry demonstrated some perivascular myelin vacuolization in rAb-10-treated animals, but myelin remained largely intact. There was no difference in the extent of CNS tissue macrophage infiltration (CD68) induced by rAbs-10, -43, and -2B4. Scale bar: 200 µM.
Spinal cords from rats transfused with rAb-10 showed no primary perivascular demyelination; instead, there was perivascular myelin vacuolization and prominent human Ig deposition in perivascular areas not yet devoid of astrocytes (Fig 6). Axonal damage, as assessed by detection of APP-positive profiles, was additionally noted (data not shown). In contrast, rAb-43, which is specific for human but not rat or murine AQP4, resulted in no astrocyte loss or additional CNS immunopathology (Fig 6). NMO CSF rAbs that did not bind AQP4, rAb-168 and rAb-51, produced no novel pathology (data not shown). Our results indicate that a human AQP4-specific rAb derived from a clonally expanded intrathecal plasma cell produces NMO-related immunopathology, perivascular astrocyte destruction and complement deposition, after intravenous transfer into EAE animals.
Discussion
Serum autoantibodies against AQP4 (NMO-IgG) are a specific biomarker that distinguishes patients with NMO from those with MS. The exact role of AQP4-specific IgG in NMO pathogenesis, however, remains unclear. Certainly, if NMO-IgG is pathogenic in NMO, then AQP4 antibodies should be present in the CNS compartment before the initial clinical presentation of disease. Nevertheless, in clinically definite NMO,8 titers of CSF NMO-IgG are nearly proportional to serum NMO-IgG, indicating that the bulk of AQP4-specific IgG is synthesized in the peripheral lymphoid compartment in affected individuals5 or is absorbed by CNS astrocytes expressing AQP4. While serum AQP4 autoantibodies may gain access to the CNS through blood brain barrier disruption in established disease, this mechanism does not adequately explain AQP4 antibody-mediated pathogenesis at disease onset. Serum AQP4 antibody could enter the CNS by endothelial transcytosis or through regions of increased BBB permeability, but AQP4-specific autoantibodies are only a minor component of the serum IgG fraction and the initial NMO lesions do not demonstrate any regional predilection.
We conducted a molecular characterization of the intrathecal CD138+ plasma cell response in an NMO patient after her first clinical demyelinating attack. We focused our analysis on CSF CD138+ cells because prior studies have demonstrated that this predominantly plasma blast population is the primary source of intrathecal Ig production in inflammatory and infectious CNS disease.11, 15, 16, 21 Consistent with the proposed role of AQP4 autoantibodies in NMO pathogenesis, our findings indicate that an intrathecal humoral immune response against AQP4 is evident at the onset of clinical disease. Sequence analysis of the VH and VL sequences in the CSF CD138+ plasma cell repertoire demonstrate that the humoral immune response in NMO is compartmental, antigen-driven and T cell dependent. Like NMO-IgG in serum, nearly all (93.2%) NMO plasma cells expressed IgG1 (see Supplemental Table). The NMO plasma cell VH repertoire displayed a remarkable degree of intraclonal diversity (Table 1). This intraclonal diversity suggests either recent or active B cell receptor maturation within germinal center reactions. It is unlikely that the excessive intraclonal diversity is merely a reflection of the timing of the repertoire analysis in relation to the demyelinating event since plasma cell repertoires derived within similar intervals from patients with optic neuritis and MS do not demonstrate this degree of intraclonal diversification.22, 23 The dynamic nature of the CSF plasma cell response in the NMO patient was particularly evident in plasma cell clone 13 (Fig 1) where intraclonal diversity was manifested by rAbs with variable affinity for AQP4 (Table 2). RAbs generated from plasma cell clone 13 demonstrated high (rAb-10), low (rAb-186), and no affinity for AQP4 (rAbs-90 and -90b). Comparison of somatic hypermutations within VH sequences composing plasma cell clone 13 indicated a distinct lineage among clone members; furthermore, the high-affinity, AQP4-specific, rAb-10 displayed a number of replacement mutations that were distinct from the other members of the clone (Fig. 1). Additional CSF repertoire analyses in NMO will be necessary to determine whether this dynamic intraclonal diversity and affinity maturation are characteristic features of the humoral immune response in active NMO.
In addition to intraclonal diversity, the CSF plasma cell repertoire in the NMO patient showed evidence of light chain receptor editing: cells with identical heavy chains expressing alternative productive VκJκ light chain rearrangements (see clone 12 in Table 1 and plasma cells #31, #69, and #93 in Supplemental Table). Receptor editing is a process that occurs in peripheral lymphoid tissue where renewed expression of recombinase genes results in secondary rearrangement of light chain loci.24 Receptor editing has been shown to reshape the murine B cell repertoire in autoimmune disease by rescuing cells containing self-reactive Ig from targeted destruction.25 Evidence of light chain receptor editing has been observed in B cell repertoires in MS10 and subacute sclerosing panencephalitis (SSPE).26 Comparison of the reactivities of rAbs generated from plasma cells #93 and #31 may help to confirm the role of receptor editing in structuring the antibody response in human autoimmune disease.
Our ability to generate rAbs from intrathecal plasma cells in early NMO has allowed us to directly test the hypothesis that AQP4-specific autoantibodies are key mediators of NMO-related tissue damage. In our early NMO patient, most rAbs generated from CSF plasma cells were directed against the human AQP4 protein. The fraction of AQP4-specific CSF plasma cells that we observed in early NMO was similar to the proportion of measles-specific CSF plasma cells observed in SSPE,11 consistent with the hypothesis that AQP4 is the primary disease-specific target antigen. The AQP4-specific rAbs that we prepared from intrathecal clonally expanded NMO plasma cells were target specific in multiple independent assays and capable of initiating downstream antibody effector functions. Furthermore, we showed that administration of a single AQP4-specific rAb in the rat MBP EAE model induced an NMO-specific immunopathology: perivascular astrocyte destruction accompanied by perivascular IgG and complement deposition. Similar changes were not observed with non-specific control rAb or rAb-43 specific to human AQP4 indicating that the pathologic changes arose directly from AQP4-specific rAb binding to the rat AQP4 protein. The experimental results provide the first definitive evidence that AQP4 autoantibodies in NMO are direct mediators of disease-specific immunopathology. Whether AQP4-specific rAbs cause additional CNS injury by modulating AQP4 function or assembly remains to be determined.
Since a significant fraction of NMO patients are NMO-IgG seronegative, determining the identity of non-AQP4 target antigens in NMO will have significant implications for disease diagnosis and prognosis. The generation and characterization of disease-specific monoclonal rAbs in NMO provides the first step towards identifying these novel antigenic targets and understanding antibody-mediated pathogenesis in human demyelinating disease.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Multiple Sclerosis Society, RG3908 (J.L.B); Canadian Institute of Health Research (CIHR) CGS Doctoral award (P.S.); Multiple Sclerosis Society of Canada (J.A.); NIH, National Institute of Neurologic Disorders and Stroke, NS32623 (D.G, G.P.O.); Deutsche Forschungsgemeinschaft, He2386/7-1 and BMBF (KKN-MS Verbund Control-MS) (S.R.K., S.N., B.H.); and DFG Centre for Molecular Physiology of the Brain (CMPB) and TR-SFB 43 “The brain as a target of inflammatory processes” (C.S.).
Footnotes
Conflict of interest: All authors report no conflict of interest.
References
- 1.Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet neurology. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 4.Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007;130:1235–1243. doi: 10.1093/brain/awm062. [DOI] [PubMed] [Google Scholar]
- 6.Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072–3080. doi: 10.1093/brain/awn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beyer AM, Wandinger KP, Siebert E, et al. Neuromyelitis optica in a patient with an early onset demyelinating episode: clinical and autoantibody findings. Clinical neurology and neurosurgery. 2007;109:926–930. doi: 10.1016/j.clineuro.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 8.Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 9.Bennett JL, Haubold K, Ritchie AM, et al. CSF IgG heavy-chain bias in patients at the time of a clinically isolated syndrome. J Neuroimmunol. 2008 doi: 10.1016/j.jneuroim.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owens G, Ritchie A, Burgoon M, et al. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol. 2003;171:2725–2733. doi: 10.4049/jimmunol.171.5.2725. [DOI] [PubMed] [Google Scholar]
- 11.Owens GP, Ritchie AM, Gilden DH, et al. Measles virus-specific plasma cells are prominent in subacute sclerosing panencephalitis CSF. Neurology. 2007;68:1815–1819. doi: 10.1212/01.wnl.0000262036.56594.7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu X, Gilden DH, Ritchie AM, et al. Specificity of recombinant antibodies generated from multiple sclerosis cerebrospinal fluid probed with a random peptide library. J Neuroimmunol. 2006;172:121–131. doi: 10.1016/j.jneuroim.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Zhou D, Srivastava R, Nessler S, et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci USA. 2006;103:19057–19062. doi: 10.1073/pnas.0607242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vincent T, Saikali P, Cayrol R, et al. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J Immunol. 2008;181:5730–5737. doi: 10.4049/jimmunol.181.8.5730. [DOI] [PubMed] [Google Scholar]
- 15.Cepok S, Rosche B, Grummel V, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. 2005;128:1667–1676. doi: 10.1093/brain/awh486. [DOI] [PubMed] [Google Scholar]
- 16.Winges KM, Gilden DH, Bennett JL, et al. Analysis of multiple sclerosis cerebrospinal fluid reveals a continuum of clonally related antibody-secreting cells that are predominantly plasma blasts. J Neuroimmunol. 2007 doi: 10.1016/j.jneuroim.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Cook GP, Tomlinson IM. The human immunoglobulin VH repertoire. Immunology today. 1995;16:237–242. doi: 10.1016/0167-5699(95)80166-9. [DOI] [PubMed] [Google Scholar]
- 18.Reiber H, Lange P. Quantification of virus-specific antibodies in cerebrospinal fluid and serum: sensitive and specific detection of antibody synthesis in brain. Clin Chem. 1991;37:1153–1160. [PubMed] [Google Scholar]
- 19.Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 21.Cepok S, von Geldern G, Nolting T, et al. Viral load determines the B-cell response in the cerebrospinal fluid during human immunodeficiency virus infection. Ann Neurol. 2007;62:458–467. doi: 10.1002/ana.21195. [DOI] [PubMed] [Google Scholar]
- 22.Haubold K, Owens GP, Kaur P, et al. B-lymphocyte and plasma cell clonal expansion in monosymptomatic optic neuritis cerebrospinal fluid. Ann Neurol. 2004;56:97–107. doi: 10.1002/ana.20152. [DOI] [PubMed] [Google Scholar]
- 23.Ritchie AM, Gilden DH, Williamson RA, et al. Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol. 2004;173:649–656. doi: 10.4049/jimmunol.173.1.649. [DOI] [PubMed] [Google Scholar]
- 24.Kouskoff V, Nemazee D. Role of receptor editing and revision in shaping the B and T lymphocyte repertoire. Life Sci. 2001;69:1105–1113. doi: 10.1016/s0024-3205(01)01219-x. [DOI] [PubMed] [Google Scholar]
- 25.Retter MW, Nemazee D. Receptor editing occurs frequently during normal B cell development. J Exp Med. 1998;188:1231–1238. doi: 10.1084/jem.188.7.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgoon MP, Keays KM, Owens GP, et al. Laser-capture microdissection of plasma cells from subacute sclerosing panencephalitis brain reveals intrathecal disease-relevant antibodies. Proc Natl Acad Sci U S A. 2005;102:7245–7250. doi: 10.1073/pnas.0502323102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.