

Abstract
The angular vestibulo-ocular reflex (aVOR) has a fast pathway, which mediates compensatory eye movements, and a slow (velocity storage) pathway, which determines its low frequency characteristics and orients eye velocity toward gravity. We have proposed that motion sickness is generated through velocity storage, when its orientation vector, which lies close to the gravitational vertical, is misaligned with eye velocity during head motion. The duration of the misalignment, determined by the dominant time constant of velocity storage, causes the buildup of motion sickness. To test this hypothesis, we studied bilateral labyrinthine-defective subjects with short vestibular time constants but normal aVOR gains for their motion sickness susceptibility. Time constants and gains were taken from rotational responses. Motion sickness was generated by rolling the head while rotating, and susceptibility was assessed by the number of head movements made before reaching intolerable levels of nausea. More head movements signified lower motion sickness susceptibility. Labyrinthine-defective subjects made more head movements on their first exposure to roll while rotating than normals (39.8 ± 7.2 vs 13.7 ± 5.5; P < 0.0001). Normals were tested eight times, which habituated their time constants and reduced their motion sickness susceptibility. Combining data from all subjects, there was a strong inverse relationship between time constants and number of head movements (r = 0.94), but none between motion sickness susceptibility and aVOR gains. This provides further evidence that motion sickness is generated through velocity storage, not the direct pathway, and suggests that motion sickness susceptibility can be reduced by reducing the aVOR time constant.
Keywords: Nystagmus, Vestibular, Velocity storage, GABA
Introduction
Motion sickness, characterized by sweating, dizziness, disorientation, hyperventilation, nausea, and vomiting, are autonomic reactions (Kucharczyk et al. 1991; Yates 1992; Yates and Miller 1998; Balaban 1999) that occur in association with movement of the subject or the visual surround (Guedry and Benson 1978; Cheung et al. 1991). Similar symptoms also occur on entry and return from space (Graybiel et al. 1975; Money 1981; Matsnev et al. 1983; Igarashi and Kobayashi 1985; Thornton and Uri 1991; Putcha et al. 1999), or with linear oscillation (Golding and Kerguelen 1992). Less severe forms of motion sickness occur with static reversal of the visual surround (Yardley et al. 1992) or from eye muscle surgery for strabismus (Mathew et al. 2004). Rolling the head while rotating is particularly nauseogenic, and has been widely used to study motion sickness susceptibility (Graybiel and Wood 1969; Guedry and Benson 1978; Lackner and Graybiel 1986). The stimulus alternately brings the vertical and horizontal canals into and out of the plane of rotation, causing subjects to feel tumbling or pitching, disorientation, and nausea, which can finally result in vomiting. The number of head movements that subjects make before overwhelming nausea has been widely used as an operational definition of motion sickness susceptibility (Lackner and Graybiel 1994; Clément et al. 2001; Young et al. 2001; Dai et al. 2003). A motion sickness score can also be calculated to characterize the level of motion sickness (Miller and Graybiel 1969; Hecht et al. 2001; Young et al. 2001).
Motion sickness most probably arises in the vestibular system, since people with a loss of vestibular function do not become motion sick (Money 1972; Yates and Miller 1998; Johnson et al. 1999) and patients with partial labyrinthine lesions also have reduced motion sickness susceptibility (Johnson et al. 1999). However, the exact vestibular mechanisms that are involved in the production of motion sickness are still unknown. It had long been thought that motion sickness is caused by a sensory conflict (Reason 1975), but the specific nature and quantification of the conflict that caused the motion sickness is obscure. Bles (1988) made the sensory conflict theory more specific: “all situations which provoke motion sickness are characterized by a condition in which the sensed vertical, as determined on the basis of integrated information from the eyes, the vestibular system, and nonvestibular proprioceptors, is at variance with the subjective vertical as predicted on the basis of previous experience”. This was supported by his experimental results with Bos et al. (2002), in which subjects were rotated around a vertical or a horizontal axis. Only horizontal axis rotation evoked motion sickness. For the first time, this idea related motion sickness to the spatial vertical.
Recently, we habituated the vestibular time constant of normal subjects with repeated exposures to roll while rotating and demonstrated that velocity storage, which encodes both the spatial vertical and the temporal aspects of vestibular responses, is the critical central vestibular mechanism involved in the production of motion sickness (Dai et al. 2003). However, although activity, which produces motion sickness arises in the vestibular system, motion sickness is finally mediated through the autonomic system. Thus, it is possible that joint habituation of the autonomic system could also have contributed to the results.
We addressed the question of the relative contributions of time constant of velocity storage and extent of habituation of the autonomic system to motion sickness reduction by studying subjects with bilateral labyrinthine loss who had short vestibular time constants but normal aVOR gains and had no prior habituation experience of the VOR. Although relatively uncommon, such bilateral vestibular loss can occur after a wide range of vestibular diseases that include aminoglycoside ototoxicity, infection, bilateral acoustic neuroma, bilateral sarcoid, and bilateral damage of the vestibular end organs of unknown etiology (Baloh et al. 1989; Brandt 1999). A prominent feature of bilateral labyrinthine damage is its insidious onset that does not involve significant vertigo or nausea. Rather the presenting complaints are usually imbalance and/or oscillopsia (Baloh et al. 1989; Brandt 1999), which would not habituate the autonomic system. A common feature of these lesions is a reduction in the high frequency aVOR gain and a reduction in the vestibular time constant (Wade et al. 1999). The reduction in the aVOR gain tested at frequencies of 2–6 Hz, can recover to normal range over about 1 year, but the time constant and low frequency aVOR gain may never recover (Fetter and Zee 1988; Black et al. 2001; Palla and Straumann 2004; Newlands et al. 2005).
In this study, therefore, we assumed that habituation of autonomic responses would not be present in bilateral labyrinthine defective subjects when they were first exposed to roll while rotating, and postulated that there would be a similar relationship between motion sickness susceptibility and the velocity storage time constant, as in our previous study (Dai et al. 2003). If the aVOR gains of these bilateral labyrinthine-defective subjects had no relation to motion sickness susceptibility, it would further strengthen our previous conclusion that it was the temporal response of the velocity storage, not the high frequency component of the aVOR that was related to motion sickness.
Methods
Subjects
Ten subjects with bilateral labyrinthine lesions were selected for this study. Six subjects (4 males, 2 females, ages 30–62; S1–S6; Table 1) had normal aVOR gains but short aVOR time constant. Three subjects (S7–S9) with absent vestibular function, and one subject with a low aVOR gain but a normal time constant (S10) were also studied. Consistent with the findings of Baloh and Brandt (Baloh et al. 1989; Brandt 1999), the major complaints of these subjects were imbalance in dimly lit conditions and visual–vestibular disorientation when in motion. All of the subjects had normal oculomotor function, and no neurological dysfunction except for the imbalance. The diagnoses in these subjects are shown in Table 1. Before being tested, the test procedure was explained in detail, after which, subjects gave informed consent. This explanation included the stresses, “apparent” disorientation, dizziness and nausea that they might encounter during the test. The study conformed to the guidelines set by the Institutional Review Board of the Mount Sinai School of Medicine and was IRB-approved.
Table 1.
Age, sex and etiology of disease in labyrinthine defective subjects
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | S10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age | 32 | 40 | 62 | 60 | 30 | 49 | 32 | 61 | 51 | 49 |
| Sex | M | F | F | M | M | M | M | F | F | F |
| Etiology | Gentamycin ototoxicity | Cisplatin chemo-therapy | Bilateral acoustic neuromas | Idiopathic vestibulo-pathy | Idiopathic vestibulo-pathy | Idiopathic vestibulo-pathy | Idiopathic vestibulo-pathy | Idiopathic vestibulo-pathy | Bilateral meniere’s disease | Idiopathic vestibulo-pathy |
Test protocol
Horizontal and vertical positions of the right eye were recorded by video-oculography (ISCAN) at 60 frames/s with an accuracy of ≈0.2° over ±30°. The eye position data were then digitally differentiated and filtered to obtain slow phase eye velocity, which was the major variable utilized in this study. An inclinometer (Seika) registered the position of the head in roll relative to gravity. The resolution of the sensor was 0.01° over ±80° of tilt with a response time constant of 0.3 s. Rotations of the chair and data acquisition were under computer control. Eye position calibrations were done in darkness while subjects watched a laser that displayed targets at visual angles of ±20° horizontally and 15° upward from the mid-position. In the recordings, eye positions and slow phase eye velocities to the left and down are positive.
Pre-testing was done with steps of constant velocity rotation at 138°/s in darkness (acc. 200/s2), with standard warm (48°C) and cold (20°C) air caloric examinations followed by visual suppression of vestibular nystagmus, with sinusoidal ocular pursuit at 0.1 Hz (peak velocity, 15.7°/s) and 0.25 Hz (peak velocity, 40°/s), and with optokinetic stimulation at 20 and 40°/s. Subjects were also tested for gaze nystagmus and for spontaneous and positional nystagmus in darkness. This identified the bilateral vestibular deficit subjects, and ensured that these subjects had no central vestibular disease and no oculomotor dysfunction. The subjects were trained to make appropriate roll head movements while they were stationary until they felt comfortable in executing the head movements. They were then seated in a standard vertical-axis rotation chair (Neurokinetics Inc. Pittsburgh) with a three-point seat belt that held their trunks firmly. A surveillance camera was used to monitor the subjects and chair operation throughout the experiment. The operator was in constant communication with the subjects through an intercom during the entire run. Practically, the communication served as an alerting stimulus.
The roll while rotating test paradigm has been described in detail previously (Dai et al. 2003). Briefly, subjects were rotated in darkness while upright from 0 to 138°/s (Fig. 1a), generating horizontal per-rotatory nystagmus. When the per-rotatory nystagmus had declined to approximately zero, the chair was stopped, producing post-rotatory nystagmus. The per- and post-rotatory nystagmus were utilized to establish baseline horizontal aVOR time constants and gains. The chair was then rotated in the opposite direction (Fig. 1b). After the nystagmus had again disappeared, subjects were instructed to tilt their head 45° to the right (Fig. 1c) and to hold it there until the nystagmus had disappeared. This usually occurred in 10–40 s. The motion sickness score was checked, and then subjects were instructed to move their head back to the upright (Fig. 1d), and so forth.
Fig. 1.

Head positions during pretesting (a) and during testing with roll while rotating (b–f). The top row shows the head position of the subject, the circular arrows shows the direction of rotation. The third trace is chair velocity, which begins with the subject stationary, i.e., at 0°/s. The chair is accelerated to 138°/s, and then stopped. It is then accelerated in the opposite direction to −138°/s with the subject upright (b). The subject then successively tilts his/her head to the right (c), to upright (d), to the left (e), and back to upright (f) until either reaching 50 head movements or intolerable nausea. Each head position from c to f is held until the nystagmus generated by the head movement has disappeared. g Simplified model of angular vestibulo-ocular reflex. SCC: semicircular canals. See text for details
Because the adaptation and habituation acquired in one head position relative to gravity does not necessarily transfer to head positions or movements in other planes (Guedry et al. 1964; Torte et al. 1997; Yakushin et al. 2000), we used a full cycle of four head tilts (Fig 1c–f) to avoid asymmetrical habituation. The head movements were analyzed by a sigmoidal function to ensure that each head movement was made appropriately (Dai et al. 2003). The time and angle of the head position in roll were displayed concurrently and the operator informed the subjects if the head movements were not executed correctly. The head was subsequently returned to the upright, tilted to the left and returned to the upright again, etc. This was continued until the subjects asked to stop because of severe motion sickness or because they had had made 50 head movements, the maximum allowed in the experimental design. The rotatory chair then was stopped with the head in an upright position, producing post-rotatory horizontal nystagmus.
The vestibular responses to the rotational velocity of 138°/s and the responses to roll while rotating were compared to those of the 16 normal subjects from our previous study that underwent the same testing (Dai et al. 2003). The tests in the 16 normal subjects have been described in detail (Dai et al. 2003). Briefly, the first set of tests was done in 1 week on four consecutive days, and after a 1 month hiatus, the subjects were retested again on four consecutive days. In contrast, the labyrinthine-defective subjects had no previous experience with this test and were tested with this paradigm only once to avoid possible habituation of the autonomic system.
Scoring of motion sickness
The level of motion sickness was reported verbally 10–30 s after a head movement, and was scaled with a simplified Pensacola scale from 0 to 20 (Hecht et al. 2001; Young et al. 2001). Zero was no reaction, 10 was a moderate gastro-intestinal reaction with dizziness, and 20 was a sense of being about to vomit or of becoming too dizzy to continue. Dizziness and nausea were produced immediately in normal subjects by roll while rotating. The symptoms generally subsided to a stable level within 5–30 s, and at this level another head movement was made and the process was repeated. The test lasted from 10 min to about 1.5 h, depending on the number of head movements that could be made and how long took subjects to recover to a stable motion sickness level after executing a head movement.
Analysis of data
Eye position data were digitally differentiated to yield eye velocity. Quick phase eye movements (saccades) were removed, leaving slow phase eye velocity for analysis (Fig. 2; gray trace). Horizontal aVOR gains and time constants were determined from the slow phase eye velocities of per- and post-rotatory nystagmus induced by rotation when subjects were upright before any roll head movements were made. The gain of the horizontal aVOR was the ratio of peak eye velocity relative to the stimulus velocity in yaw (138°/s). Because the labyrinthine-defective subjects had relatively shorter time constants that were close to the time constant of the semicircular canals (SSC) (cupula/endolymph time constant), we used a single exponential to obtain the time constant of the overall aVOR.
Fig. 2.
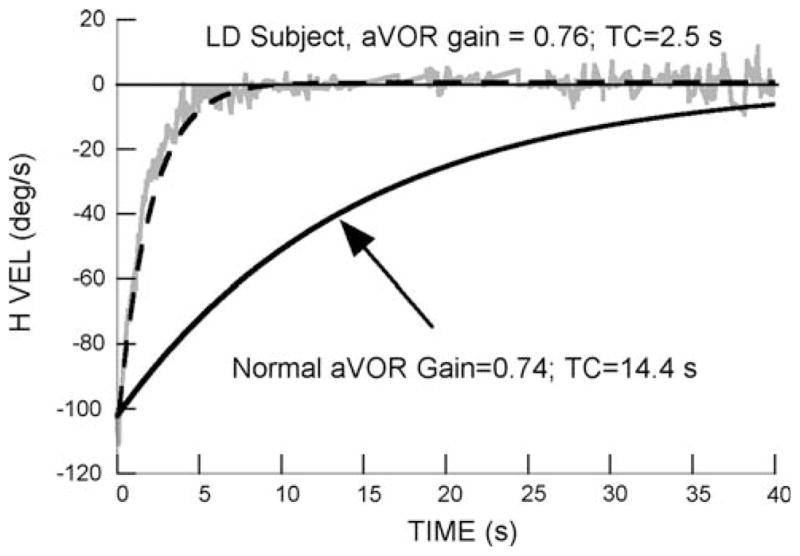
Comparison of the per-rotatory response of a labyrinthine-defective subject (S1) with the average responses to rotation of 16 normal subjects (Dai et al. 2003). All subjects were rotated at 138°/s. The desaccaded slow phase eye velocity (H VEL) of the labyrinthine-defective subject is shown by the gray trace, and is fit by a first order exponential function (dashed line). The averaged normal aVOR responses are shown by the black line (upward arrow)
Statistical analysis
The number of head movements made during roll while rotating was used as the major measure of motion sickness susceptibility. If more head movements were made before being too sick to continue, it signaled a higher resistance to motion sickness. The mean number of head movements, the aVOR gains and the time constants of the labyrinthine-defective subjects were compared with the corresponding means from each of the eight tests of the normal subjects using an unpaired student t-test, assuming unequal variance. Linear regressions were used to correlate the number of head movements with the aVOR time constants.
Theoretical basis for study of motion sickness
Insight into neural processing for motion sickness in the vestibular system has come from a model of the aVOR, which simulates visual and vestibular responses related to rotation of the head or the visual surround (Raphan et al. 1979; Raphan and Cohen 2002). A simplified version of this model is shown in Fig. 1g. The aVOR is organized with a direct and an indirect (velocity storage) pathway. The direct pathway (g1), which essentially represents Lorente de Nó’s three neuron arc (Lorente de Nó 1933), receives input from the Semicircular canals (SCC) (Fernández and Goldberg 1971; Büttner and Waespe 1981; Correia et al. 1992), to produce the high frequency response of the aVOR. In humans, the gain of the direct pathway response (head velocity/initial eye velocity) measured in darkness is about 0.6 (Dai et al. 1999). The indirect pathway, which has much slower response characteristics, has been simulated by an imperfect (leaky) integrator with a characteristic rise and fall time. Because it receives velocity input from the vestibular and visual systems and discharges this stored activity over its own time course, it has been called, a “velocity storage” integrator (Cohen et al. 1977; Raphan et al. 1979). The time constant of velocity storage, which can be estimated from this model (Dai et al. 1999), is normally about 16 s in naïve human subjects. A critical characteristic of the velocity storage integrator is its orienting properties, which tend to align the spatial response vector of eye velocity during rotation with the velocity storage orientation vector, which is close to the gravitational vertical (Dai et al. 1991; Raphan and Sturm 1991; Raphan et al. 1992; Raphan and Cohen 2002). Based on data obtained from experiments utilizing roll while rotating, we postulated that motion sickness susceptibility is dependent on the indirect velocity storage pathway, i.e., on the aVOR time constant and is not reliant on the direct pathway, i.e., on the gain of the aVOR (Cohen et al. 2003; Dai et al. 2003).
Results
All ten labyrinthine-defective subjects had normal oculomotor function, including saccadic amplitudes, latencies and velocities. Their optokinetic and pursuit gains were normal; none had spontaneous, positional or gaze nystagmus. Four of the six subjects (S1–S4) with short time constants (2.8–5 s) had absent caloric responses bilaterally. The other two (S5, S6) had longer time constants (6.5 and 7 s) and small but symmetrical caloric responses (≈5°/s). The three subjects (S7–S9) with absent vestibular function had no response to either steps of rotational velocity or to caloric stimuli. The single subject (S10) with a low aVOR gain (0.22) and a normal time constant (12 s) also had symmetrical caloric responses with a peak velocity of 15°/s for warm and 25°/s for cold caloric stimuli.
A typical per-rotatory vestibular response of S1 to a rotational velocity of 138°/s is shown in Fig. 2, and is compared to the averaged per- and post-responses of the 16 normal subjects from our previous study (Fig. 2, black line). The labyrinths of this LD subject had been damaged by gentamycin administration. He had a normal aVOR gain (0.76), but a short aVOR time constant (2.5 s). He made 50 head movements before experiencing mild motion sickness (Score 12). He did not have sweating after completing the test, as was commonly present in the normal subjects, and he recovered quickly.
The grouped results are shown in Fig. 3 and Table 2. The labyrinthine-defective subjects made significantly more head movements during their first exposure to roll while rotating than the normals (39.8 ± 7.2 vs 13.7 ± 5.5; P < 0.0001; Fig. 3a, filled circles vs filled square, first). The labyrinthine-defective subjects also had significantly shorter time constants than the normal subjects at the time of their first exposure (4.9 ± 1.6s vs 14.4 ± 3.8s; P < 0.0001). A striking finding of the previous study was that the 16 normal subjects were capable of making more head movements on each successive test day; concurrently their aVOR time constants became progressively shorter (Dai et al. 2003). The average reduction in aVOR time constant from the first to the eighth day of testing in the normal subjects is shown by the black squares and upward diagonal arrow in Fig. 3a. We correlated the number of head movements made by the labyrinthine-defective subjects and the normal subjects as a function of time constant from all tests (Fig. 3a). There was a statistically significant difference between the number of head movements made by the labyrinthine defective subjects and by the normal subjects on all subsequent tests except the eighth (Table 2). When all of the subjects’ responses were combined, there was a linear relationship between the number of head movements and the time constants (Fig. 3a, heavy black line; r = 0.94). Thus, the number of head movements that subjects could make in both studies was tightly coupled to their aVOR time constants.
Fig. 3.

a Number of head movements made during roll while rotating as a function of aVOR time constant in labyrinthine-defective subjects (filled circles) and by normal subjects in eight tests (filled squares). The diagonal upward arrow shows the direction of change in the time constants of the normal subjects from the first to the eighth test. b aVOR gains of labyrinthine-defective (circles) and normal subjects (squares). The upward arrow shows the gains from the first to the eighth tests of the normal subjects
Table 2.
Average aVOR gains, time constants (TC) and number of head movements in the six labyrinthine-defective subjects and the 16 normal subjects
| 6 LDs | Normal 1 | Normal 2 | Normal 3 | Normal 4 | Normal 5 | Normal 6 | Normal 7 | Normal 8 | |
|---|---|---|---|---|---|---|---|---|---|
| Gain | 0.65 ± 0.13 | 0.74 ± 0.14 | 0.74 ± 0.11 | 0.75 ± 0.08 | 0.75 ± 0.08 | 0.74 ± 0.11 | 0.74 ± 0.08 | 0.74 ± 0.10 | 0.73 ± 0.10 |
| P > 0.05 | P > 0.05 | P > 0.05 | P > 0.05 | P > 0.05 | P > 0.05 | P > 0.05 | P > 0.05 | ||
| TC(s) | 4.9 ± 1.6 | 14.4 ± 3.8 | 14.2 ± 4.4 | 12.0 ± 3.4 | 11.0 ± 3.2 | 13.1 ± 3.6 | 12.2 ± 3.1 | 11.9 ± 3.3 | 10.2 ± 2.2 |
| P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 | P < 0.0001 | ||
| No. Head Movts | 39.8 ± 7.2 | 13.7 ± 5.5 | 17.7 ± 7.1 | 23.5 ± 10.9 | 25.1 ± 11.4 | 20.9 ± 9.6 | 26.5 ± 10.1 | 31.2 ± 10.4 | 35.4 ± 11.2 |
| P < 0.0001 | P = 0.0001 | P = 0.001 | P = 0.003 | P = 0.003 | P = 0.004 | P = 0.04 | P = 0.23 |
The normals were tested eight times; the test number is given next to the title in each of the “normal” columns
There were no changes in the aVOR gains of the normal subjects from the first to the eighth test, and the gains were uncorrelated with the number of head movements made by the normal subjects during roll while rotating (Fig. 3b, upward arrow). Similarly, there was no correlation between the aVOR gains of the labyrinthine-defective subjects with the number of head movements they made during roll while rotating. Furthermore, there was no difference in aVOR gain between labyrinthine defective and normal subjects (Table 2). Thus, only the vestibular time constants, and not the aVOR gains were associated with the number of head movements that subjects could make during RWR, both in normal and in labyrinthine-defective subjects.
Three additional labyrinthine-defective subjects who had absent vestibular function (S7–S9) were tested with roll while rotating. They were completely immune to the stimulus, and did not experience turning, tumbling or the dizziness that was common in the normal or other labyrinthine-defective subjects. They were able to make 50 head movements during their first exposure to roll while rotating without motion sickness symptoms, and could have made more if they were not confined by the test limits. One additional labyrinthine-defective subject (S10) had a lower aVOR gain (0.22) but a relatively normal vestibular time constant (12 s). This subject could only make six head movements on her first exposure to roll while rotating before experiencing overwhelming nausea. This was even less than the average of 13.7 head movements made by the 16 normal subjects on their first exposure to roll while rotating. Thus, a low aVOR gain alone had no effect in reducing motion sickness susceptibility in this subject.
Discussion
This study has provided further evidence that motion sickness is intimately related to velocity storage in the vestibular nuclei. It also demonstrates that there is no relation between the high frequency component of the aVOR that produces compensatory eye movement against head movement and motion sickness susceptibility. These conclusions were supported by the findings from three other bilateral labyrinthine-defective subjects (S7–S10). Those with no vestibular response or very short vestibular time constants (S1, S7–S9) were able to make 50 head movements on their first exposure to roll while rotating, whereas the subject with a reduced aVOR gain but a normal time constant (S10) became motion sick after only six head movements. The normal subjects in our previous study were tested on eight separate occasions (Dai et al. 2003). There was a progressive increase in the number of head movements they could make as the aVOR time constant decreased, and the two sets of data could be combined into a single linear relationship characterizing aVOR time constants and motion sickness susceptibility. This supports the hypothesis that motion sickness is generated though activation of velocity storage.
In our previous study of motion sickness susceptibility (Dai et al. 2003), repetitive head movements that reduced the aVOR time constant evoked nausea and activated the autonomic system were the driving stimulus that produced the reduction in the vestibular time constant. The habituation of the time constant was most probably mediated through the nodulus and uvula (Cohen et al. 1992) since electrical stimulation of the nodulus shortens the vestibular time constant (Solomon and Cohen 1994; Sheliga et al. 1999), and nodulus/uvula lesions cause a loss of habituation of the time constant (Singleton 1967; Cohen et al. 1992). Furthermore, there is no motion sickness after the nodulus and uvula have been ablated (Bard 1945; Wang and Chinn 1956), showing that these portions of the vestibulo-cerebellum participate in producing the motion sickness state.
High frequency aVOR contributions to motion sickness susceptibility
The conclusion that activation of the high frequency aVOR does not contribute to motion sickness is at variance with earlier studies, in which motion sickness susceptible subjects were reported to have higher than normal aVOR gains in response to angular accelerations of 90°/s2 (Bos et al. 2002) and to rotation in a low frequency range (<0.08 Hz) (Gordon et al. 1996). At these accelerations and frequencies both the fast (direct) and indirect (velocity storage) pathways would be activated and would contribute to the induced eye velocity and the measured “aVOR gain”. When subjects were rotated with a higher acceleration (200°/s2) in this and a previous study (Dai et al. 2003), there was no relation between the high frequency aVOR gain and motion sickness susceptibility. Consistent with this, Nachum et al (2002) did not find a difference between the aVOR gain of motion sickness susceptible and non-susceptible subjects at higher frequencies of stimulation (2–5 Hz), and Lee et al. (2004) also concluded that the aVOR gain is not a parameter that can be used as a measure of adaptation to motion sickness.
Moreover, motion sickness can be induced by visual stimuli that activate velocity storage in the vestibular nuclei (Dichgans et al. 1973; Cohen et al. 1977; Raphan et al. 1979), without involvement of the direct vestibular pathway, although visually induced motion sickness susceptibility is reduced in labyrinthine defective subjects (Cheung et al. 1991; Johnson et al. 1999). Consistent with this, Bouyer and Watt (1996) found that motion sickness induced by torso rotation could be adapted in 3–4 days, but the gain of aVOR was unaffected. Additionally, off vertical axis rotation (OVAR) is a potent motion sickness stimulus in the steady state when all canal input and response has ceased. In this steady state condition, the OVAR nystagmus is driven by sequential activation of the otolith organs and the bias eye velocity is mediated through velocity storage (Raphan and Cohen 1981; Schnabolk 1989; Fanelli et al. 1990). Each of these examples is compatible with the conclusion that motion sickness is generated through velocity storage, and not through the high frequency direct aVOR pathway.
Possible mechanisms for reduction of motion sickness susceptibility due to changes in aVOR time constants
When this and our previous studies are compared, there was no stimulus similar for habituation in the present study that could have provided a reduction in the vestibular time constant. It is more likely that there was a reduction in some aspect of the afferent input in the labyrinthine-defective subjects. It can be argued that since peripheral lesions decrease both the aVOR gain and time constant (Fetter and Zee 1988; Hain and Buettner 1990; Wade et al. 1999), the reason that labyrinthine-defective subjects are less prone to motion sickness is that fewer impulses are generated by the partially damaged peripheral vestibular end organs. The fact that the high frequency gains return to normal levels in the labyrinthine-defective subjects is not uncommon after bilateral labyrinthine lesions (Baloh et al. 1984; Black et al. 2001; Galiana et al. 2001; Palla and Straumann 2004; Newlands et al. 2005) and may be accomplished either centrally or peripherally (Palla and Straumann 2004).
Maintenance and restoration of the velocity storage mechanism after lesions appears to be more complex than restoration of the direct pathway. Velocity storage is not only lost in response to angular acceleration after bilateral labyrinthine lesions, but also cannot be activated by optokinetic stimulation (Cohen et al. 1973; Uemura and Cohen 1973). Similarly, Fetter and Zee (1988) have shown that although the gain of the aVOR can be reestablished after unilateral labyrinthectomy, the aVOR time constants remain short permanently. This has led to experiments to determine if velocity storage is mediated through a feedback pathway from the peripheral labyrinth, but no single cell activity has been recorded that could demonstrate this (Goldberg and Fernandez 1971; Büttner and Waespe 1981; Lisberger et al. 1981; Dickman and Correia 1989).
A mechanism that controls coupling of peripheral afferent input to the neurons responsible for production of velocity storage, however, could result in a decreased velocity storage time constant. Our original model of the aVOR (Raphan et al. 1979) predicted that there is a coupling gain that connects afferent activity to the velocity storage integrator, which has been estimated to be ≈0.13 ± 0.005 in normal humans (Dai et al. 1999). Such an input has been demonstrated anatomically (Holstein et al. 1992, 1999); presynaptic terminals, presumably originating from neurons in the flocculus, end on labyrinthine afferents to central vestibular neurons in the rostral medial vestibular nucleus, which appear to be involved in the production of velocity storage (Holstein et al. 1992; Reisine and Raphan 1992; Yokota et al. 1992). In monkeys and humans, this coupling gain is responsive to the administration of baclofen, a GABAB agonist, and increased baclofen blood levels are associated with a reduction in the aVOR time constant (Cohen et al. 1987; Dai et al. 2005). In our previous study (Dai et al. 2003), the mean coupling gain of the 16 subjects was originally 0.12 ± 0.004 on the first test and this gain fell to 0.09 ± 0.004 on the eighth test, associated with a progressive fall in the vestibular time constant. If such coupling were disrupted after labyrinthine lesions, it could cause transient or permanent reduction in the afferent input to velocity storage. Modification of activity in such afferent terminals could also mediate some of the habituation of the aVOR time constant produced by the nodulus. Unfortunately, it was not possible to use a model-based technique to determine the coupling gain of afferent activity to velocity storage in this study. The aVOR time constants of the labyrinthine defective subjects were too short to use a two time constant approximation of the vestibular response (Dai et al. 1999) in order to get an accurate estimate of the coupling gain to velocity storage in the labyrinthine-defective subjects.
While there was a close relationship between motion sickness susceptibility and the velocity storage horizontal time constant in the normal and labyrinthine defective subjects, regardless of how this reduction in time constant was produced, this may be part of a more global mechanism related to spatial orientation of velocity storage. Horizontal time constants, for example, increase to 60–70 s after lesions or ablations of the nodulus and uvula (Waespe et al. 1985; Hain et al. 1988; Wearne et al. 1998), yet nodulo-uvular lesions causes dogs to be immune to motion sickness (Bard 1945; Wang and Chinn 1956). The question then arises: What distinguishes subjects with nodulo-uvular lesions and long time constants but no motion sickness susceptibility from the normal individuals with long time constants who are highly susceptible to motion sickness?
The answer is likely to lie in the difference in the orientation properties of velocity storage in the two conditions. We have postulated that motion sickness is triggered by a disparity between the eye velocity vector generated through velocity storage in response to an angular stimulus and the yaw axis orientation vector associated with velocity storage, which in normal subjects is closely aligned with the spatial or gravitational vertical (Cohen et al. 2003; Dai et al. 2003). Any disparity in orientation is presumably accumulated in an integrator in the autonomic system, and as more disparity is produced by each head movement, there is an incremental increase in motion sickness. A reduced velocity storage time constant would constrain this buildup and allow for an increased number of head movements before reaching an intolerable nauseogenic state. According to this theory, the linear relationship between the time course of velocity storage and motion sickness susceptibility was due to a reduction in the duration of the disparity between the eye velocity and the yaw axis orientation vector. This provides a common neural basis for the reduction in motion sickness susceptibility in the subjects of our previous study, in which reduced motion sickness susceptibility was due to habituation of the velocity storage time constant, and for subjects in the present study who had reduced time constants and motion sickness susceptibility due to peripheral lesions.
The reason why subjects with lesions of the nodulus and uvula do not experience motion sickness despite their long horizontal time constants is most probably because they have lost the ability to orient eye velocity toward the spatial (gravitational) vertical. After nodulo-uvular ablations (Wearne et al. 1998) or lesions (Hain et al. 1988), there is no longer cross-coupling of eye velocity from horizontal to vertical when the head is tilted, and vertical time constants, which are short in the upright position and become long when side down (Matsuo and Cohen 1984; Waespe et al. 1985; Raphan and Cohen 1988; Dai et al. 1991), remain very short, regardless of head orientation following nodulo-uvulectomy. This would cause the yaw axis orientation vector of the velocity storage integrator to remain along the head yaw axis, regardless of head position relative to gravity (Wearne et al. 1998). Thus, the eye velocity generated by velocity storage in the cerebellar patients will always be aligned with the velocity storage yaw axis orientation vector, which is now permanently along the head yaw axis. Since the orientation discrepancy is zero, there should be no motion sickness. The effects of time constant reduction on motion sickness susceptibility, as in this study, are only apparent when there is orientation discrepancy.
There are still unanswered questions with regard to vestibular input to motion sickness. It has not been possible to predict the level of motion sickness susceptibility in particular subjects from the absolute value of the VOR time constant since two subjects with the same time constant may have different levels of susceptibility (Shupak et al. 1990; Gordon et al. 1996; Bos et al. 2002; Hoffer et al. 2003). However, this and our previous study (Dai et al. 2003) show that whatever the level of susceptibility in a single individual, reduction in their time constant reduces their motion sickness sensitivity. We also do not have much information about the pathways or characteristics of the mechanisms in the autonomic system that are producing the motion sickness (Yates 1992). Intersubject variability in the autonomic system is undoubtedly responsible for some of the variability between subjects. Furthermore, we do not know whether the axis of eye rotation may normally deviate from the axis of rotation in susceptible subjects, nor do we know how the vertical time constants of such subjects are altered with the head in different positions relative to gravity. Such studies, however, are beyond the scope of the present work.
In summary, this study shows that regardless, of whether the mechanism of reduction in the aVOR time constant was related to repetitive training with roll while rotating or occurred after bilateral labyrinthine lesions, the end result was the same: subjects could make more head movements during roll while rotating when their vestibular time constants were short. This provides further evidence that the velocity storage system has an intimate connection with the production of motion sickness. This is consistent with our general theory that motion sickness susceptibility is dependent on orientation discrepancies sensed through velocity storage, and that shorter horizontal time constants reduce this discrepancy.
Acknowledgments
This work was supported by NASA NCC 9-58-25, NSBRI NA00406, NIDCD DC5222, NIDCD DC05204, and EY01867.
Contributor Information
Mingjia Dai, Email: mingjia.dai@mssm.edu, Department of Neurology, Mount Sinai School of Medicine, 1 East 100th Street, Box 1135, New York, NY 10029-6574, USA.
Theodore Raphan, Department of Computer and Information Science, Brooklyn College, CUNY, Brooklyn, NY 11210, USA.
Bernard Cohen, Department of Neurology, Mount Sinai School of Medicine, 1 East 100th Street, Box 1135, New York, NY 10029-6574, USA.
References
- Balaban CD. Vestibular autonomic regulation (including motion sickness and the mechanism of vomiting) Curr Opin Neurol. 1999;12:29–33. doi: 10.1097/00019052-199902000-00005. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Hess K, Honrubia V, Yee RD. Low and high frequency sinusoidal rotational testing in patients with vestibular lesions. Acta Otolaryngol Suppl. 1984;406:189–193. doi: 10.3109/00016488309123031. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Jacobson K, Honrubia V. Idiopathic bilateral vestibulopathy. Neurology. 1989;39:272–275. doi: 10.1212/wnl.39.2.272. [DOI] [PubMed] [Google Scholar]
- Bard P. Subcommittee on motion sickness; Committee on aviation medicine. National Research Council, National Academy of Science; 1945. [Google Scholar]
- Black FO, Gianna-Poulin C, Pesznecker SC. Recovery from vestibular ototoxicity. Otol Neurotol. 2001;22:662–671. doi: 10.1097/00129492-200109000-00018. [DOI] [PubMed] [Google Scholar]
- Bles W. Coriolis effects and motion sickness modeling. Brain Res Bull. 1988:543–549. doi: 10.1016/s0361-9230(98)00089-6. [DOI] [PubMed] [Google Scholar]
- Bos JE, Bles W, de Graaf B. Eye movements to yaw, pitch, and roll about vertical and horizontal axes: adaptation and motion sickness. Aviat Space Environ Med. 2002;73:436–434. [PubMed] [Google Scholar]
- Bouyer LJ, Watt DG. “Torso rotation” experiments; 1: adaptation to motion sickness does not correlate with changes in VOR gain. J Vestib Res. 1996;6:367–375. [PubMed] [Google Scholar]
- Brandt T. Vertigo. Springer; London: 1999. [Google Scholar]
- Büttner U, Waespe W. Vestibular nerve activity in the alert monkey during vestibular and optokinetic nystagmus. Exp Brain Res. 1981;41:310–315. doi: 10.1007/BF00238888. [DOI] [PubMed] [Google Scholar]
- Cheung BS, Howard IP, Money KE. Visually induced sickness in normal and bilaterally labyrinthine-defective subjects. Aviat Space Environ Med. 1991;62:527–531. [PubMed] [Google Scholar]
- Clément G, Deguine O, Parant M, Costes-Salon MC, Vasseur-Clausen P, Pavy-LeTraon A. Effects of cosmonaut vestibular training on vestibular function prior to spaceflight. Eur J Appl Physiol. 2001;85:539–545. doi: 10.1007/s004210100494. [DOI] [PubMed] [Google Scholar]
- Cohen B, Uemura T, Takemori S. Effects of labyrinthectomy on optokinetic nystagmus (OKN) and optokinetic after-nystagmus (OKAN) Int J Equilb Res. 1973;3:88–93. [PubMed] [Google Scholar]
- Cohen B, Matsuo V, Raphan T. Quantitative analysis of the velocity characteristics of optokinetic nystagmus and optokinetic after-nystagmus. J Physiol (Lond) 1977;270:321–344. doi: 10.1113/jphysiol.1977.sp011955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen B, Helwig D, Raphan T. Baclofen and velocity storage: a model of the effects of the drug on the vestibulo-ocular reflex in the rhesus monkey. J Physiol. 1987;393:703–725. doi: 10.1113/jphysiol.1987.sp016849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Cohen B, Raphan T, Waespe W. Habituation and adaptation of the vestibulo-ocular reflex: a model of differential control by the vestibulo-cerebellum. Exp Brain Res. 1992;90:526–538. doi: 10.1007/BF00230935. [DOI] [PubMed] [Google Scholar]
- Cohen B, Dai M, Raphan T. The critical role of velocity storage in production of motion sickness. Ann NY Acad Sci. 2003;1004:359–376. doi: 10.1196/annals.1303.034. [DOI] [PubMed] [Google Scholar]
- Correia MJ, Perachio AA, Dickman JD, Kozlovskaya IB, Sirota MG, Yakushin SB, Beloozerova IN. Post-flight changes in yaw responses of horizontal semicircular canal afferents in rhesus monkey following 14 days of microgravity. XVI-Ith Barany Society Meeting; Czechoslovakia. June 1–5; 1992. pp. 188–189. [Google Scholar]
- Dai M, Raphan T, Cohen B. Spatial orientation of the vestibular system: dependence of optokinetic after nystagmus on gravity. J Neurophysiol. 1991;66:1422–1438. doi: 10.1152/jn.1991.66.4.1422. [DOI] [PubMed] [Google Scholar]
- Dai M, Klein A, Cohen B, Raphan T. Model-based study of the human cupular time constant. J Vestib Res. 1999;9:293–301. [PubMed] [Google Scholar]
- Dai M, Kunin M, Raphan T, Cohen B. The relation of motion sickness to the spatial-temporal properties of velocity storage. Exp Brain Res. 2003;151:173–189. doi: 10.1007/s00221-003-1479-4. [DOI] [PubMed] [Google Scholar]
- Dai M, Raphan T, Cohen B. Effects of baclofen on the angular vestibulo-ocular reflex. Exp Brain Res. 2005:1–10. doi: 10.1007/s00221-005-0264-y. [DOI] [PubMed] [Google Scholar]
- Dichgans J, Schmidt CL, Graf W. Visual input improves the speedometer function of the vestibular nuclei in the gold-fish. Exp Brain Res. 1973;18:319–322. doi: 10.1007/BF00234602. [DOI] [PubMed] [Google Scholar]
- Dickman JD, Correia MJ. Responses of pigeon horizontal semicircular canal afferent fibers. II. High-frequency mechanical stimulation. J Neurophysiol. 1989;62:1102–1112. doi: 10.1152/jn.1989.62.5.1102. [DOI] [PubMed] [Google Scholar]
- Fanelli R, Raphan T, Schnabolk C. Neural network modelling of eye compensation during off-vertical axis rotation. Neural Netw. 1990;3:265–276. [Google Scholar]
- Fernández C, Goldberg JM. Physiology of peripheral neurons innervating semicircular canals of the squirrel monkey. II. Response to sinusoidal stimulation and dynamics of peripheral vestibular system. J Neurophysiol. 1971;34:661–675. doi: 10.1152/jn.1971.34.4.661. [DOI] [PubMed] [Google Scholar]
- Fetter M, Zee DS. Recovery from unilateral labyrinthectomy in rhesus monkey. J Neurophysiol. 1988;59:370–393. doi: 10.1152/jn.1988.59.2.370. [DOI] [PubMed] [Google Scholar]
- Galiana HL, Smith HL, Katsarkas A. Modelling non-linearities in the vestibulo-ocular reflex (VOR) after unilateral or bilateral loss of peripheral vestibular function. Exp Brain Res. 2001;137:369–386. doi: 10.1007/s002210000667. [DOI] [PubMed] [Google Scholar]
- Goldberg JM, Fernandez C. Physiology of peripheral neurons innervating semicircular canals of the squirrel monkey. I. Resting discharge and response to angular accelerations. J Neurophysiol. 1971;34:635–660. doi: 10.1152/jn.1971.34.4.635. [DOI] [PubMed] [Google Scholar]
- Golding JF, Kerguelen M. A comparison of the nauseogenic potential of low-frequency vertical versus horizontal linear oscillation. Aviat Space Environ Med. 1992;63:491–497. [PubMed] [Google Scholar]
- Gordon CR, Spitzer O, Doweck I, Shupak A, Gadoth N. The vestibulo-ocular reflex and seasickness susceptibility. J Vestib Res. 1996;6:229–233. [PubMed] [Google Scholar]
- Graybiel A, Wood CD. Rapid vestibular adaptation in a rotary environment by means of controlled head movements. Aerosp Med. 1969;40:638–643. [PubMed] [Google Scholar]
- Graybiel A, Miller EF, II, Homick JL. Individual differences in susceptibility to motion sickness among six Skylab astronauts. Acta Astronaut. 1975;2:155–174. doi: 10.1016/0094-5765(75)90051-x. [DOI] [PubMed] [Google Scholar]
- Guedry FE, Benson AJ. Coriolis cross-coupling effects: disorienting and nauseogenic or not? Aviat Space Environ Med. 1978;49:29–35. [PubMed] [Google Scholar]
- Guedry FE, Collins WE, Graybiel A. Vestibular habituation during repetitive complex stimulation: a study of transfer effects. J Appl Physiol. 1964;19:1005–1015. doi: 10.1152/jappl.1964.19.5.1005. [DOI] [PubMed] [Google Scholar]
- Hain T, Buettner UE. Static roll and the vestibulo-ocular reflex (VOR) Exp Brain Res. 1990;82:463–471. doi: 10.1007/BF00228788. [DOI] [PubMed] [Google Scholar]
- Hain TC, Zee DS, Maria BL. Tilt suppression of vestibulo-ocular reflex in patients with cerebellar lesions. Acta Otolaryngol. 1988;105:13–20. doi: 10.3109/00016488809119440. [DOI] [PubMed] [Google Scholar]
- Hecht H, Kavelaars J, Cheung CC, Young LR. Orientation illusions and heart-rate changes during short-radius centrifugation. J Vestib Res. 2001;11:115–127. [PubMed] [Google Scholar]
- Hoffer ME, Gottshall K, Kopke RD, Weisskopf P, Moore R, Allen KA, Wester D. Vestibular testing abnormalities in individuals with motion sickness. Otol Neurotol. 2003;24:633–636. doi: 10.1097/00129492-200307000-00017. [DOI] [PubMed] [Google Scholar]
- Holstein GR, Martinelli GP, Cohen B. L-balcofen-sensitive GABAb binding sites in the medial vestibular nucleus localized by immunocytochemistry. Brain Res. 1992;581:175–180. doi: 10.1016/0006-8993(92)90361-c. [DOI] [PubMed] [Google Scholar]
- Holstein GR, Martinelli GP, Wearne S, Cohen B. Ultra-structure of vestibular commissural neurons related to velocity storage in the monkey. Neuroscience. 1999;93:155–170. doi: 10.1016/s0306-4522(99)00142-6. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Kobayashi K. Space motion sickness and space vestibulology. J UOEH. 1985;7(Suppl):228–236. [PubMed] [Google Scholar]
- Johnson WH, Sunahara FA, Landolt JP. Importance of the vestibular system in visually induced nausea and self-vection. J Vestib Res. 1999;9:83–87. [PubMed] [Google Scholar]
- Kucharczyk J, Stewart DJ, Miller AD. Nausea And Vomitting, Recent Research And Clinical Advances. CRC Press; Boca Raton, Ann Arbor, Boston, London: 1991. [Google Scholar]
- Lackner JR, Graybiel A. The effective intensity of Coriolis, cross-coupling stimulation is gravitoinertial force dependent: implications for space motion sickness. Aviat Space Environ Med. 1986;57:229–235. [PubMed] [Google Scholar]
- Lackner JR, Graybiel A. Use of promethazine to hasten adaptation to provocative motion. J Clin Pharmacol. 1994;34:644–648. doi: 10.1002/j.1552-4604.1994.tb02018.x. [DOI] [PubMed] [Google Scholar]
- Lee MY, Kim MS, Park BR. Adaptation of the horizontal vestibuloocular reflex in pilots. Laryngoscope. 2004;114:897–902. doi: 10.1097/00005537-200405000-00021. [DOI] [PubMed] [Google Scholar]
- Lisberger SG, Miles FA, Optican LM, Eighmy BB. Opto-kinetic response in monkey: underlying mechanisms and their sensitivity to long-term adaptive changes in vestibuloocular reflex. J Neurophysiol. 1981;45:869–890. doi: 10.1152/jn.1981.45.5.869. [DOI] [PubMed] [Google Scholar]
- Lorente de No R. Vestibular ocular reflec arc. Arch Neurol Psychiatry. 1933;30:245–291. [Google Scholar]
- Mathew PJ, Madan R, Subramaniam R, Bhatia A, Mala CG, Soodan A, Kaul HL. Efficacy of low-dose dexamethasone for preventing postoperative nausea and vomiting following strabismus repair in children. Anaesth Intensive Care. 2004;32:372–376. doi: 10.1177/0310057X0403200312. [DOI] [PubMed] [Google Scholar]
- Matsnev EI, Yakovleva IY, Tarasov IK, Alekseev VN, Kornilova LN, Mateev AD, Gorgiladze GI. Space motion sickness: phenomenology, countermeasures, and mechanisms. Aviat Space Environ Med. 1983;54:312–317. [PubMed] [Google Scholar]
- Matsuo V, Cohen B. Vertical optokinetic nystagmus and vestibular nystagmus in the monkey: up-down asymmetry and effects of gravity. Exp Brain Res. 1984;53:197–216. doi: 10.1007/BF00238150. [DOI] [PubMed] [Google Scholar]
- Miller EF, Graybiel A. A standardized laboratory means of determining susceptibility to coriolis (motion) sickness. Aerospace Medical Institute NAMI-1058; 1969. [Google Scholar]
- Money KE. Motion sickness. Physiol Rev. 1972;50:1–39. doi: 10.1152/physrev.1970.50.1.1. [DOI] [PubMed] [Google Scholar]
- Money KE. Biological effects of space travel. Can Aeronaut Space J. 1981;27:195–201. [PubMed] [Google Scholar]
- Nachum Z, Gordon CR, Shahal B, Spitzer O, Shupak A. Active high-frequency vestibulo-ocular reflex and seasickness susceptibility. Laryngoscope. 2002;112:179–182. doi: 10.1097/00005537-200201000-00031. [DOI] [PubMed] [Google Scholar]
- Newlands SD, Dara S, Kaufman GD. Relationship of static and dynamic mechanisms in vestibuloocular reflex compensation. Laryngoscope. 2005;115:191–204. doi: 10.1097/01.mlg.0000154718.80594.2e. [DOI] [PubMed] [Google Scholar]
- Palla A, Straumann D. Recovery of the high-acceleration vestibulo-ocular reflex after vestibular neuritis. J Assoc Res Otolaryngol. 2004;5:427–435. doi: 10.1007/s10162-004-4035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha L, Berens KL, Marshburn TH, Ortega HJ, Billica RD. Pharmaceutical use by US astronauts on space shuttle missions. Aviat Space Environ Med. 1999;70:705–708. [PubMed] [Google Scholar]
- Raphan T, Cohen B. The role of integration in oculomotor control. In: Zuber BL, editor. Models of Oculomotor Behavior and Control. CRC Press; Boca Raton, Fla: 1981. pp. 91–109. [Google Scholar]
- Raphan T, Cohen B. Organizational principles of velocity storage in three dimensions: The effect of gravity on cross-coupling of optokinetic after-nystagmus. Ann NY Acad Sci. 1988;545:74–92. doi: 10.1111/j.1749-6632.1988.tb19556.x. [DOI] [PubMed] [Google Scholar]
- Raphan T, Cohen B. The vestibulo-ocular reflex (VOR) in three dimensions. Exp Brain Res. 2002;145:1–27. doi: 10.1007/s00221-002-1067-z. [DOI] [PubMed] [Google Scholar]
- Raphan T, Sturm D. Modelling the spatiotemporal organization of velocity storage in the vestibuloocular reflex by optokinetic studies. J Neurophysiol. 1991;66:1410–1420. doi: 10.1152/jn.1991.66.4.1410. [DOI] [PubMed] [Google Scholar]
- Raphan T, Dai M, Cohen B. Spatial orientation of the vestibular system. Ann NY Acad Sci. 1992;656:140–157. doi: 10.1111/j.1749-6632.1992.tb25205.x. [DOI] [PubMed] [Google Scholar]
- Raphan T, Matsuo V, Cohen B. Velocity storage in the vestibulo-ocular reflex arc (VOR) Exp Brain Res. 1979;35:229–248. doi: 10.1007/BF00236613. [DOI] [PubMed] [Google Scholar]
- Reason JT, Brand JJ. Motion sickness. Academic; London: 1975. [Google Scholar]
- Reisine H, Raphan T. Neural basis for eye velocity generation in the vestibular nuclei during off-vertical axis rotation. Exp Brain Res. 1992;92:209–226. doi: 10.1007/BF00227966. [DOI] [PubMed] [Google Scholar]
- Schnabolk C, Raphan T. Modelling 3-D slow phase velocity estimation during off-vertical axis rotation (OVAR) J Vest Res. 1992;2:1–14. [PubMed] [Google Scholar]
- Sheliga BM, Yakushin SB, Silvers A, Raphan T, Cohen B. Control of spatial orientation of the angular vestibulo-ocular reflex by the nodulus and uvula of the vestibulocerebellum. Ann NY Acad Sci. 1999;871:94–122. doi: 10.1111/j.1749-6632.1999.tb09178.x. [DOI] [PubMed] [Google Scholar]
- Shupak A, Kerem D, Gordon C, Spitzer O, Mendelowitz N, Melamed Y. Vestibulo-ocular reflex as a parameter of seasickness susceptibility. Ann Otol Rhinol Laryngol. 1990;99:131–136. doi: 10.1177/000348949009900211. [DOI] [PubMed] [Google Scholar]
- Singleton GT. Relationships of the cerebellar nodulus to vestibular function: a study of the effects of nodulectomy on habituation. Laryngoscope. 1967;77:1579–1620. doi: 10.1288/00005537-196709000-00001. [DOI] [PubMed] [Google Scholar]
- Solomon D, Cohen B. Stimulation of the nodulus and uvula discharges velocity storage in the vestibulo-ocular reflex. Exp Brain Res. 1994;102:57–68. doi: 10.1007/BF00232438. [DOI] [PubMed] [Google Scholar]
- Thornton WE, Uri JJ. Oculomotor function during space flight and susceptibility to space motion sickness. Acta Astronaut. 1991;23:53–61. doi: 10.1016/0094-5765(91)90099-q. [DOI] [PubMed] [Google Scholar]
- Torte MP, Clément G, Courjon JH, Magenes G. Absence of habituation of the vertical vestibulo-ocular reflex in the cat. Exp Brain Res. 1997;116:73–82. doi: 10.1007/pl00005746. [DOI] [PubMed] [Google Scholar]
- Uemura T, Cohen B. Effects of vestibular nuclei lesion on vestibulo-ocular reflexes and posture in monkeys. Acta Otolaryngol Suppl. 1973;315:1–71. doi: 10.3109/00016487409129565. [DOI] [PubMed] [Google Scholar]
- Wade SW, Halmagyi GM, Black FO, McGarvie LA. Time constant of nystagmus slow-phase velocity to yaw-axis rotation as a function of the severity of unilateral caloric paresis. Am J Otol. 1999;20:471–478. [PubMed] [Google Scholar]
- Waespe W, Cohen B, Raphan T. Dynamic modification of the vestibulo-ocular reflex by the nodulus and uvula. Science. 1985;228:199–201. doi: 10.1126/science.3871968. [DOI] [PubMed] [Google Scholar]
- Wang SC, Chinn HI. Experimental motion sickness in dogs. Importance of labyrinth and vestibular cerebellum. Am J Physiol. 1956;185:617–623. doi: 10.1152/ajplegacy.1956.185.3.617. [DOI] [PubMed] [Google Scholar]
- Wearne S, Raphan T, Cohen B. Control of spatial orientation of the angular vestibuloocular reflex by the nodulus and uvula. J Neurophysiol. 1998;79:2690–2715. doi: 10.1152/jn.1998.79.5.2690. [DOI] [PubMed] [Google Scholar]
- Yakushin SB, Raphan T, Cohen B. Context-specific adaptation of the vertical vestibuloocular reflex with regard to gravity. J Neurophysiol. 2000;84:3067–3071. doi: 10.1152/jn.2000.84.6.3067. [DOI] [PubMed] [Google Scholar]
- Yardley L, Lerwill H, Hall M, Gresty M. Visual destabilisation of posture in normal subjects. Acta Otolaryngol. 1992;112:14–21. doi: 10.3109/00016489209100777. [DOI] [PubMed] [Google Scholar]
- Yates BJ. Vestibular influences on the sympathetic nervous system. Brain Res Brain Res Rev. 1992;17:51–59. doi: 10.1016/0165-0173(92)90006-8. [DOI] [PubMed] [Google Scholar]
- Yates BJ, Miller AD. Physiological evidence that the vestibular system participates in autonomic and respiratory control. J Vestib Res. 1998;8:17–25. [PubMed] [Google Scholar]
- Yokota JI, Reisine H, Cohen B. Nystagmus induced by electrical microstimulation of the vestibular and prepositus hypoglossi nuclei in the monkey. Exp Brain Res. 1992;92:123–138. doi: 10.1007/BF00230389. [DOI] [PubMed] [Google Scholar]
- Young LR, Hecht H, Lyne L, Sienko K, Cheung C, Kavelaars J. Artificial gravity: head movements during short-radius centrifugation. Acta Astronaut. 2001;49:215–226. doi: 10.1016/s0094-5765(01)00100-x. [DOI] [PubMed] [Google Scholar]