

Abstract
The transcription factor, Nuclear Factor kappa B (NF-κB) is a critical regulator of inflammation and immunity, and is negatively regulated via S-glutathionylation. The inhibitory effect of S-glutathionylation is overcome by glutaredoxin-1 (Grx1), which under physiological conditions catalyses deglutathionylation and enhances NF-κB activation. The mechanisms whereby expression of the Glrx1 gene is regulated remain unknown. Here we examined the role of NF-κB in regulating activation of Glrx1. Transgenic mice which express a doxycyclin-inducible constitutively active version of inhibitory kappa B kinase-beta (CA-IKKβ) demonstrate elevated expression of Grx1. Transient transfection of CA-IKKβ also resulted in significant induction of Grx1. A 2kb region Glrx1 promoter that contains two putative NF-κB binding sites was activated by CA-IKKβ, RelA/p50, and lipopolysaccharide (LPS). Chromatin immunoprecipitation experiments confirmed binding of RelA to the promoter of Glrx1 in response to LPS. Stimulation of C10 lung epithelial cells with LPS caused transient increases in Grx1 mRNA expression, and time-dependent increases in S-glutathionylation of IKKβ. Overexpression of Grx1 decreased S-glutathionylation of IKKβ, prolonged NF-κB activation, and increased levels of pro-inflammatory mediators. Collectively, this study demonstrates that the Glrx1 gene is positively regulated by NF-κB, and suggests a feed forward mechanism to promote NF-κB signaling by decreasing S-glutathionylation.
Keywords: S-glutathionylation, Nuclear Factor kappa B, Glutaredoxin, Lung, Inhibitory kappa B kinase
Introduction
The transcription factor, Nuclear Factor kappa B (NF-κB) is a cardinal regulator of cell survival, and immune and inflammatory responses. Its activation has been linked to a wide variety of chronic inflammatory, and immune diseases, as well as cancer [1]. NF-κB is comprised of diverse subunits of NF-κB/Rel proteins and inducibly regulates transcription of over 100 target genes. The canonical NF-κB pathway is activated following stimulation of various receptors including toll like receptors and cytokine receptors and results in activation of the Inhibitory kappa B kinase (IKK) complex which consists of the catalytic subunits IKKα and IKKβ, and the regulatory protein, IKKγ. The catalytic activity of IKKβ mediates phosphorylation of the NF-κB inhibitor IκBα, leading to its proteasomal degradation. Non canonical signaling is activated by agonists such as CD40 and B cell activating factor receptor, stabilization of NF- κB inducing kinase, which in turn phosphorylates and activates homodimers of IKKα (Fig. 1A). IKKα and IKKβ also mediate additional phosphorylation events and collectively lead to enhanced transcriptional activation of target genes [2–4].
Fig. 1.

Overview of the NF-κB activation pathways and the impact of S-glutathionylation. A) Schematic representation of activation of classical (top) and alternative (bottom) NF-κB activation pathways, and outcomes. The classical pathway is activated by diverse ligands, such as LPS, Tumor Necrosis Factor-alpha (TNFα), Interleukin 1-beta (IL-1β), among many others, which results in the activation of Inhibitory kappa B kinase beta (IKKβ) which in turn mediates degradation of IκBα, resulting in the nuclear translocation and activation of RelA/p50 NF-kB subunits. The alternative NF-κB pathway is activated by distinct subsets of ligands, such as B cell Activating Factor (BAFF), CD40 ligand (CD40L) etc. which result in NF-κB Inducing Kinase (NIK) dependent activation of I kappa B kinase alpha (IKKα) which phosphorylates p100, and resultant proteolytic processing to p52. RelB/p52 dimeric complexes then are translocated to the nucleus, to activate transcription of unique sets of genes. Note that this schematic is an oversimplification, as additional regulatory post-translational modifications, and chromatin remodeling events occur to enable transcriptional activation of genes. Cross talk between classical and alternative NF-κB pathways also occurs, and is not illustrated here. B) Impact of H2O2 of IKKβ and NF-κB signaling. Stimulation of cells with LPS or TNFα leads to activation of IKKβ, and downstream NF-κB signaling. In the presence of H2O2 (100–200 μM) or following overexpression of NOX1, IKKβ is inhibited via S-glutathionylation (-SSG) of Cys179. Overexpression of glutaredoxin-1 (Grx1) reverses S-glutathionylation of IKKβ (-SH), and permits NF-κB signaling in the presence of H2O2. This schematic is a summary of previously published data [10].
Redox-based regulation of cell signaling is receiving increased attention as a result of the demonstration that diverse pathways and transcription factors, including NF-κB can be dynamically regulated through reversible cysteine oxidations [5, 6]. Reactive, low pKa, cysteines in the thiolate state can be reversibly oxidized in diverse manners, which include S-nitrosylation, sulfenic acid formation, disulfide formation, and S-glutathionylation [5]. Protein S-glutathionylation (also referred to as S-glutathiolation or mixed disulfides) causes functional alterations in target proteins, and both activation and inhibition of physiological function has been observed [7–10]. We and others have previously demonstrated that the NF-κB pathway is inhibited via S-glutathionylation, and S-glutathionylation of IKKβ, RelA, and p50 have been documented [10–12]. However, the exact proportion of S-glutathionylation of NF-κB members that occurs in intact cells, and impact for the strength of NF-κB signaling remains unknown to date.
Steady state levels of S-glutathionylated proteins are controlled by glutaredoxins, members of the thioredoxin family of oxidoreductases. Glutaredoxins (Grx) under physiological conditions act to efficiently, and specifically deglutathionylate proteins [13]. We previously demonstrated that ablation of Grx1 enhances S-glutathionylation of IKKβ-Cys179 induced by hydrogen peroxide (H2O2), and inhibits cytokine-induced NF-κB activation and pro-inflammatory mediators, while overexpression of Grx1 decreases S-glutathionylation of IKKβ-Cys179 and enhances NF-κB activation following oxidation by H2O2, collectively demonstrating that the cellular content of Grx1 regulates the extent to which NF-κB becomes activated under conditions of oxidative stress [10] (Fig. 1B).
We recently demonstrated that the cellular content of Grx1 is modulated by diverse pro-inflammatory stimuli. In mice with allergic airway inflammation, Grx1 content was increased in bronchial epithelial cells [14], which also show activation of NF-κB [15]. Upregulation of Grx1 has also been observed in retinal glial cells cultured in high glucose medium, concomitant with activation of NF-κB [16]. These findings suggest that NF-κB and Grx1 may be regulated in a coordinate fashion. In the present study we sought to determine whether the glutaredoxin (Glrx1) gene is regulated via the NF-κB pathway, and to determine the functional implications of such regulation.
Materials and methods
Plasmids and reagents
Constitutively active IKKβ (CA-IKKβ) in which serines 177 and 181 are mutated to glutamic acid residues, dominant negative IκBα (dnIκBα) in which serines 32 and 36 are mutated to alanines, and flag tagged glutaredoxin-1 (Grx1) constructs were used as previously described [9, 17, 18]. All transfections were performed using the DharmaFECT reagent (Thermo Scientific) according to the manufacturer’s instructions. Grx1 targeting SiRNA, and SiRNA controls (Invitrogen) were used as previously described, with modification [19]. Herein we will refer to glutaredoxin 1 using the commonly used abbreviation, Grx1. When referring to the mouse glutaredoxin 1 gene, we will use the Glrx1 as the abbreviation.
Cell culture
Murine type two lung epithelial (C10), and macrophage (RAW 264.7) cell lines were propagated in CMRL medium (C10), or Dulbecco’s minimal essential medium (RAW 264.7), supplemented with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin. For stimulation experiments, RAW 264.7 cells were plated and allowed to adhere for 1 h prior to treatment. Cells were stimulated with 5 ng/mL IL-1β (Axxora), 10 ng/mL TNF-α (Calbiochem), or 0.5–10 μg/mL LPS (List Biological Laboratories) for the times indicated.
Immunoblotting
Cells were lysed in buffer containing 137 mM Tris·HCl (pH 8.0), 130 mM NaCl, and 1% NP-40. Proteins were resolved by SDS-PAGE and blotted onto PVDF membranes (Millipore) prior to immunoblotting. In selected experiments, nuclear extracts were prepared according to previously published procedures [20], and nuclear proteins resolved by SDS-PAGE. The following antibodies were used for Western Blotting, Grx1 (American Diagnostica), β-actin (Sigma), IKKβ (Santa Cruz), RelA (Santa Cruz), and Histone H3 (Millipore).
Glutaredoxin-1 activity assay
Cells were lysed in buffer containing 137 mM Tris·HCl (pH 8.0), 130 mM NaCl, and 1% NP-40, lysates were then cleared by centrifugation, and 100 μg of protein incubated with reaction buffer containing 137 mM Tris-HCl, pH 8.0, 0.5 mM glutathione (Roche), 1.2 U glutathione disulfide reductase (Roche), 0.35 mM NADPH (Sigma), 1.5 mM EDTA, and 2.5 mM cysteine-SO3 (Sigma) for 10 min. Consumption of NAPDH was determined spectrophotometrically at 340 nm and data are expressed as Units, in which 1 Unit equals the oxidation of 1 μmol NADPH/min/mg protein.
Assessment of protein-S-glutathionylation (PSSG)
Protein S-glutathionylation in cells was determined using the glutathione/glutathione reductase/NADPH/5,5′-dithiobis (2-nitrobenzoic acid) recycling assay, according to procedures as described elsewhere [21] with minor modifications. Cells were lysed in 137 mM Tris-HCl, pH 8.0, 130 mM NaCl, and 1% NP-40. Protein content was determined, and samples equalized for protein content. 200 μg of protein was precipitated with acetone. The pellet was resuspended in 0.1% Triton-X100, 0.6% sulfosalicylic acid containing buffer, and freeze thawed twice. Protein-associated glutathione was released with sodium borohydride, and GSH determined. The sodium borohydride sensitive fraction of GSH was calculated, and expressed as nmol GSH/mg of protein. S-glutathionylation of IKKβ was assessed in cells lysed in buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 0.25% SDS, 1% NP-40, 0.5% CHAPS, and 20 mM N-ethylmaleimide with protease inhibitor cocktail (Sigma-Aldrich), via immunoprecipitation using and antibody directed against GSH (Virogen). As a reagent control, lysates were incubated in the presence of 1 mM DTT to decompose S-glutathionylated proteins prior to immunoprecipitation [9].
Chromatin Immunoprecipitation (ChIP)
ChIP assays were carried out as described elsewhere with minor modifications [22]. Briefly, at the appropriate times of harvest, formaldehyde (Sigma) was added to the culture medium to a final concentration of 1% and cells incubated for 10 min. Glycine was then added to a final concentration of 125 mM. Cells were washed with ice cold PBS and scraped into buffer containing 25 mM Hepes (pH 7.8), 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, and protease inhibitor (Sigma). A Dounce homogenizer was used to isolate nuclei, which were then resuspended in buffer containing 50mM Hepes pH (7.8), 140 mM NaCl, 1 mM EDTA, 1% SDS, and protease inhibitor (Sigma). Isolated nuclei were sonicated (VibraCell Sonicator) to produce chromatin fragments of 200–1000 nucleotides in length. Following sonication, isolated chromatin was diluted 1:10 and precleared with protein-G linked magnetic Dynabeads (Invitrogen). Immunoprecipitations were performed using 2 μg anti-RelA antibody (Millipore), 2 μg anti-RNA Polymerase 2 antibody (Millipore), 2 μg Acetylated histone H4 antibody (Millipore), or IgG isotype control antibody, at 4°C overnight with constant agitation. Protein-G linked magnetic Dynabeads (Invitrogen) were added and immunoprecipitates isolated using a DynaMag (Invitrogen) magnet rack and washed four times according to protocol. Isolated chromatin was then incubated with 20 μg of Proteinase K (Roche) for 2 h at 55°C, and then 65°C overnight to reverse the cross-links. Isolated DNA was then purified by QAIquick column (Qiagen) purification according to the manufacturer’s protocol, prior to PCR analysis. Primer sequences used for PCR were as follows, forward 5′-aacaggagtggcaaatattgaga-3′ and reverse 5′-ctttctggcaaccttctgatg-3′.
Glutaredoxin Promoter Analysis
The online promoter mining algorithm PROMO3.0 was used to analyze the 2kb sequence upstream of the Glrx1 locus derived from the NCBI MGI build 37 of the Mus musculus genome [23]. Forward primer, 5′-ctcgagtaggagagcttggctattccatgt and reverse primer 3′-agatctgctgacaggctgcagcttctccag, were designed using NCBI MGI build 37 to clone the genomic sequence 2kb upstream of the Glrx1 locus, introducing an Xho1 site 5′ and Bgl2 site 3′. The resultant amplicon was inserted into the pGL4.0 (Promega) vector to create Glrx1-luc. C10 cells were transfected with Glrx1-luc, β-galactosidase (β-gal), in the presence or absence of CA-IKKβ. 24 h post transfection, cells were lysed in luciferase lysis buffer (Promega), and luciferase (Promega) and β-gal (Applied Biosystems) activity measured according to the manufacturer’s instructions. Luciferase units were expressed as relative light units (RLU) after correction for β-gal. In select experiments, cells pRL-TK was employed to correct for differences in transfection efficiency, and cells were analyzed with the dual-luciferase reporter assay system (Promega) according to manufacturer’s protocol instructions.
Mice
Bi-transgenic mice that inducibly express constitutively active IKKβ (CA-IKKβ) under the control of the rat clara cell secretory protein, 10 kDa promoter were used. In these mice, expression of the CA-IKKβ transgene is induced in epithelial cells of the conducting airways, upon administration of doxycycline, as previously described [18]. For all experiments, two month old CA-IKKβ transgenic mice, or transgene negative littermates were maintained on doxycycline containing chow (6g/kg) (Purina Diet Tech) for 1 week prior to analyses. All studies were approved by the Institutional Animal Care and Use Committee at the University of Vermont.
mRNA analyses
Total RNA was isolated from C10 cells or lung tissue using the RNeasy mini-kit (Qiagen, Valencia, CA), subjected to reverse transcription and DNase treatment to produce cDNA for Taqman gene analysis using SYBR green (Biorad, Hercules, CA) or Assays on Demand for the individual target genes (Applied Biosystems, Foster City, CA). Results were normalized to house keeping genes, cyclophilin, or HPRT. Primer sequences are: Glutaredoxin-1: forward; TTT ACA ACA GCT CAC CGG AG, reverse; TCA CTG CAT CCG CCT ATG (accession number: NM 053108.4) Cyclophilin: forward; TTC CTC CTT TCA CAG AAT TAT TCC A, reverse; CCA GTG CCA TTA TGG (accession number: NM 008907.1), HPRT: forward; AGA ATG TCT TGA TTG TGG AAG A, reverse; ACC TTG ACC ATC TTT GGA TTA (accession number NM 13556.2)
Scratch assays
Scratch assays were performed as previously described with modifications [24]. On Day 1 C10 were transfected with pcDNA or CA-IKKβ expression plasmid, on Day 2 cells were transfected with Grx1 SiRNA or control SiRNA, on Day 3 medium was changed and cells were allowed to recover for 24 h. On Day 5 cells were scratched in a linear fashion using a p1000 micropippette tip, immediately following the scratch a photograph was taken of the cells using an Olympus IX70 inverted light microscope with QImaging Retiga 2000R digital camera. Twenty four h later, an identical photograph was taken of the same location to assess scratch closure. Scratch closure analysis was performed using ImageJ software (http://rsb.info.nih.gov/ij). All experiments were performed in triplicate, with additional samples prepared simultaneously for biochemical analysis of gene over expression and SiRNA knockdown.
Statistics
Analyses of all data were performed using the Graph Pad Prism software (Graphpad, Inc.) by one way ANOVA or Student’s t test where appropriate. Data from each experiment is presented plus/minus the standard error of the mean (SEM). All experiments were repeated twice. Analyses with resultant p values of < 0.05 were accepted as significant.
Results
Activation of the NF-κB pathway in lung epithelial cells in vitro and in vivo results in enhanced Grx1 expression
Previous reports have demonstrated that within the lung, Grx1 is highly expressed in airway epithelial cells, in addition to macrophages [14, 25], and in mice with allergic airway inflammation, NF-κB activation and increases in Grx1 are apparent in lung epithelium [14, 15]. In order to determine whether Glrx1 is regulated by NF-κB, we took advantage of a transgenic mouse model wherein NF-κB activation is selectively induced in lung epithelial cells, following doxycyclin-inducible expression of CA-IKKβ [18]. Results in Fig. 2A demonstrate that Grx1 content was increased in homogenized lung tissue of mice that express the CA-IKKβ transgene, while in the absence of doxycyclin, Grx1 expression was equal to controls (not shown). Increases in Grx1 content were accompanied by increases in Grx1 mRNA (Fig. 2B). Since CA-IKKβ-expressing mice display marked neutrophilic inflammation [18], it is difficult to ascertain whether increases in Grx1 expression are the direct result of NF-κB activation, or a consequence of the inflammatory process. In order to directly determine whether Grx1 expression in epithelial cells is regulated by NF-κB activation, we over expressed CA-IKKβ in a line of mouse type II alveolar epithelial cells (C10). In pcDNA vector-transfected cells, Grx1 content increased over time in culture (Fig. 2C). Following expression of CA-IKKβ, Grx1 content and mRNA levels were further increased (Fig. 2C and D). In CA-IKKβ expressing lung epithelial cells, the overall content of protein-S-glutathionylation was decreased (Fig. 2E), consistent with the observed increases in Grx1 content, and the physiological role of Grx1 in catalyzing protein-deglutathionylation. Lipopolysaccharide (LPS) is a well known activator of NF-κB, and administration of LPS to airways results in activation of NF-κB in lung epithelium, and consequently neutrophilic inflammation. Moreover, in response to administration of LPS, increases in Grx1 content were observed in the lung [26]. We therefore assessed whether LPS also increased Grx1 expression in C10 cells. Results in Fig. 2F demonstrate that LPS induced transient increases in Grx1 mRNA expression after 30 min and 2 h of exposure. At later time points, no increased in Grx1 mRNA were apparent. Collectively, these findings demonstrate that Grx1 expression can be induced in an NF-κB-dependent mechanism.
Fig. 2.

Increases of Grx1 expression following activation of the NF-κB pathway in lung epithelial cells. (A) Transgenic mice that express CA-IKKβ within the conducting airways, in a doxycycline inducible manner, or transgene negative littermate controls were maintained on doxycycline for 1 week. Mice were euthanized, and whole lung homogenates prepared for assessment of Grx1 expression by immunoblot analysis. IKKβ blot is shown to verify expression of trangenic IKKβ. β-actin is shown as a loading control. (B) Assessment of Grx1 mRNA content by real time PCR is lung tissues from mice expressing the CA-IKKβ transgene, compared to C57B/6 littermates. Results were normalized to the housekeeping gene, HPRT, and expressed as fold increases compared to transgene negative littermate controls that were fed doxycycline containing food. Data reflect mean +SEM of 4 mice/group. * p < 0.05 (ANOVA) compared to C57B/6 group. (C) Mouse alveolar type II cells (C10) were transfected with 1 μg or pcDNA3 or CA-IKKβ plasmids. After 24 and 48 h, whole cell lysates were evaluated for Grx1 expression by immunoblot analysis. β-actin: loading control. (D) Assessment of Grx1 mRNA expression via real time PCR in C10 cells transfected with 1 μg or pcDNA3 or CA-IKKβ plasmids. Results were normalized to the housekeeping gene, cyclophilin, and expressed as fold increases compared to pcDNA controls. * p<0.05 (Student T Test) compared to pcDNA3 controls. (E) Assessment of protein S-glutathionylation in C10 cells following expression of CA-IKKβ. 24 or 48 post transfection with PcDNA3 or CA-IKKβ, cells were lysed and proteins precipitated for assessment of PSSG. The sodium borohydride dependent release of GSH was measured. Results are normalized to cellular protein content. * p<0.05 (ANOVA) compared to pcDNA3 controls. (F) Assessment of Grx1 mRNA expression in C10 cells exposed to 1 μg/ml of LPS for the indicated times. Results were normalized to the housekeeping gene, cyclophilin, and expressed as fold increases compared to pcDNA controls. * p<0.05 (ANOVA) compared to pcDNA3 controls.
Stimulation of RAW264.7 cells with LPS induces expression of Grx1 in an NF-κB dependent manner
In addition to lung epithelial cells, Grx1 expression is also robust in alveolar macrophages [26]. We therefore next determined the impact of pro-inflammatory cytokines on Grx1 content in RAW264.7 macrophage like cells. RAW264.7 cells were stimulated for 24 h with LPS, IL-1β, or TNF-α, agonists known to induce NF-κB activation. Exposure to IL-1β or TNF-α, resulted in no or modest increases in Grx1 expression or activity. However, stimulation of cells with LPS resulted in a significant increase in Grx1 protein expression and activity (Fig. 3A–C).
Fig. 3.

Expression of Grx1 in RAW264.7 macrophage like cells following stimulation with known NF-κB agonists. (A) RAW264.7 cells were stimulated with IL-1β (5ng/mL), TNF-α (10 ng/mL), or LPS (1μg/mL) for 24 h. Whole cell lysates were resolved by SDS-PAGE and immunoblotted for Grx1, and β-actin. (B) Assessment of Grx1 activity RAW264.7 cells, 24 h post stimulation with agonists, as in A. Data are expressed as mean (±SEM) units. * p < 0.05 (ANOVA) compared to sham controls. (C) Dose dependent modulation of Grx1 content in RAW264.7 cells 24 h after stimulation with LPS. RAW264.7 cells were stimulated with the indicated concentrations of LPS, and after 24 h, whole cell lysates were prepared for Western Blot analysis. (D) RAW264.7 cells were transfected with vector control (pcDNA3.0), constitutively active IKKβ (CA-IKKβ), or dominant negative IκBα (dn-IκBα). 24 h later, cells were exposed to LPS (1 μg/ml) for an additional 24 h before immunoblot analysis for Grx1. Actin is shown as a loading control.
We next sought to determine whether NF-κB activity was directly involved in LPS-induced increases in Grx1 content, by over expression of a dominant negative version of IκBα in RAW264.7 cells. As is demonstrated in Fig. 3D, over expression of dominant negative IκBα markedly inhibited LPS-induced increases in Grx1 content. As was demonstrated earlier in C10 lung epithelial cells (Fig. 2C), overexpression of CA-IKKβ, also enhanced Grx1 expression in RAW264.7 cells in the presence or absence of LPS (Fig. 3D). Collectively these results demonstrate that LPS induces Grx1 expression in an NF-κB-dependent manner.
Identification of NF-κB binding sites within the promoter of the Glrx1 gene locus
In order to further examine the molecular mechanisms regulating Grx1 expression in RAW264.7 cells, we analyzed the 2 kb sequence of genomic DNA upstream of the Glrx1 start codon for potential NF-κB binding sites. This sequence of genomic DNA was previously described and determined to be a transcriptionally competent promoter sequence [27]. Using the PROMO3.0 promoter mining software revealed of two regions containing putative NF-κB1 (p50) binding sites at −1247–1256 kb and −1307–1316 kb upstream of the transcriptional start site (Fig. 4A). We next assessed the transcriptional activity of the 2 kb region of the Glrx1 promoter containing the putative NF-κB sites, and assessed the impact of CA-IKKβ, RelA/p50, or LPS on Glrx1 promoter activation. Transfection of C10 cells with Glrx1-luc resulted in enhanced luciferase expression over the PGL4 vector control (Fig. 4B). Co-transfection of cells with constitutively active IKKβ resulted in a concentration dependent increase in luciferase compared to vector controls (Fig. 4B). Similarly, transfection of RelA/p50 or exposure to LPS increased Glrx1-luciferase activity (Fig. 4C). In order to determine whether NF-κB could directly bind to the Glrx1 promoter, we next conducted chromatin immunoprecipitation analyses in RAW264.7 cells stimulated with LPS. Results in Fig. 4D demonstrate that binding of RelA to the Glrx1 promoter occurred between 4–24 h post stimulation of cells with LPS, which coincided with occupancy of the Glrx1 promoter with RNA polymerase II. In contrast, no RelA or RNA polymerase II was bound in unstimulated cells. Acetylated histone H4 was constitutively bound to the Glrx1 promoter, suggesting that this genomic site is competent with respect to transcription factor binding [28]. In aggregate, these findings demonstrate that the Glrx1 gene can be activated by canonical NF-κB signaling.
Fig. 4.

Assessment of activation of the glrx1 promoter by NF-κB. (A) Schematic depiction of the glrx1 promoter highlighting two putative NF-κB1 (p50) binding sites at −1250 base pairs (bp) and −1310 bp. Arrows indicate the primer sequences used for ChIP analysis. (B) C10 cells were transfected with vector encoding β-galactosidase, empty PGL4.0 vector, or PGL4.0 vector containing the 2000 bp sequence up stream of the glrx1 gene locus (Glrx1-luc), in the presence or absence of increasing amounts of PcDNA3, or CA-IKKβ. Cells were incubated 24 h prior to luciferase activity analysis. All data are expressed as mean (±SEM) relative light units (RLU) normalized to β-galactosidase activity. * p < 0.05 (ANOVA) compared to Glrx1-luc controls. (C) Cells were transfected with Glrx1-luc and renilla luciferase (pRL-TK), and where either co-transfected with 1 μg, pcDNA3, CA-IKKβ, or 0.5 μg RelA plus 0.5 μg p50. After 24 h, pcDNA3-transfected cells were stimulated with 1 μg/ml LPS. All cells were harvested 24 h later using the dual-luciferase reporter assay system (Promega) according to manufacturer’s instructions. Data are expressed as mean (±SEM) relative light units (RLU) normalized to Renilla activity. * p < 0.05 (ANOVA) compared to PcDNA3 controls. (D) Assessment of RelA binding to the Glrx1 promoter via ChIP analysis. RAW264.7 cells were stimulated with 1 μg/ml of LPS for the indicated times. Chromatin was crosslinked, sheared, and precipitated with antibodies recognizing RelA, RNA polymerase II (Pol II), or aceylated Histone H4. Pre-immune IgG antibody was used at a control. Immmunoprecipitated DNA was subjected to PCR analysis, using primer sequences indicated in Fig. 3A.
Expression of Grx1 promotes LPS-induced NF-κB signaling in lung epithelial cells
Activation of IKKβ is the pre-requisite signal in NF-κB activation by LPS, and previously we determined that H2O2- induced S-glutathionylation of IKKβ inhibits is activity [10]. We next determined whether exposure of lung epithelial cells to LPS leads to S-glutathionylation of IKKβ. Indeed, results in Fig. 5A demonstrate that stimulation of C10 cells with LPS results in increases of S-glutathionylation of IKKβ. Incubation of cell lysates with 1 mM dithiotreitol prior to immunoprecipitation with anti-glutathione antibody resulted in a complete loss of immunoprecipitation of IKKβ (data not shown). S-glutathionylation of IKKβ occurred at protracted time points relative to phosphorylation of RelA, degradation of IκBα, and increases in nuclear RelA content, which are all reflective of activation of IKKβ. We next assessed whether Grx1 overexpression would reverse increases in S-glutathionylation of IKKβ in response to LPS, and the impact for NF-κB activation. Consistent with its physiological role in de-glutathionylation, overexpression of Grx1 prevented LPS-induced increases in S-glutathionylation of IKKβ (Fig. 5, top panel). Assessment of IκBα content, which is degraded upon IKKβ-induced phosphorylation in response to LPS (Fig. 1A), demonstrated a second wave of IκBα degradation in cells overexpressing Grx1, in particular at the 4 and 6 h time points. Prolonged degradation of IκBα corresponded with increases in phoshorylation of RelA and nuclear content of RelA at those times, in Grx1 expressing cells, compared to pcDNA3 control cells exposed to LPS (Fig. 5). The content of LPS-induced NF-κB dependent pro-inflammatory cytokines, interleukin-6 (Il-6), keratinocyte-derived chemokine (KC), granulocyte monocyte-colony stimulating factor (GM-CSF), and Regulated on Activation Normal T Cell Expressed and Secreted (RANTES) in supernatants was markedly enhanced in Grx1 overexpressing cells, comparison to pcDNA3-transfected vector controls, in particular at the later time points (Table 1), demonstrating that increased expression of Grx1 enhances LPS-induced NF-κB dependent pro-inflammatory signaling.
Fig. 5.

Assessment of the impact of over expression of Grx1 on LPS-induced NF-κB activation and S-glutathionylation of IKKβ (IKKβ-SSG) in C10 lung epithelial cells. Top panel: S-glutathionylation of IKKβ. At the indicated times, S-glutathionylated proteins were immunoprecipitated (IP) with anti-GSH antibody, and subjected to Western Blotting to detect IKKβ. No immunoreactivity occurred in IgG control immunoprecipitations or following decomposition of protein-S-glutathionylation with DTT (data not shown). Whole cell lysates (WCL); Assessment of IKKβ content as a control in samples used for IP, and IκBα content, and phoshorylation of RelA at serine 536 (pRelA) as measures of NF-κB activation. Total RelA: loading control, Grx1: confirmation of Grx1 overexpression. Bottom panels: Assessment of nuclear content (Nucl) of RelA in response to LPS in cells transfected with PcDNA3 (left), or Grx1 (right). H3: histone H3 as a loading control.
Table 1.
Impact of overexpression of Grx1 on content of NF-κB dependent pro-inflammatory cytokines in C10 cells stimulated with LPS. C10 cells were transfected with 1 μg of Flag-Grx1 plasmid, or PcDNA3 control, and 24 h later stimulated with 10 μg/ml of LPS for 24, 48 or 72 h. Cytokine content in medium was assessed via ELISA assays, * p< 0.05, ANOVA, compared to PcDNA group at the same time.
| PcDNA3 | Flag GRX1 | |
|---|---|---|
| hr | RANTES
|
|
| - | 207 ± 28 | 286 ± 46 |
| 24 | 906 ± 97 | 1282 ± 114 |
| 48 | 1210 ± 206 | 2079 ± 482* |
| 72 | 1375 ± 242 | 1787 ± 289 |
| IL-6 | ||
|
|
||
| - | 16.1 ± 1.4 | 20.4 ± 3.9 |
| 24 | 19.5 ± 3.4 | 33.5 ± 3* |
| 48 | 19.0 ± 4.1 | 33.5 ± 3* |
| 72 | 16.5 ± 2.4 | 26.6 ± 4* |
| GM-CSF | ||
|
|
||
| - | 2.2 ± 0.1 | 2.3 ± 0.4 |
| 24 | 4.0 ± 1.0 | 3.4 ± 1.1 |
| 48 | 6.2 ± 0.9 | 10.9 ± 1.7* |
| 72 | 2.9 ± 0.9 | 9.9 ± 2.3* |
| KC | ||
|
|
||
| - | 386 ± 12 | 369.7 ± 98 |
| 24 | 927 ± 121 | 941.0 ±67 |
| 48 | 3179 ± 297 | 3261.2 ± 265 |
| 72 | 3471 ± 492 | 4644.4 ± 401* |
In addition to its role in inflammation, activation of NF-κB has been demonstrated to be important in wound healing [29]. We recently demonstrated that S-glutathionylation is prominent in cells at the leading edge of a wound [19]. We therefore conducted scratch assays to determine the impact of CA-IKKβ expression of wound closure in lung epithelial cells, and the role of Grx1 therein. Results of Fig. 6 demonstrate that CA-IKKβ expressing cells showed an enhanced ability to close the wound area. However, following siRNA-mediated knock down of Grx1, the enhanced ability of CA-IKKβ expressing cells to close the wound area was completely abolished. These results suggest that Grx1 induction following CA-IKKβ mediated activation of NF-κB is critical in promoting wound repair. In aggregate, these findings suggest that NF-κB-dependent induction of Grx1 represents a feed forward regulatory mechanism to promote NF-κB signaling, by decreasing levels of protein-S-glutathionylation, which inhibit the NF-κB pathway (Fig. 7).
Fig. 6.

Enhanced wound closure in CA-IKKβ expressing cells requires the presence of Grx1. C10 cells were transfected with control SiRNA, or Grx1 SiRNA, and 24 h thereafter transfected with PcDNA3 or CA-IKKβ. 24 h later, a scratch was made with a pipet tip, and 24 h thereafter, the % closure of the wound area quantified. Results are representative of 6 observations conducted in two separate experiments. * p < 0.05 (ANOVA) compared to the pcDNA group; ‡ p < 0.05 (ANOVA) compared to the control siRNA, CA-IKKβ-transfected group.
Fig. 7.
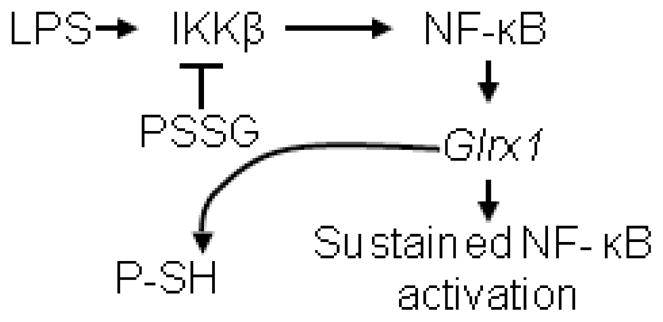
Model depicting the potential impact of Grx1 on prolonging activation of NF-κB. In response to stimulation with LPS, S-glutathionylation (PSSG) of IKKβ is important to shut down the activity of NF-κB. Activation of the Glrx1 gene via canonical NF-κB activation prevents the accumulation of IKKβ-SSG, thereby prolonging activation of the NF-κB pathway, and the production of pro-inflammatory mediators. Note that Grx1-catalyzed deglutathionylation results in the formation of protein sulfhydryl groups (P-SH). It is plausible that in addition to IKKβ, other members of the NF-κB pathway are regulated via S-glutathionylation and Grx1-catalyzed deglutationylation (not shown).
Discussion
NF-κB has been considered a prototypic redox-sensitive transcription factor that is induced following oxidative stress. While convincing studies exist which document activation of NF-κB following activation of NADPH oxidases or oxidative stress [30, 31], other studies have demonstrated that NF-κB is inhibited following oxidative insults [32]. The exact oxidative events that regulate the activity of NF-κB have remained elusive. Our laboratory recently demonstrated that canonical NF-κB signaling is inhibited via S-glutathionylation. Specifically we demonstrated that S-glutathionylation of cysteine 179 of the IKKβ following exposure to hydrogen peroxide (H2O2, 100–200 μM) resulted in the reversible inactivation of IKKβ [10]. Importantly, the thioltransferase Grx1 effectively reversed the H2O2-induced S-glutathionylation of IKKβ, and permitted activation of NF-κB in the presence of H2O2 [10]. Other members of the NF-κB pathway have also been identified as targets for S-glutathionylation, including p50 and RelA (p65), in association with impaired DNA binding and transcriptional activation [11, 12]. A consensus cysteine has been identified in rel homology domains of all members of the NF-κB family [33], suggesting that other members of the NF-κB family also may be susceptible to redox modification.
The present study expands upon previous observations in that we demonstrate that a physiological ligand of NF-κB, LPS, results in S-glutathionylation of IKKβ. S-glutathionylation of IKKβ occurred at relatively protracted times relative to IKKβ-mediated phosphorylation, suggesting that S-glutathionylation may be a negative feedback mechanism in order to decrease kinase activity. We were not able to accurately determine this using in vitro kinase assays due to the requirement of reducing agents in these assays which reverse S-glutathionylation, and additional studies are needed to determine the exact mechanism whereby S-glutathionylation inhibits the activity of IKKβ. Additional studies are also needed to elucidate the exact stoichiometry of S-glutathionylation of NF-κB family members in intact cells, with consideration of formation of IKK signalsomes, subcellular localization, and unique pools of NF-κB complexes in those settings. The link between S-glutathionylation of IKKβ and activation of NADPH oxidases also needs further study. Furthermore, the specificity of S-glutathionylation of IKKβ also will need to be unraveled, in light of the existence of many proteins with reactive cysteines that are potential targets for oxidation. Nonetheless, it is worthy of mention that glutathione S-transferase P was recently unraveled as a catalyst of S-glutathionylation reactions [34, 35], and could be a major determinant for which proteins constitute biologically relevant targets for S-glutathionylation, together with Grx enzymes.
In C10 cells, which transiently increased Grx1 mRNA expression in response to LPS, overexpression of Grx1 largely prevented the LPS-induced increases in S-glutathionylation of IKKβ, and prolonged degradation of IκBα, phoshorylation of RelA, nuclear localization of RelA, and led to further increases in expression of diverse NF-κB dependent pro-inflammatory cytokines. In addition to its role in inflammation, NF-κB also plays a role in wound healing [29]. Results from our present study indeed demonstrate enhanced wound closure in epithelial cells expressing active IKKβ, and that the ability of IKKβ to facilitate wound closure required the presence of Grx1, findings which suggest a role for Grx1 in wound healing. Using a technique of Grx1-based cysteine derivatization, we previously demonstrated that S-glutathionylation was preferentially apparent in cells at the leading edge of the wound [19], which potentially is due to activation of NADPH oxidases [36]. We did not unravel whether NF-κB subunits or IKKβ are S-glutathionylated during wound healing nor do we know the functional implications of such events. Alternatively, S-glutathionylation of actin has been shown to occur and interferes with its ability to polymerize [37]. Grx1-catalysed de-glutathionylation may be required to facilitate actin remodeling, and cell migration.
Given the functional significance of Grx1 in prolonging the activation of the NF-κB pathway, and its role in CA-IKKβ-induced wound closure we sought to further explore the molecular mechanisms by which Grx1 expression is regulated. The results of the present study demonstrate that Glrx1 expression is increased by activation of the canonical NF-κB pathway itself, through the direct interaction of the NF-κB subunit RelA (p65) with the glrx1 promoter. To date, little information exists regarding the transcriptional regulation of Glrx1. The human Glrx1 gene contains putative activator protein-1 (AP-1) sites in its promoter, which links expression of Glrx1 to signaling pathways that control Fos and Jun family members [27]. Indeed, the chicken Glrx1 gene was demonstrated to be a direct target of oncogenic Jun [38], and similarly, under conditions of oxidative stress, in lens epithelial cells the human Glrx1 gene was induced in an AP-1 dependent manner [39]. Results from the present study demonstrate that both in RAW 264.7 macrophages and C10 lung epithelial cells, Grx1 protein expression was increased following activation of NF-κB through expression of CA-IKKβ. The present data also clearly demonstrate that Glrx1 induction is agonist specific. Despite its well-known ability to activate NF-κB, TNF-α failed to increase Grx1 expression or activity in RAW 264.7 macrophages (Fig. 2A and B). These data suggest that besides canonical NF-κB pathway activation, other pathways may either enhance or dampen Glrx1 gene activation. Computational analysis of the Glrx1 promoter, revealed a putative PU.1 binding site adjacent to the NF-κB binding sites. PU.1 is an ETS family transcription factor associated with hematopoietic differentiation and maturation, which has been described to antagonize NF-κB signaling in macrophages [40]. Of relevance to our findings, silencing of PU.1 using short interfering RNA resulted in enhanced NF-κB signaling following stimulation of RAW264.7 cells with LPS, while conversely, overexpression of PU.1 dampened NF-κB-dependent signaling [40]. Additional studies are necessary to formally determine the repressive role of PU.1 in the activation of the Glrx1 gene, and to unravel the other transcription factors or signaling events that either enhance or dampen activation of the Glrx1 gene in response to different ligands.
Canonical NF-κB signaling is critical to the initiation of innate immune responses following exposure to bacterial toxins such as LPS. Our laboratory has demonstrated that over expression of a dominant negative version of IκBα specifically within the airway epithelium is sufficient to inhibit influx of neutrophils into the lung and block inflammatory cytokine production following exposure to LPS [17]. Furthermore, we and others have demonstrated that activation of canonical NF-κB signaling within the airway epithelium is sufficient to induce an inflammatory response in the lungs, which is associated with neutrophil influx and enhanced production of inflammatory cytokines [18]. Results from the present study suggest a direct link between Grx1 expression and a feed forward mechanism for the propagation of NF-κB signaling (Fig. 7). These results would suggest that under conditions wherein Grx1 expression is increased, inflammatory responses in the lung are potentiated, while conversely, in the absence of Grx1, NF-κB-dependent inflammatory responses would be attenuated. A recent study from our laboratory demonstrated that in Glrx1 deficient mice the ability of LPS to induce acute inflammation was identical to WT mice exposed to LPS. However, a clear trend toward more rapid resolution of LPS-induced inflammation was apparent in Glrx1−/− mice, which corresponded with time-dependent increases in protein-S-glutathionylation [26]. Studies examining patients with chronic obstructive pulmonary disease have correlated increases in expression of Grx1 in alveolar macrophage with disease progression and decreased lung function. In contrast, patients with sarcoidosis and allergic alveolitis show decreased expression of Grx1 in alveolar macrophages [25]. Based upon those observations, additional studies are needed to unravel the impact of Grx1 status in lung tissue on the extent and resolution of inflammatory responses, and to functionally link these associations with S-glutathionylation of NF-κB. Such endeavors will be important, given the documented roles of LPS, and Toll like receptor 4 signaling in the orchestration not only of acute inflammatory responses, and lung injury, but also in promoting allergic airways disease.
In summary, results from the present study demonstrate that activation of the Glrx1 gene by canonical NF-κB signaling represents a feed forward mechanism to prolong NF-κB activation (Fig. 7). These findings suggest that Grx1-based control of protein-S-glutathionylation represents a post-translational mechanism to control the timing of the NF-κB activation, and point to Grx1 as a possible target to combat diseases characterized by NF-κB-driven chronic inflammation.
Acknowledgments
This work was supported by grant T32 HL076122 and R01 HL060014 from the National Institutes of Health.
List of Abbreviations
- NF-κB
Nuclear factor kappa B
- Grx1
Glutaredoxin-1
- Glrx1
Glutaredoxin-1 gene
- IKKβ
Inhibitory kappa B Kinase beta
- CA-IKKβ
Constitutively Active Inhibitory kappa B Kinase beta
- LPS
Lipopolysaccharide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 3.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 5.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 7.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 8.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 9.Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS, Budd RC, Janssen-Heininger YM. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol. 2009;184:241–252. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 12.Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 13.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 14.Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol. 2007;36:147–151. doi: 10.1165/rcmb.2006-0259RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poynter ME, Irvin CG, Janssen-Heininger YM. Rapid activation of nuclear factor-kappaB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol. 2002;160:1325–1334. doi: 10.1016/s0002-9440(10)62559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shelton MD, Distler AM, Kern TS, Mieyal JJ. Glutaredoxin regulates autocrine and paracrine proinflammatory responses in retinal glial (muller) cells. J Biol Chem. 2009;284:4760–4766. doi: 10.1074/jbc.M805464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poynter ME, Irvin CG, Janssen-Heininger YM. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol. 2003;170:6257–6265. doi: 10.4049/jimmunol.170.12.6257. [DOI] [PubMed] [Google Scholar]
- 18.Pantano C, Ather JL, Alcorn JF, Poynter ME, Brown AL, Guala AS, Beuschel SL, Allen GB, Whittaker LA, Bevelander M, Irvin CG, Janssen-Heininger YM. Nuclear factor-kappaB activation in airway epithelium induces inflammation and hyperresponsiveness. Am J Respir Crit Care Med. 2008;177:959–969. doi: 10.1164/rccm.200707-1096OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760:380–387. doi: 10.1016/j.bbagen.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Janssen YM, Driscoll KE, Howard B, Quinlan TR, Treadwell M, Barchowsky A, Mossman BT. Asbestos causes translocation of p65 protein and increases NF-kappa B DNA binding activity in rat lung epithelial and pleural mesothelial cells. Am J Pathol. 1997;151:389–401. [PMC free article] [PubMed] [Google Scholar]
- 21.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 22.Carey MF, Peterson CL, Smale ST. Chromatin immunoprecipitation (ChIP) Cold Spring Harb Protoc. 2009;2009:pdb prot5279. doi: 10.1101/pdb.prot5279. [DOI] [PubMed] [Google Scholar]
- 23.Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18:333–334. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- 24.Bove PF, Hristova M, Wesley UV, Olson N, Lounsbury KM, van der Vliet A. Inflammatory levels of nitric oxide inhibit airway epithelial cell migration by inhibition of the kinase ERK1/2 and activation of hypoxia-inducible factor-1 alpha. J Biol Chem. 2008;283:17919–17928. doi: 10.1074/jbc.M709914200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peltoniemi M, Kaarteenaho-Wiik R, Saily M, Sormunen R, Paakko P, Holmgren A, Soini Y, Kinnula VL. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum Pathol. 2004;35:1000–1007. doi: 10.1016/j.humpath.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, Ho YS, Janssen-Heininger YM. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am J Respir Cell Mol Biol. 2011;44:491–499. doi: 10.1165/rcmb.2009-0136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JB, Levine M. The human glutaredoxin gene: determination of its organization, transcription start point, and promoter analysis. Gene. 1997;197:189–193. doi: 10.1016/s0378-1119(97)00262-x. [DOI] [PubMed] [Google Scholar]
- 28.Vettese-Dadey M, Grant PA, Hebbes TR, Crane-Robinson C, Allis CD, Workman JL. Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- 29.Egan LJ, de Lecea A, Lehrman ED, Myhre GM, Eckmann L, Kagnoff MF. Nuclear factor-kappa B activation promotes restitution of wounded intestinal epithelial monolayers. Am J Physiol Cell Physiol. 2003;285:C1028–1035. doi: 10.1152/ajpcell.00167.2003. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. J Biol Chem. 2007;282:30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 34.Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxidants & redox signaling. 2011;15:233–270. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. The Journal of biological chemistry. 2009;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wesley UV, Bove PF, Hristova M, McCarthy S, van der Vliet A. Airway epithelial cell migration and wound repair by ATP-mediated activation of dual oxidase 1. J Biol Chem. 2007;282:3213–3220. doi: 10.1074/jbc.M606533200. [DOI] [PubMed] [Google Scholar]
- 37.Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- 38.Goller ME, Iacovoni JS, Vogt PK, Kruse U. Glutaredoxin is a direct target of oncogenic jun. Oncogene. 1998;16:2945–2948. doi: 10.1038/sj.onc.1201819. [DOI] [PubMed] [Google Scholar]
- 39.Krysan K, Lou MF. Regulation of human thioltransferase (hTTase) gene by AP-1 transcription factor under oxidative stress. Invest Ophthalmol Vis Sci. 2002;43:1876–1883. [PubMed] [Google Scholar]
- 40.Zeng H, Ornatowska M, Joo MS, Sadikot RT. TREM-1 expression in macrophages is regulated at transcriptional level by NF-kappaB and PU.1. Eur J Immunol. 2007;37:2300–2308. doi: 10.1002/eji.200737270. [DOI] [PubMed] [Google Scholar]