

Abstract
Allyl glycidyl ether, polymerized from potassium alkoxide/naphthalenide initiators under both neat and solution conditions was shown to be a highly-controlled process. In both cases, molar masses (10–100 kg/mol) were determined by the reaction stoichiometry, and low polydispersity indices (1.05–1.33) could be obtained with a full understanding of the dominant side reaction, isomerization of the allyl side chain, being developed. The degree of isomerization of allyl to cis-prop-1-enyl ether groups (0 – 10 % mol.) was not correlated to the molar mass or polydispersity of the polymer but was dictated by the polymerization temperature. This allows the extent of isomerization to be reduced to essentially zero under either melt or solution conditions at polymerization temperatures of less than 40 °C.
Keywords: polyethers, ring-opening polymerization, anionic polymerization
Polyethers, such as poly(ethylene glycol) (PEG), are widely used materials in commercially established fields such as drug-delivery,1 and control of biocompatibility,2 and are becoming increasingly important in emerging technologies such as dye-sensitized solar cells,3 and lithium-polymer batteries.4 A fundamental challenge with all PEG-based systems is the lack of functional handles along the polymer backbone which limits the modification and tunability of this valuable materials platform.5 As a functional alternative to PEG in some applications, low-Tg poly(allyl glycidyl ether) (PAGE) has been examined due to its inherent chemical flexibility stemming from the pendant allyl groups. For example, the allyl-ethers along the PAGE backbone are amenable to thiol-ene radical coupling chemistry which enables the elaboration of PAGE with a wide variety of functionalities without the need for tedious protection-deprotection chemistries.6 Such modular and facile reactivity increases the relevance of PAGE for applications in therapeutics, bioconjugation, and polymer-supported catalysis.7,8 This combination of latent chemical functionality, and inherent physical properties (low Tg, lack of crystallinity) makes a compelling case to systematically explore the synthesis and reactivity of this potentially useful and inexpensive polyether materials platform.
Examples of the polymerization of AGE,9,10 and the selective functionalization of its pendant allyl groups by thiol-ene radical coupling can be found in the literature.11,12,13 Erberich et al. carried out an extensive analysis of the polymerization chemistry of AGE and the functionalization, protection, and allyl-deprotection to linear polyglycidol.10 However, the authors concluded that the polymerization of allyl glycidyl ether (AGE) was only controlled to 80% conversion with termination by abstraction of an allylic proton competing with propagation. Hrubý et al. also investigated the application of poly(ethylene oxide)–b–poly(allyl glycidyl ether) (PEO-PAGE) as a micellar drug delivery vehicle with doxorubicin units being randomly attached, via pH-sensitive hydrazone linkages, along the PAGE block.11 Similarly, Persson and Jannasch synthesized a poly(allyl glycidyl ether)–b–poly(ethylene oxide)–b–poly(allyl glycidyl ether) (PAGE-PEO-PAGE) triblock copolymer and graft copolymers of PAGE on a poly(p-hydroxy styrene) backbone. In these cases, thiol-ene coupling of benzimidazole units to the PAGE blocks creates robust proton-exchange membrane materials for hydrogen fuel-cell applications.12 The challenge with these studies is that the polymerization of AGE under the reported conditions results in significant termination and other side reactions with impure block copolymers being obtained that required removal of PEO and PAGE homopolymer contaminants. Alternatively, Hu et al. have polymerized AGE to low molar masses (2–4 kg/mol) using a sodium ethoxide initiator and xylene as the polymerization solvent.14 The molar masses agreed well with the reaction stoichiometry, however the resultant polydispersity indices were low (1.04–1.08) only at low molecular weights. Obermeier and Frey investigated the copolymerization of ethylene oxide and AGE.13 However, their analysis was limited to low molecular weights of less than 10 kg/mol and utilized cesium alkoxide initiators that were generated by the deprotonation of an alcohol with cesium hydroxide, followed by removal of water. The molar masses they reported were consistently 10% above that defined by the reaction stoichiometry.
The significant promise shown by PAGE-based materials, coupled with unresolved issues concerning the polymerization of AGE, prompted a thorough reinvestigation of the homopolymerization of AGE. A driving force for this focus on homopolymerization is the ease of handling AGE compared to ethylene oxide with its associated low boiling point and high toxicity which would increase the complexity of the polymerization. The use of alternate initiating systems under a wide variety of bulk and solution conditions resulted in optimized polymerization conditions and the development of a fundamental understanding for controlling side reactions in this useful materials platform.
Results and Discussion
In examining the methods to synthesize poly(allyl glycidyl ether), many of the approaches have relied on the utilization of a strong, non-nucleophilic base and removal of the conjugate acid (e.g., water, or tert-butanol) to generate an alkoxide initiator followed by the addition of allyl glycidyl ether (AGE) and polymerization at high temperatures (ca. 100 °C).9–12 A challenge with these strategies is that protic impurities are introduced during generation of the initiating system and these impurities may be difficult to quantitatively remove, leading to subsequent interference with the polymerization. Moreover, the use of high polymerization temperatures can be detrimental to the polymerization leading to unwanted side-reactions.
To overcome these issues, the radical-anion potassium naphthalenide was used to generate a potassium alkoxide initiator which was employed for the polymerization of allyl glycidyl ether (AGE).15,16 Significantly, the byproducts of deprotonation of the benzyl alcohol initiator with potassium naphthalenide are naphthalene and dihydronaphthalene13 which are innocuous to the polymerization, and the introduction and removal of residual water or alcohol characteristic of other initiation strategies such as sodium or cesium alkoxides is unnecessary.9–12 Scheme 1 shows the generation of potassium alkoxide initiator and subsequent polymerization of AGE.
Scheme 1.
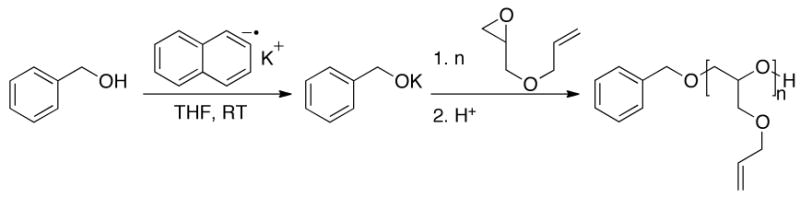
Polymerization of allyl glycidyl ether from potassium benzoxide.
In developing a fundamental understanding of the effect of polymerization conditions on the resulting PAGE polymers, polymerizations were conducted over the temperature range, 30–80 °C, both neat and in solution. All polymerizations were carried out for 20 hours, which was sufficient time for quantitative conversion of AGE except for the highest molar masses (> 50 kg/mol) for which 144 hours was used to reach completion. It should be noted that the polymerization of AGE was controlled over an order of magnitude in molar mass (10–100 kg/mol) and under a variety of polymerization conditions. Figure 1 details the observed number-averaged molar mass (Mnobs.) plotted versus the molar mass as defined by the reaction stoichiometry (Mnstoich.) with number-averaged molar masses being determined by 1H NMR spectroscopy using end-group analysis based on the benzylic protons (2H) on the initiator and either allyl (1H) or backbone ether protons (5H) to determine the number of repeat units. In solution and melt polymerizations carried out between 30–80 °C, the resultant molar masses compared well with the monomer to initiator feed ratios as indicated by the close agreement between Mnobs. and Mnstoich.. Polydispersity indices (PDIs) were measured by size exclusion chromatography (SEC) relative to polystyrene standards and were typically between 1.05 and 1.20 with higher polydispersities only being observed for high molecular weight materials (Figure 1). These increases in PDI are due to chain coupling occurring after complete consumption of AGE or radical coupling of backbone allyl substituent, rather than any transfer or termination reactions competing with propagation at high conversion as noted by Erberich et al.10
Figure 1.
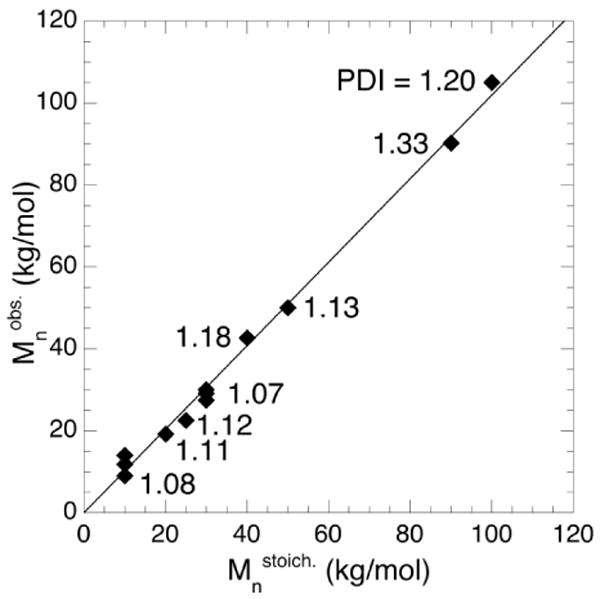
Number-average molar mass (Mobs.) as measured by 1H NMR spectroscopy versus the molar mass defined by the reaction stoichiometry (Mstoich.). The line plotted alongside the data has a slope of one and an intercept of zero. Polydispersity indices as measured by size-exclusion chromatography are shown adjacent to each data-point. In the case of several closely-space data points, the average polydispersity is shown. Detailed polymerization results are shown in Table 1.
Hans et al. reported a chain-transfer reaction in ethoxy ethyl glycidyl ether (EEGE) polymerizations beginning with the abstraction of a methylene proton adjacent to the epoxide ring by an active alkoxide chain-end. Subsequently, the epoxide ring opened introducing a new alkoxide which reinitiated polymerization.17 This chain-transfer to monomer generated PEEGE materials with allylic-ether end-groups exhibiting characteristic resonances in the 1H NMR spectra, and molecular weight distributions with long, low-molecular weight tails in the size-exclusion chromatograms. No chain-transfer to monomer was detected in polymerizations of AGE either by 1H NMR spectroscopy or size-exclusion chromatography (Figure 2).
Figure 2.
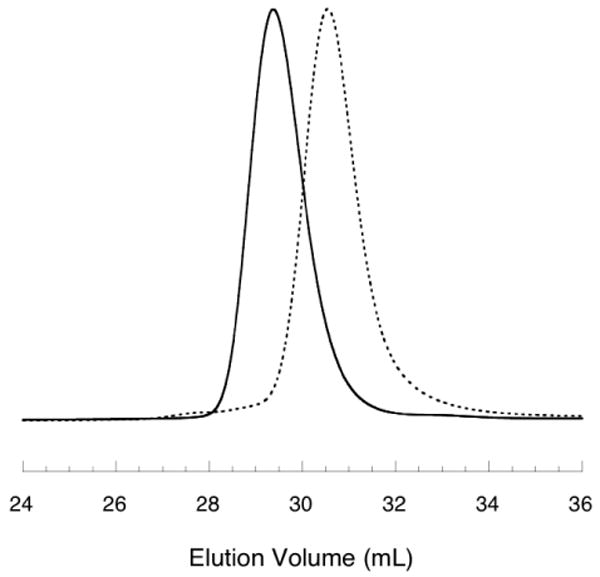
Size-exclusion chromatographs of polymers 2 (dashed line, Mn = 11.8 kg/mol, PDI = 1.08) and 6 (solid line Mn = 27.5 kg/mol, PDI = 1.07) as detailed in Table 1.
The controlled nature of this synthetic approach to PAGE then allowed a detailed examination of the relationship between polymer structure and polymerization conditions. A representative 1H NMR spectrum for AGE polymerized at 30 °C (Table 1, entry 5) is shown in Figure 3 (top) along with peak assignments. At lower polymerization temperatures, the repeat-unit structure is derived from the AGE monomer feedstock without isomerization as can be seen by the defined resonances corresponding to the allyl substituent in Figure 3, inset (a). However, at higher polymerization temperatures the repeat-unit structure can no longer be assigned to a single isomer. PAGE polymerized above 40 °C both neat and in solution exhibits 1H and 13C NMR spectra consistent with isomerization of the allyl repeat unit (Figure 3, bottom and Figure 4, respectively). Significantly, peak assignments for the three possible isomers (allyl; cis-prop-1-enyl, and trans-prop-1-enyl) clearly reveal the presence of the cis-isomer and the absence of the trans-isomer.18 Sunder et al. and Obermeier and Frey reported on the presence of prop-1-enyl isomers but identified the isomer as trans, but as shown above only the cis-isomer is present.9,13 Finally, Erberich et al. reported the termination of AGE polymerizations as possibly occurring through the abstraction of the allylic proton, but did not report the presence or formation of the 1-propenyl isomer.10
Table 1.
Polymerization results carried out under a variety of polymerization conditions.
| Sample | Mnobs,a | Mnstoich,b | PDIc | %isomerd | solvente | TPolym.( °C) |
|---|---|---|---|---|---|---|
| 1 | 9.0 | 10.0 | 1.09 | 0.0 | neat | 30 |
| 2 | 11.8 | 10.0 | 1.08 | 8.8 | diglyme | 80 |
| 3 | 14.0 | 10.0 | 1.08 | 0.6 | diglyme | 40 |
| 4 | 19.2 | 20.0 | 1.11 | 1.3 | neat | 60 |
| 5 | 22.5 | 25.0 | 1.12 | 0.0 | neat | 30 |
| 6 | 27.5 | 30.0 | 1.07 | 6.3 | neat | 80 |
| 7 | 29.1 | 30.0 | 1.10 | 0.0 | diglyme | 40 |
| 8 | 30.0 | 30.0 | 1.05 | 2.8 | neat | 80 |
| 9 | 42.7 | 40.0 | 1.18 | 0.2 | neat | 40 |
| 10 | 50.0 | 50.0 | 1.13 | 5.0 | diglyme | 80 |
| 11 | 90.2e | 90.0 | 1.33 | 0.0 | neat | 30 |
| 12 | 105 | 100 | 1.20 | 4.0 | diglyme | 80 |
Mnobs. measured by NMR spectroscopy.
Mnstoich was defined by the monomer to initiator ratio.
Polydispersity indices were determined by SEC in chloroform relative to polystyrene standards.
The percent cis-prop-1-enyl isomer incorporation was determined by 1H NMR spectroscopy.
Solution polymerizations were carried out at 20wt% monomer in diglyme.
Polymerization time of 144 hours.
Figure 3.
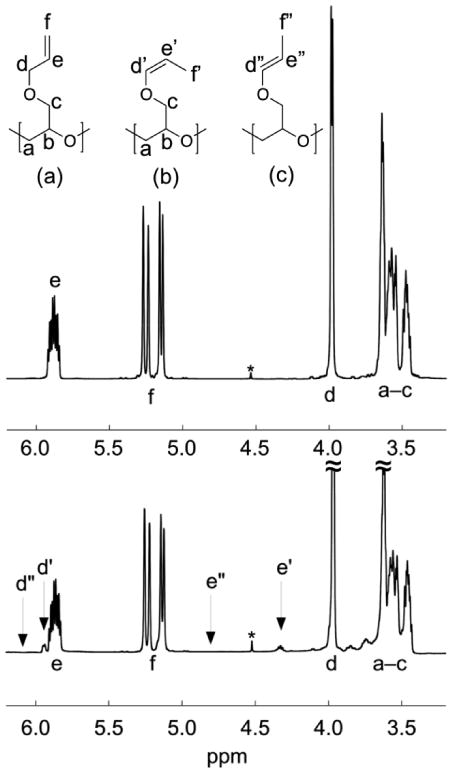
(Top spectrum) 1H NMR spectrum of poly(allyl glycidyl ether) polymerized neat at 30 °C (Table 1, entry 5). Peak assignments are shown in the inset. The resonance near 4.5 ppm marked with an asterisk is due to the benzyl (2H) end-group protons used for determination of molar mass. (Bottom spectrum) 1H NMR spectrum of PAGE polymerized neat at 120 °C (see Table 2) with inset peak assignments showing the presence of extra resonances due to the presence of cis-propenyl isomers. Three possible isomers exist: (a) allyl, (b) cis-prop-1-enyl, (c) trans-prop-1-enyl. Resonances are observed for the monomer-derived allyl, and cis-prop-1-enyl isomers only. No evidence of trans isomerization is present.
Figure 4.
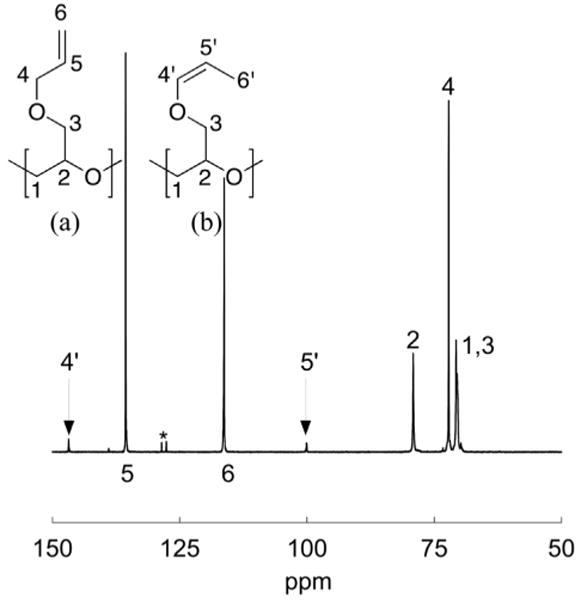
13C NMR spectrum of PAGE polymerized neat at 120 °C (see Table 2). Repeat unit isomers are shown with assignments in (a) and (b). Resonances due to carbon atoms in the benzyl end-group are indicated with an asterisk.
Based on the work of Prosser, the isomerization of allyl ethers along the backbone may occur according to Scheme 2.19 The living potassium alkoxide chain-end abstracts an allylic proton from an AGE repeat unit and the alkyl potassium formed from this proton abstraction reaction coordinates with the potassium counter-ion forming a five-membered ring, stabilizing the cis-isomer. The alkyl potassium then deprotonates a dormant alcohol creating a potassium alkoxide chain-end and propagation of AGE continues resulting in a cis-propenyl repeat-unit isomer. As a result, this process does not constitute a termination reaction with isomerization being fast relative to propagation since no evidence of premature termination or chain-transfer to monomer is seen in the SEC traces (see Figure 2). In addition, the amount of cis-isomer is not correlated with the molar mass or polydispersity, instead there is a strong correlation with reaction temperature (see Table 1 and Figure 1). No significant isomerization occurs below a polymerization temperature of 40 °C and the mole fraction of isomerized cis-prop-1-enyl gradually increases as the reaction temperature increases. Interestingly, at the same temperature, the nature of the reaction conditions did not significantly affect the extent of isomerization with similar values being obtained for polymerizations in solution, and under melt conditions. Finally, there was no evidence from either 1H or 13C NMR spectroscopy that the deprotonated allyl-ether side-chains react nucleophilically with AGE monomer leading to branching which leads to the conclusion that the deprotonated allyl side-chains are transient and non-nucleophillic species.
Scheme 2.
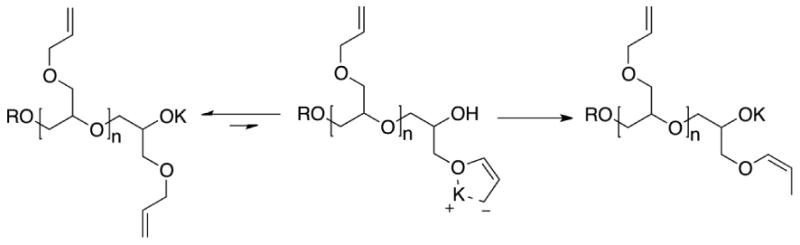
Proposed isomerization of allyl-substituents to cis-prop-1-enyl units.
Having noted that the extent of cis-prop-1-enyl isomer formation was related to reaction temperature, several neat polymerizations were carried out at fixed polymerization times (20h) between 40–140 °C in 20 °C increments with the degree of isomerization being characterized by 1H NMR spectroscopy (Figure 5). For neat polymerizations carried out at lower temperatures (40 °C), very low levels of isomerization are observed (1.5 % mol.) which increases on progressing to 80 °C (3.7 % mol.) followed by a significant increase in the level of isomerization for neat polymerizations carried out above 100 °C (8.3–16.6 % mol.). The mole percent of cis-prop-1-enyl isomers that resulted from neat polymerizations carried out at 40–140 °C are shown in Table 2.
Figure 5.
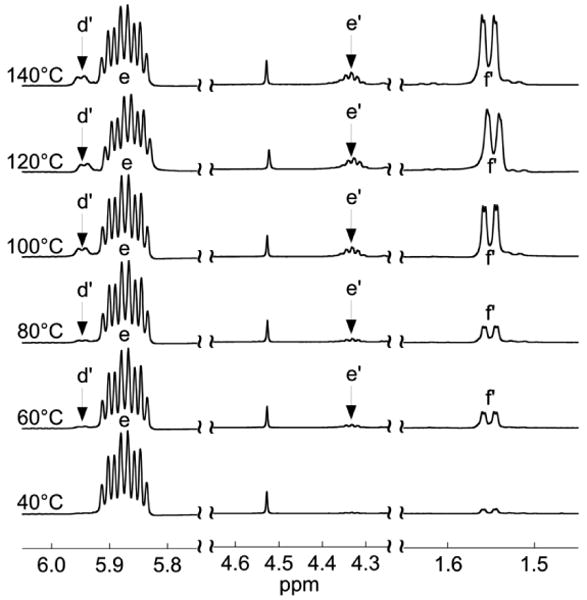
1H NMR spectra resulting from polymerizations carried out at various temperatures (40–140 °C). All spectra are normalized to the intensity of the benzyl resonance near 4.5 ppm (2H) and peak-assignments are shown in Figure 3. The mole percent incorporation of cis-prop-1-enyl isomers increases with polymerization temperature for polymerizations of equivalent duration as evidenced by the increase in intensity of the peaks due to the d' (1H), e' (1H), and f' (3H) protons.
Table 2.
Isomerization of allyl groups during in neat polymerization of AGE at 40–140 °C.
| Tpolym.a | Mnb | Mnc | PDId | %isomere |
|---|---|---|---|---|
| 40 | 17.2 | 17.4 | 1.09 | 1.5 |
| 60 | 18.7 | 19.2 | 1.11 | 3.9 |
| 80 | 16.8 | 17.2 | 1.11 | 3.7 |
| 100 | 14.0 | 14.4 | 1.14 | 8.3 |
| 120 | 15.7 | 17.5 | 1.19 | 16.3 |
| 140 | 14.8 | 16.4 | 1.20 | 16.6 |
Polymerization temperature in °C.
Molar mass in kg/mol, determined by the allyl protons using 1H NMR spectroscopy.
Molar mass in kg/mol determined by the backbone-ether protons using 1H NMR spectroscopy.
Determined by RI-SEC in chloroform relative to polystyrene standards.
Mole percent of cis-prop-1-enyl ether units determined by 1H NMR spectroscopy.
Heatley et al. investigated the isomerization of allyl ethers to propenyl ethers that occurs during the oxyanionic polymerization of propylene oxide as a side-reaction.20 Through a detailed analysis by 1H NMR spectroscopy, the authors came to the conclusion that the activation energy for the allyl to cis-prop-1-enyl isomerization was 116 kJ/mol. For a bimolecular reaction between a living chain-end and an allyl group, the rate of isomerization is given by –d[Allyl]/dt = k [Allyl] [ROK], with the temperature dependent rate constant given by:
| (1) |
Using the integrated rate law, and assuming the density of PAGE is 1.0 g/mL, for a fixed reaction time of 20 hours, the incorporation of isomers would be expected to increase precipitously above 100 °C based on the rate constant determined by Heatley et al. Although the concentration of allyl groups Heatley et al. used in their analysis was significantly lower than that for PAGE, their kinetics for allyl to cis-prop-1-enyl isomerization agree qualitatively with the temperature dependence of allyl to cis-prop-1-enyl isomerization occurring in AGE during neat polymerization.
Obermeier and Frey briefly addressed the issue of allyl-ether isomerization in copolymerizations of AGE and ethylene oxide.13 A very broad range of isomerization values (0-10 mol%) was reported for polymerizations carried out even at 40 °C. This is in contrast to our results where a polymerization temperature of 100 °C resulted in 8 mol% cis-prop-1-enyl isomers, and 30 °C results in undetectable levels of isomerization along the PAGE backbone. It is difficult to speculate on the origins of this difference. However, the potassium naphthalenide system that we employ for the polymerization of AGE yields reproducibly undetectable levels of isomerization for melt polymerization carried out below 40 °C up to molar masses as high as 90 kg/mol explored in this study which suggests another advantage for this polymerization system when compared to other alkoxide (cesium) systems.
A detailed understanding of the isomerization of AGE during polymerization at elevated temperatures opens the opportunity to exploit the reactivity of the resulting cis-prop-1-enyl groups which are hydrolytically unstable and can give rise to hydroxyl groups. Indeed, examination of the 1H and 13C NMR spectra of PAGE materials containing cis-prop-1-enyl isomers revealed an apparent loss in the number of prop-1-enyl groups relative to backbone ether protons on exposure to hydrolytic conditions. This discrepancy was correlated to the degree of isomerization of each sample and in most cases surpassed the amount of remaining cis-prop-1-enyl isomer. Vutukuri et al. and Ishizaki et al. reported that allyl ethers can be deprotected to alcohols via catalytically generating in situ the labile cis- and trans-prop-1-enyl isomers in a protic solvent (e.g., methanol) under mildly basic conditions.21,22 These conditions are similar to those present during termination of the polymerization reaction with protic solvents.
The flexibility of PAGE as a molecular platform is enhanced by this controlled formation of cis-prop-1-enyl groups and can be specifically exploited.19 For example, reaction of PAGE containing approximately 13% (mol.) cis-1-propenyl isomer in methanol over a polymer-supported sulfonic acid resin (DOWEX) results in quantitative cleavage of the cis-1-propenyl groups to hydroxy functionalities. This affords a linear random copolymer of glycidol and AGE and provides a mechanism for orthogonal modification of the PAGE backbone via the hydroxy- and allyl-functionalities. Significantly, the amount of cis-1-propenyl isomerization may be tuned by the reaction conditions; lower temperatures generally result in lower levels of isomerization (see Tables 1, 2, and Figure 5) for the same polymerization time.
Experimental
Materials
All chemicals were used as received from Sigma-Aldrich unless otherwise specified. THF was collected from a dry solvent system and used immediately thereafter. Benzyl alcohol was dried over calcium hydride and distilled prior to titration with potassium naphthalenide in THF. Allyl glycidyl ether (TCI-America) was degassed through several freeze-pump-thaw cycles and distilled from butyl magnesium chloride to a pre-weighed and flame-dried buret immediately prior to use. Potassium naphthalenide was prepared from potassium metal and recrystallized naphthalene in dry THF and allowed to stir with a glass-coated stir-bar for 24h at room temperature before use.
Characterization
1H NMR spectroscopy was carried out on a Bruker AC 500 spectrometer in deuterated chloroform. 13C NMR spectroscopy was carried out on neat PAGE containing a sealed D2O capillary containing for locking and shimming the spectrometer. Size exclusion chromatography (SEC) was performed on a Waters chromatograph with four Viscotek colums (two I-MBHMW-3078, I-series mixed bed high molecular weight columns and two I-MBLMW-3078, I-series mixed bed low molecular weight columns) for fractionation, a Waters 2414 differential refractometer and a 2996 photodiode array detector for detection of eluent, and chloroform with 0.1% tetraethylamine at room temperature was used as the mobile phase. Gas chromatography was carried out on a Shimadzu GC-2014 using a flame ionization detector and a Restek column (SHRXI-5MS) for separation.
Polymerizations and modifications
All polymerizations were carried out on a Schlenk line in custom thick-walled glass reactors fitted with threaded ACE-threads under an argon atmosphere. The reactors were dried under vacuum then refilled with argon five times. Under an argon atmosphere, benzyl alcohol initiator was added by gas-tight syringe through a 6mm puresep septum. Potassium alkoxide initiators were formed by titration of benzyl alcohol with potassium naphthalenide under argon until a green color persisted in solution indicating the deprotonation of all alcohols. Two polymerization procedures were followed: A. Bulk polymerizations were carried out between 30–140 °C for 20h and terminated with methanol. B. Solution polymerizations were carried out in diglyme between 30-140 °C for 20 hours and terminated with methanol. Polymerizations carried out at higher temperatures (> 100 °C) were carried out on small scales (ca. 1 g). Deprotection of the cis-prop-1-enyl ether isomers was carried out in methanol using five equivalents of DOWEX resin by mass. Complete conversion of the cis-prop-1-enyl ethers to hydroxyls was observed by complete disappearance of all 1H NMR signals consistent with the cis-prop-1-enyl ethers. For the synthesis of PAGE with undetectable levels of isomerization, controlled molar masses, and low polydispersity indices, polymerization in the melt at 30 °C was carried out for 20–144h depending on the target molecular weight.1H NMR of PAGE (500 MHz, CDCl3): δ 1.55 (d, –O–CH=CH–CH3), 3.47–3.72 (broad m, –O–CH2–CH(CH2–O–CH2-CH=CH2)–O– and –CH2–CH(CH2–O–CH=CH-CH3)–O–), 3.79/3.87 (two broad peaks, –CH2–CH(CH2–O–CH=CH-CH3)–O–), 4.01 (d, –O–CH2–CH=CH2), 4.38 (m, –O–CH=CH-CH3), 4.56 (s, Ph-CH2–O–), 5.18/5.28 (doublet of doublets, –O–CH2–CH=CH2), 5.91 (m, –O–CH2–CH=CH2), 5.97 (d, –O–CH=CH–CH3), 7.30 (overlap with residual CHCl3, 1H on Ph–CH2–O–), 7.36 (s, 4H on Ph–CH2–O–). 13C NMR of PAGE (500 MHz, neat PAGE, D2O capillary for shimming): δ 70.9 (–O–CH2–CH(CH2–O–CH2-CH=CH2)–O–), 72.0 (–O–CH2-CH=CH2), 79.4 (–O–CH2–CH(CH2–O–CH2-CH=CH2)–O–), 100.3 (–O–CH=CH-CH3), 116.3 (–O–CH2–CH(CH2–O–CH2-CH=CH2)–O–), 127.5/128.6 (5C, Ph–CH2–O–), 135.6 (–O–CH2–CH(CH2–O–CH2-CH=CH2)–O–), 138.8 (1C, Ph–CH2–O–), 146.8 (–O–CH2–CH(CH2–O–CH=CH–CH3)–O–).
Conclusion
The polymerization of allyl glycidyl ether using potassium alkoxide initiators has been shown to result in low polydispersity, controlled molecular weight materials under both solution and melt conditions. Depending on the specific polymerization temperature, either no isomerization of the allyl groups was observed (30 °C) or the fraction of allyl groups along the backbone isomerized into labile cis-prop-1-enyl groups was found to increase with increasing reaction temperature. Significantly, hydrolysis of the isomerized cis-prop-1-enyl groups to hydroxyl groups could be achieved by a post-polymerization treatment giving orthogonally reactive hydroxy and allyl groups along the backbone. The properties and inherent chemical functionality of the resultant PAGE material makes it amenable to application as a polymeric platform with potential applications in a wide range of technological areas.
Acknowledgments
This project has been funded in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN268201000046C. This work was also supported by the Los Alamos National Laboratory Institute for Multiscale Materials Studies, and the National Science Foundation (MRSEC Program DMR-05204156 (MRL-UCSB)). Materials Research Laboratory Central Facilities are supported by the MRSEC Program of the NSF under Award No. DMR11-21053; a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org).
References
- 1.Knop K, Hoogenboom R, Fischer D, Schubert US. Angew Chem Int Ed. 2010;9:2–23. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 2.Kainthan RK, Janzen J, Levin E, Devine DV, Brooks DE. Biomacromolecules. 2006;7:703–709. doi: 10.1021/bm0504882. [DOI] [PubMed] [Google Scholar]
- 3.De Freitas JN, Nogueira AF, De Paoli MA. J Mater Chem. 2009;19:5279–5294. [Google Scholar]
- 4.a) Armand M. Adv Mater. 1990;2:278–286. [Google Scholar]; b) Meyer WH. Adv Mater. 1998;10:439–448. doi: 10.1002/(SICI)1521-4095(199804)10:6<439::AID-ADMA439>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; Wright PV. Electrochimica Acta. 1998;43:1137–1143. [Google Scholar]; c) Zhuang X, Xiao C, Oyaizu K, Chikushi N, Chen X, Nishide H. J Polym Sci Part A: Polym Chem. 2010;48:5404–5410. [Google Scholar]
- 5.a) Kojima C, Yoshimura K, Harada A, Sakanishi Y, Kono K. J Polym Sci Part A: Polym Chem. 2010;48:4047–4054. [Google Scholar]; b) Du W, Li Y, Nyström AM, Cheng C, Wooley KL. J Polym Sci Part A: Polym Chem. 2010;48:3487–3496. [Google Scholar]; c) Ren Y, Jiang X, Liu R, Yin J. J Polym Sci Part A: Polym Chem. 2009;47:6353–6361. [Google Scholar]; d) Rahm M, Westlund R, Eldsäter C, Malmström E. J Polym Sci Part A: Polym Chem. 2009;47:6191–6200. [Google Scholar]; e) Keul H, Möller M. J Polym Sci Part A: Polym Chem. 2009;47:3209–3231. [Google Scholar]; f) Saville PM, Reynolds PA, White JW, Hawker CJ, Frechet JMJ, Wooley KL, Penfold J, Webster JRP. J Phys Chem. 1995;99:8283–8289. [Google Scholar]
- 6.a) Kade MJ, Burke DJ, Hawker CJ. J Polym Sci Part A: Polym Chem. 2010;48:743–750. [Google Scholar]; b) Nilsson C, Malmström E, Johansson M, Trey SM. J Polym Sci Part A Polym Chem. 2009;47:5815–5826. [Google Scholar]; c) Rosen BM, Lligadas G, Hahn C, Percec V. J Polym Sci Part A Polym Chem. 2009;47:3931–3939. [Google Scholar]; d) Yu B, Chan JW, Hoyle CE, Lowe ABJ. J Polym Sci Part A Polym Chem. 2009;47:3544–3557. [Google Scholar]
- 7.Haag R, Kratz F. Angw Chem Int Ed. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 8.Madhavan N, Jones CW, Weck M. Acc Chem Res. 2008;41:1153–1165. doi: 10.1021/ar800081y. [DOI] [PubMed] [Google Scholar]
- 9.Sunder A, Türk H, Haag R, Frey H. Macromolecules. 2000;33:7682–7692. [Google Scholar]
- 10.Erberich M, Keul H, Möller M. Macromolecules. 2007;40:3070–3079. [Google Scholar]
- 11.Hrubý M, Koňák Č, Ulbrich K. J Appl Polym Sci. 2005;95:201–211. [Google Scholar]; Hrubý M, Koňák Č, Ulbrich K. Controlled Release. 2005;103:137–148. doi: 10.1016/j.jconrel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 12.Persson JC, Jannasch P. Solid State Ionics. 2006;177:653–658. [Google Scholar]; Persson JC, Jannasch P. Chem Mater. 2006;18:3096–3102. [Google Scholar]
- 13.Obermeier B, Frey H. Bioconjugate Chem. 2011;22:436–444. doi: 10.1021/bc1004747. [DOI] [PubMed] [Google Scholar]
- 14.Hu Z, Fan X, Zhang G. Carbohydrate Polymers. 2010;79:119–124. [Google Scholar]
- 15.Garst JF. Acc Chem Res. 1971;4:400–406. [Google Scholar]
- 16.Giguére G, Zhu XX. Biomacromolecules. 2010;11:201–206. doi: 10.1021/bm9010694. [DOI] [PubMed] [Google Scholar]
- 17.Hans M, Keul H, Moeller M. Polymer. 2009;50:1103–1108. [Google Scholar]
- 18.Crivello JV, Kim WG. J Polym Sci Part A: Polym Chem. 1994;32:1639–1648. [Google Scholar]
- 19.Prosser TJ. J Am Chem Soc. 1971;4:400–406. [Google Scholar]
- 20.Heatley F, Booth C, Blease T. J Polym Sci Part A: Polym Chem. 1994;32:1131–1135. [Google Scholar]
- 21.Ishizaki M, Yamada M, Watanabe S, Hoshino O, Nishitani K, Hayashida M, Tanaka A, Hara H. Tetrahedron. 2004;60:7973–7981. [Google Scholar]
- 22.Wuts PGM, Greene TW. Greene's Protecting Groups in Organic Synthesis. 4th. John Wiley & Sons; Hoboken, NJ: 2006. [Google Scholar]