

Summary
Unselective histone deacetylase (HDAC) inhibitors are a promising novel therapy for lymphoid malignancies. However, these treatments remain empiric as the pattern of HDAC enzymes in different types of cancer, including lymphoid malignancies, remains unknown. We examined the expression of class I and class II HDACs in a panel of cell lines and tissue sections from primary lymphoid tumors. Class I enzymes were highly expressed in all cell lines and primary tumors studied, including the non-malignant reactive cells in the Hodgkin lymphoma (HL) microenvironment. The most frequently altered HDAC expression was HDAC6, as it was either weakly expressed or undetected in 9/14 (64%) of lymphoid cell lines and in 83/89 (93%) of primary lymphoma tissue specimens, including 50/52 (96%) cases of diffuse large B-cell lymphoma, and 18/22 (82%) cases of classical HL. Cell lines that had low expression level of HDAC6 demonstrated aberrant expression of hyper-acetylated tubulin, and were found to be more sensitive to the growth inhibitory effects of the class I HDAC inhibitor MGCD0103. Collectively, our data demonstrate that HDAC6 is rarely expressed in primary lymphoma cases, suggesting that it may not be an important therapeutic target in these lymphoid malignancies.
Keywords: Hodgkin Lymphoma, Reed Sternberg cells, HDAC, HDAC inhibitors, gene expression profile
Introduction
Histone deacetylases (HDACs) are enzymes that regulate diverse cellular functions, including cell cycle progression, survival, and proliferation(Glozak and Seto 2007, Xu, et al 2007). To date, 18 human HDACs have been identified and grouped into two major families; zinc-dependent and the NAD+-dependant enzymes(Haberland, et al 2008). The zinc-dependent HDACs are further classified into three classes, class I (HDAC1, 2, 3, and 8), class II (IIa; HDAC4, 5, 7, 9, and IIb; HDAC6 and 10), and class IV (includes only one enzyme, HDAC11). Class III are the NAD+-dependant sirtuins (SIRT1-7)(Yamamoto, et al 2007). In addition to their histone substrates, HDACs also deacetylate non-histone proteins, including transcription factors, chaperone proteins, signal transduction mediators, structural proteins, and inflammation mediators(Haberland, et al 2008). Because HDACs modulate a variety of cellular functions that are involved in carcinogenesis, cell growth, survival, and homologous recombination, they are considered a promising target for cancer therapy(Adimoolam, et al 2007, Claus and Lubbert 2003, Glozak and Seto 2007, Khan and La Thangue 2008). Current pharmacological inhibitors primarily target the zinc-dependant HDACs, but remain rather non-specific as they inhibit either class I (e.g; MGCD-0103, SNDX-275) or class I and class II (e.g; vorinostat, panobinostat) HDACs(Itoh, et al 2008, Khan, et al 2008, Witt, et al 2008, Xu, et al 2007). The unselectivity of the currently available HDAC inhibitors results in modulating the acetylation status of a wide range of protein targets leading to a therapeutic response, but also to undesired toxic effects(Buglio, et al 2008, Fournel, et al 2008, Haberland, et al 2008). Thus, it is widely believed that developing selective isotype-specific HDAC inhibitors will be needed to enhance their therapeutic value while decreasing their side effects. To develop these inhibitors, improved understanding of the functional role of each HDAC and its expression pattern in different cancers will be required. Thus, in the absence of pharmacological inhibitors that selectively target one HDAC enzyme at a time, a debate continues regarding whether class-I inhibitors or pan HDAC inhibitors are more effective for the treatment of cancer. While this question is currently under intensive investigation, data from ongoing clinical trials demonstrate that both strategies can induce clinical remissions in patients with lymphoma. However, these treatments remain empiric as the pattern of HDAC enzymes expression in different types of cancer, including lymphoid malignancies, remains unknown.
To gain more insight into the pattern of HDAC enzymes expression in lymphoma, and to better explore the potential utility of isotype-specific HDAC inhibitors in the treatment of lymphoid malignancies, we examined the expression of class I and class II HDACs in a panel of cell lines and tissue sections from primary lymphoid tumors. Consistent with their ubiquitous distribution in normal tissues, class I enzymes were highly expressed in all cell lines and primary tumors studied, including the non-malignant reactive cells in the Hodgkin lymphoma (HL) microenvironment. In contrast, the class II enzyme HDAC6 was infrequently expressed, compared with HDACs 5, 8, and 10. Our results suggest that HDAC6 may not be an important therapeutic target in selected lymphoid malignancies.
Materials and Methods
Cell lines and cell culture
The human Hodgkin and Reed-Sternberg (HRS)-derived cell lines HD-LM2, L-428, KM-H2 and L1236 were obtained from the German Collection of Microorganisms and Cell Cultures, Department of Human and Animal Cell Cultures (Braunschweig, Germany). All cell lines were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Gibco BRL, Gaithersburg, MD), 1% L-glutamine, and penicillin/streptomycin in a humid environment of 5% CO2 at 37°C. The diffuse large cell non-Hodgkin lymphoma (NHL) cell line SKI-DLCL-1, the mantle cell lymphoma cell lines (Jeko-1, Mino, and SP53), the anaplastic large cell lymphoma cell lines (Karpas, SUP-M2 and SU-DHL-1) and the multiple myeloma cell lines (RPMI 8226, U266-B1, and Molp 8) were cultured in a similar way, except that the SKI-DLCL-1 cells were incubated with 20% heat-inactivated fetal bovine serum. The phenotypes and genotypes of these cell lines have been previously published(Amin, et al 2003, Drexler 1993, Gogusev, et al 2002, Goy, et al 2001, Matsuo, et al 2004, Morgan, et al 1989).
Reagents, antibodies and recombinant proteins
The HDAC inhibitor suberoylanilidehydroxamic acid (vorinostat, SAHA) was purchased from Biovision, Inc. (Mountain View, CA). The HDAC inhibitor MGCD0103 was provided by MethylGene (Quebec, Canada)(Duvic, et al 2005, Fournel, et al 2008, O’Connor, et al 2006). For Western blot and immunohistochemistry experiments, antibody to HDAC3 was purchased from BD Bioscience (San Diego, CA). Antibodies to HDAC4, HDAC5 and HDAC7 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies to HDAC1, HDAC2, HDAC6 were purchased from Abcam Inc. (Cambridge, MA). Antibodies to HDAC8, HDAC9, alpha-1 tubulin and acetylated alpha-1 tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to HDAC10, HDAC11 and β-actin were from Sigma Chemicals Co. (St. Louis, MO).
Western blot analysis
Total cellular proteins were extracted by sonication and incubation in lysis buffer (Cell Signaling Technology, Beverly, MA) for 40 min on ice and then centrifuged to remove cellular debris. The protein in the resulting supernatant was quantified by using the bicinchoninic acid (BCA) method (Pierce Chemical Co., Rockford, IL) according to the manufacturer’s instructions. Then, protein was diluted 1:2 in protein sodium dodecyl sulfate (SDS) loading buffer (Cell Signaling Technology, Beverly, MA), and heated to 95°C for 5 min. A total of 30 μg of protein was loaded onto 12% tris-HCl SDS polyacrylamide electrophoresis Ready Gels (Bio-Rad, Hercules, CA), transferred to a nitrocellulose transfer membrane (Bio-Rad, Hercules, CA), and detected by using SuperSignalWest Dura Extended Duration Substrate (Pierce Chemical Co., Rockford, IL), as previously described(Georgakis, et al 2006, Zheng, et al 2003).
Immunohistochemistry
Table 1 lists the anti HDACs antibodies used for immunohistochemical studies, together with their clone designation, source, and working dilution. For HDAC1, 2, 3, 6, 8, and 11, immunostaining was performed on an automated immunostainer (BenchMark XT, Ventana Medical Systems, Inc, Tucson, AZ) according to the company’s instructions. For HDAC5 and 10, sections were first pretreated in autoclave for 10 min in ethylenediaminetetraacetic acid (EDTA) solution (1 mM pH 8), and then manually immunostained by using SuperPicture Polymer Detection Kit (Zymed Laboratories, Invitrogen Corporation, Carlsbad, CA). As a negative control, phosphate-buffered saline was substituted for the primary antibody.
Table 1.
HDAC antibodies used for immunohistochemical studies
| Antibody (clone) | Source | Working dilution |
|---|---|---|
| HDAC1 (rabbit polyclonal) | Abcam Inc., Cambridge, MA | 1:100 |
| HDAC2 (HDAC2-62) | Abcam Inc., Cambridge, MA | 1:1000 |
| HDAC3 (40) | BD Biosciences Pharmingen, San Diego, CA | 1:200 |
| HDAC5 (rabbit polyclonal) | Cell Signaling Technology, Inc., Danvers MA | 1:100 |
| HDAC6 (rabbit polyclonal) | Abcam Inc., Cambridge, MA | 1:200 |
| HDAC8 (E5) | Santa Cruz Biotechnology, Inc., Santa Cruz, CA | 1:100 |
| HDAC10 (rabbit polyclonal) | Sigma, Saint Louis, MO | 1:200 |
| HDAC11 (HDAC11-31) | Sigma, Saint Louis, MO | 1:1000 |
The expression of the HDACs was measured by using immunohistochemical analysis of paraffin embedded tissue sections obtained from reactive non-neoplastic lymph nodes (n=5), diffuse large B-cell lymphomas (DLBCL, n=171) and other B- (n=152) and T-cell (n=5) NHL and a series of classic HL (cHL, n=22). A HL cell line (i.e. HDLM2) and an anaplastic large cell lymphoma CD30+ cell line (i.e. Karpas 299) were also included in the study. Cases were interpreted as positive only when HDACs positive cells (>1%) could be morphologically identified as neoplastic cells. The percentage of positive cells was given only when the positivity was displayed by a number lower than 90%. For control purposes, tissue sections from normal tissues and solid tumours including breast carcinomas, colon carcinomas, hepatocellular carcinomas, undifferentiated carcinomas of nasopharyngeal type, leiomyosarcomas) were also analyzed.
In vitro proliferation assay
Cells were cultured in 12-well plates at a concentration of 0.5 × 106 cells/ml. Cell viability was assessed with the non-radioactive cell proliferation MTS [3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium] assay by using CellTiter96AQueous One Solution Reagent (Promega, Madison, WI), as previously published.(Buglio, et al 2008) Briefly, 80 μl of cell suspension was incubated with 20 μl of CellTiter96AQueous One Solution Reagent in 96-well plates for 1 h at 37°C and 5% CO2, and formazan absorbance was measured at 490 nm on a μQuant plate reader equipped with Gen5 software (Biotek Instruments, Winooski, VT). Each measurement was made in triplicate and the mean value was determined. Results represent mean value from three independent experiments.
Results
Expression of HDAC isotype enzymes in lymphoid cell lines
The level of class I and class II HDACs expression was initially measured in a panel of 14 well characterized lymphoid cell lines. All cell lines expressed class I HDACs (Fig 1A). In contrast, class II HDACs were variably expressed. Specifically, HDAC6 was weakly expressed in the anaplastic large cell lymphoma cell lines (SU-DHL-1, SUP-M2, and KARPAS 299), the B-cell derived HL cell lines (KM-H2, L428, and L1236) and the multiple myeloma cell lines (RPMI8226, U266-B1, and MOLP8), but was expressed at a higher level in the T-cell derived HL cell line HD-LM2, the mantle cell lymphoma cell lines (Jeko-1, Mino, and SP53), and the diffuse large cell lymphoma cell line SKI-DLCL-1. (Fig 1A and B). Collectively, these data demonstrate that the expression of HDAC6 is the most frequently altered HDAC enzyme in lymphoid cell lines.
Fig 1. Expression of class I and class II HDACs in lymphoid cell lines by Western blot analysis.

A) Expression of class I, II, and IV HDACs in 9 lymphoma cell lines demonstrating the variable expression of HDAC6. B) Selective class I and class II HDAC expression in large cell lymphoma and multiple myeloma cell lines. Three HL cell lines were included for comparison.
Low expression of HDAC6 is associated with increased level of acetylated α-tubulin
HDAC6 has been reported to deacetylase several non-histone targets, including α-tubulin(Hubbert, et al 2002). pharmacologicinhibition of HDAC6, or HDAC6 knockout have been shown to increase acetylation of α-tubulin in a variety of benign and malignant cells (Khan, et al 2008, Zhang, et al 2008). Therefore, to further confirm the low expression status of HDAC6, the level of acetylated α-tubulin expression was examined in the same cells. As predicted, cells that expressed high levels of HDAC6 demonstrated no detectable levels of acetylated α-tubulin (Fig 2). In contrast, cells that expressed low levels of HDAC6 demonstrated the highest level of acetylated α-tubulin (Fig 2). Collectively, these data confirm that HDAC6 is frequently expressed at low levels in lymphoid cell lines, resulting in aberrant hyper-acetylated α-tubulin.
Fig 2. Regulation and correlation of α-tubulin and acetylated α-tubulin expression in lymphoid cell lines.

A) HL and anaplastic large cell lymphoma cell lines had low expression of HDAC6 and high expression of acetylated tubulin. B) In contrast, the mantle cell lymphoma cell lines demonstrated high levels of HDAC6 and no detectable level of acetylated tubulin. C) The pan-HDAC inhibitor vorinostat increased tubulin acetylation within 24 h of incubation, indicative of HDAC6 inhibition. D) The class I HDAC inhibitor MGCD0103 failed to increase tubulin acetylation, even at higher concentrations, indicating its inability to inhibit HDAC6.
Effect of HDAC6 expression on the antiproliferative activity of HDAC inhibitors
The contribution of HDAC6 to cell growth and viability remains unclear. HDAC6-deficient mice show hyperacetylatedtubulin in most tissues, but are viable, fertile, and maintain normal lymphoid development(Zhang, et al 2008). On the other hand, HDAC6 inactivation in several cancer cell lines has been reported to reduce anchorage-independent growth and the ability to form tumors in mice(Lee, et al 2008). To shed more light on the role of HDAC6 in the oncogenic process of lymphoid malignancies, we examined the antiproliferative effect of HDAC inhibitors in cell lines that expressed low or high levels of HDAC6.
First, we examined whether the pan HDAC inhibitor vorinostat can further inhibit HDAC6 activity in the cell lines that expressed low levels of HDAC6, as measured by a further increase in α-tubulin acetylation. For these experiments, the HL cell lines HD-LM2, L428, and KM-H2 were incubated with dimethylsulfoxide (DMSO) or vorinostat (1μM) for 24 or 48 hours, and whole cell lysates were examined by Western blot analysis for tubulin acetylation. As shown in Fig 2, vorinostat increased α-tubulin acetylation in all three of the cell lines, indicative of HDAC6 inhibition. In contrast, and as expected, the class I HDAC inhibitor MGCD0103 (0.1–2 μM for 24 h) had no effect on α-tubulin acetylation in the same cell lines (Fig 2).
Next, we compared the antiproliferative effect of the class I HDAC inhibitor MGCD0103 with that of the pan HDAC inhibitor vorinostat in the HL cell lines that expressed low levels of HDAC6 and the mantle cell lymphoma cell lines that expressed high levels of HDAC6 (Fig 3). For these experiments, cells were incubated with DMSO, or with 1 μM of either MGCD0103 or vorinostat for 48 h before viable cell numbers were determined by the MTS assay. Results were reported as the mean of three independent experiments. MGCD0103 was effective in all HL cell lines, killing at least 75% of the cells within 48 h. In contrast, only one mantle cell lymphoma cell line (Mino) was sensitive to MGCD0103, suggesting that overexpression of HDAC6 may attenuate the activity of the class I HDAC inhibitor MGCD0103. Interestingly, the pan HDAC inhibitor vorinostat was less effective than MGCD0103 in all cell lines, irrespective of their HDAC6 expression status, perhaps reflecting its weak anti HDAC properties.
Fig 3. Antiproliferative effects of vorinostat and MGCD0103 in cell lines that differentially express HDAC6.

Cells were incubated with 1 μM of MGCD0103 or vorinostat for 72 h before cell viability was determined by the MTS assay. MGCD0103 was effective in all three HL cell lines that weakly expressed HDAC6 but was also effective in the mantle cell lymphoma cell line Mino, which expressed a higher level of HDAC6. MGCD0103 was less effective in Jeko-1 and SP53 mantle cell lymphoma cells that expressed high levels of HDAC6. In contrast, vorinostat was less effective than MGCD0103 in all cell lines, irrespective of HDAC6 expression status. Results are mean of 3 independent experiments (± standard error of the mean).
In view of the fact that 1 μM of vorinostat inhibited basal HDAC6 activity in HL cell lines (Fig 2), we examined whether the inhibition of HDAC6 by vorinostat may further enhance the antiproliferative effect of MGCD0103 in HL cells. For these experiments, we incubated the same HL cell lines with increasing concentrations (0.1, 0.2, 05, and 1 μM) of MGCD0103, vorinostat, or both for 24 or 48 h, before determining cell viability using the MTS assay. The addition of vorinostat to MGCD0103 showed no additive or synergistic effect (data not shown). Thus, pharmacological inhibition of HDAC6 did not potentiate the effect of class I HDAC inhibitors in HL cell lines.
Expression of HDAC enzymes in benign reactive lymph nodes and primary lymphoma tumors
We recently reported that the class IHDAC inhibitor MGCD0103 was more effective in achieving clinical responses in patients with relapsed HL than in those with relapsed DLBCL (Younes, et al 2007a, Younes, et al 2007b). On the basis of our observation that HDAC6 is differentially expressed in lymphoid cell lines, and that HDAC6 overexpression was more associated with a decreased sensitivity to MGCD0103, we determined whether primary DLBCL lymph node sections have a higher expression level of HDAC6 compared with HL primary tissue sections.
First, we evaluated a panel of antibodies (Table 1) in normal and tumor tissues that are known to express specific HDAC enzymes. In particular, we tested normal and neoplastic colon tissues for HDAC1,2, and 3, normal and neoplastic liver for HDAC10, undifferentiated carcinomas of nasopharyngeal type for HDAC5 (unpublished observations), normal and neoplastic breast tissues for HDAC6, and leiomyosarcoma for HDAC8(Ishihama, et al 2007, Kaler, et al 2008, Nakagawa, et al 2007, Park, et al 2007, Spurling, et al 2008, Waltregny, et al 2004, Wilson, et al 2006, Zhang, et al 2004). Representative control tissue sections that were used for evaluating the expression of HDAC1,6,8, and 10 are shown in Figure 4.
Fig 4. Immunoreactivity of HDAC1 in colon carcinoma, HDAC6 in breast cancer, HDAC8 in leiomyosarcoma, and HDAC10 in hepatocellularcarcinoma.

These tissues were used as positive control for the specified antibodies.
Next, we studied HDACs expression in five reactive lymph nodes. Class I HDACs 1, 2, and 3 were expressed in all cellular compartments (Table 2), but the intensity of staining for HDAC1 and 2 was variable in non-lymphoid cells, ranging from weak to strong. Consistent with its role in promoting cell proliferation, HDAC1 expression was more intense in the proliferating germinal centre lymphocytes compared with other cellular compartments (Fig 5). Similar to our observation with the cell lines, class II enzymes were more differentially expressed; HDAC6 expression was restricted to the plasma cells (Fig. 5), whereas HDAC10 was widely expressed in all cellular compartments.
Table 2.
Immunoreactivity of HDAC antibodies in reactive lymph nodes*
| Cell type | HDAC 1 | HDAC 2 | HDAC 3 | HDAC 6 | HDAC 10 |
|---|---|---|---|---|---|
| GC B-cells | + | + | + | − | + |
| MZ B-cells | + | + | + | − | + |
| Plasma cells | + | + | + | + | + |
| T-cells | + | + | + | − | + s |
| FDC | + w | + s | + | − | + |
| IDC | + w | + s | + | − | + w |
| FRC | + w | + s | + | − | + |
| Macrophages | + w | + s | + | − | + |
| Sinus histiocytes | + | + s | + | − | + |
| Endothelial cells§ | + | + s | + | − | + |
In the table are listed results with selected HDAC antibodies that showed more effective immunoreactivity in formalin fixed paraffin embedded tissue sections.
Lymph vessels, capillaries, venules
GC, germinal centre; MZ, mantle zone; FDC, follicular dentritic reticulum cells; IDC, Interdigitating reticulum cells; FRC, fibroblastic reticulum cells; w, weak; s, strong; +, positive; −, negative
Fig 5. Immunoreactivity of HDAC1 (A, B) and HDAC6 (C) in a reactive lymph node.

(A) HDAC1 was expressed in germinal center B-cells and mantle zone cells of secondary follicles. (B) HDAC1 reactivity was also seen in primary lymphoid follicles, the paracortical area, endothelial cells of lymphatic vessels, and sinus histiocytes. (C) HDAC6 was expressed only in plasma cells (arrows). Immunostaining for HDAC1 was nuclear (A, B), whereas that for HDAC6 was either nuclear or cytoplasmic (C). GC: germinal centre of a secondary follicle; MZ: mantle zone; S: marginal sinus; PC: paracortical area; PF: primary follicle; LV: intracapsular lymphatic vessel.
Finally, we examined the pattern of expression of class I HDACs, selected class II HDACs, and the class IV HDAC in tissue sections from a panel of 171 primary NHL (Table 3 and Fig 6A) and 22 classical HL cases (Table 3 and Fig 6B). As shown in Table 3, Class I HDAC1, 2 and 3 were expressed in all NHL and classical HL cases tested. All tumor cells were immunostained and the intensity of staining was strong. HDAC8 was expressed in most of the DLBCL cases tested (142/147; 96.6%) and all of the classical HL tested (3/3; 100%). On the other hand, class II and IV enzymes were differentially expressed. HDAC5, 10 and 11 were expressed in all the NHL cases tested. HDAC10 was expressed in all cases of classical HL, whereas HDAC5 was expressed in 2 of 3 of classical HL cases tested and HDAC11 was not expressed in any of the 4 classical HL cases tested. In HL, HDAC expression was observed in both the malignant Hodgkin and Reed-Sternberg (HRS) cells and the surrounding reactive cells (Fig 6B). In contrast, HDAC6 was expressed only in two out of 52 DLBCL cases tested (3.8%). Among the other B-cell lymphomas tested, HDAC6 was expressed only in cases exhibiting plasmacytoid/plasmablastic differentiation. Finally, HDAC6 expression was absent in all 22 cases of cHL tested, although in 4 cases a few HRS cells were weakly stained.
Table 3.
Immunoreactivity of HDAC antibodies* in lymphomas
| Lymphoma type/cell line studied | HDAC
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 5 | 6 | 8 | 10 | 11 | |
| (number of positive cases/number of tested cases) | ||||||||
| Non-Hodgkin Lymphomas | ||||||||
| Diffuse large B-cell lymphoma† | 152/152 | 60/60 | 60/60 | 60/60# | 2/52 | 142/147 | 39/39 | 56/56 |
| Other B-cell lymphomas | ||||||||
| Follicular B-cell | 1/1 | 1/1 | 1/1 | NA | 0/1 | 1/1 | 1/1 | 1/1 |
| Mantle cell lymphoma | 2/2 | 2/2 | 2/2 | NA | 0/5 | NA | 2/2 | 2/2 |
| Plasmablasticlymphoma | 1/1 | 1/1 | 1/1 | NA | 1/1 | NA | 1/1 | 1/1 |
| Plasmacytoma | 1/1 | 1/1 | 1/1 | NA | 1/1 | NA | 1/1 | 1/1 |
| Anaplastic ALK+ | 6/6 | 6/6 | 6/6 | NA | 2/6§ | NA | 6/6 | 6/6 |
| Peripheral T-cell lymphoma | 1/1 | 1/1 | 1/1 | NA | 0/1 | NA | 1/1 | 1/1 |
| Classic Hodgkin lymphomas | 40/40 | 22/22 | 22/22 | 2/3 | 4/22‡ | 3/3 | 8/8 | 0/4 |
| Cell lines | ||||||||
| HDLM2 | + | + | + | − | + | + | + | + |
| Karpas 299 | + | + | + | − | + | + | + | + |
In the table are listed unambiguous results obtained using antibodies more effective in formalin-fixed paraffin embedded tissue sections
immunohistochemistry performed on tissue microarrays;
Percentage of positive cells varying from <10% to 50%;
<5% of neoplastic cell positive;
20% of Reed-Sternberg cells weakly positive; +, positive; −, negative
Fig 6. Immunoreactivity of HDACs in primary lymphoma tissue sections. A) diffuse large cell lymphoma.
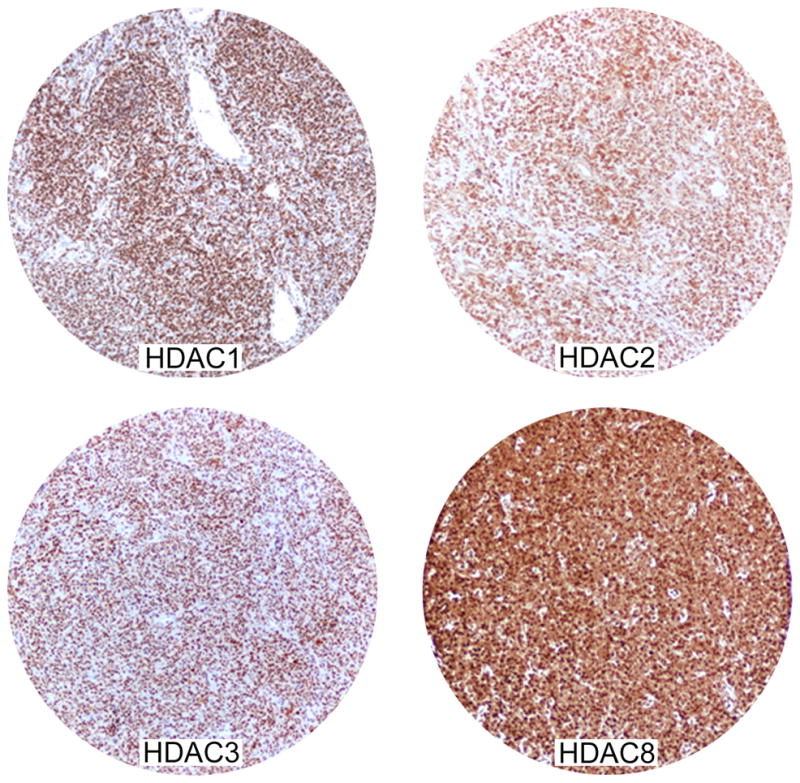

Most tumour cells stained positive for HDAC1,2,3 and 8 in all cases. The staining for HDAC1, 2, and 3 is nuclear with a strong intensity; the staining for HDAC8 was either nuclear or cytoplasmic with strong intensity. B) classical Hodgkin lymphoma. HRS cells (arrow) and the surrounding reactive cells expressed HDAC1, 2, 3, 8 and 10, but not HDAC6.
Discussion
The currently used HDAC inhibitors in the clinical setting are unselective as they inhibit several or all HDACs. Some of these unselective HDAC inhibitors have already demonstrated a promising clinical activity in patients with lymphoid malignancies, especially T-cell lymphoma (cutaneous and peripheral) and HL (Duvic, et al 2005, Haberland, et al 2008, O’Connor, et al 2006, Prince, et al 2007, Spencer, et al 2008, Younes, et al 2007a, Younes, et al 2007b). However, because of their unselectivity, these HDAC inhibitors also cause a wide range of side effects, including hematological, gastrointestinal, and cardiac toxicity. Thus, the future direction of developing HDAC inhibitors for cancer therapy is to define the cancer relevant HDAC enzyme(s) in a specific tumor type to enable the development of selective inhibitors that preferentially target cancer cells while sparing normal cells, and to pre-identify patients who are likely to benefit from this novel therapy(Su, et al 2008).
We have recently examined the efficacy of the class I HDAC inhibitor MGCD0103 in patients with relapsed HL, diffuse large cell lymphoma, and follicular lymphoma(Younes, et al 2007a, Younes, et al 2007b). Patients with relapsed HL had the highest response rate (35%). In contrast, patients with follicular lymphoma had a 15% response rate and those with DLBCL had a 12% response rate. Thus, our initial hypothesis was that the difference in the response rate among these lymphoma subtypes is the result of differential expression of class I enzymes, the targets for MGCD0103. Surprisingly, we did not observe any differences in the expression of HDACs 1, 2, 3, and 8 among the lymphoid cell lines or the primary lymphoma tumors.
We next examined whether the lower response rate observed in DLBCL patients, when compared with HL patients, is related to differential expression of class II enzymes. Based on our preclinical data, which suggested that cell lines that expressed high levels of HDAC6 were more resistant to MGCD0103, we focused our study on examining the level of HDAC6 protein in primary tumor sections from DLBCL and HL. We expected to see higher frequency of HDAC6 expression in patients with DLBCL, which is not a target for MGCD0103(Fournel, et al 2008, Zhou, et al 2008). HDAC6, a cytoplasmic class IIb HDAC, is primarily expressed in non-lymphoid organs, including the kidneys, liver, heart, and pancreas. HDAC6-deficient mice have been reported to be viable with no major defects and have normal lymphoid development, but they demonstrate hyperacetylatedtubulin.(Zhang, et al 2008) Aberrant expression of HDAC6 has been reported in human breast carcinoma and oral squamous cell carcinoma, and has been associated with the oncogenic process (Lee, et al 2008, Sakuma, et al 2006, Zhang, et al 2004). In hematological malignancies, HDAC6 has also been reported to be overexpressed in primary and cultured multiple myeloma cells, myeloid leukemia, and T-cell lymphoma(Fiskus, et al 2008, Hideshima, et al 2005, Marquard, et al 2008). This study demonstrated, for the first time, that HDAC6 expression in benign hyperplastic nodes is restricted to plasma cells. Furthermore, we also demonstrated for the first time that HDAC6 is the most variably expressed HDAC enzyme in different types of lymphoid cell lines. However, this variable expression was not evident in the primary lymphoma sections; only 2/52 (4%) of primary DLBCL and 4/22 (18%) of HL cases demonstrated detectable levels of HDAC6.
Our study included only a few cases of follicular lymphoma, mantle cell lymphoma, T-cell lymphoma, and plasmacytomas, and therefore the expression pattern of HDAC6 in these histological subsets remains undetermined. Interestingly, MGCD0103 was highly effective in HL cell lines that had low levels of HDAC6 expression, but also remained effective in the mantle cell lymphoma cell line Mino, which expressed high levels of HDAC6. Thus, because the majority of primary lymphoma cases tested had low expression of HDAC6, and because HDAC6 expression did not confer an absolute resistance to the class I HDAC inhibitor MGCD0103, our data raise questions about the clinical relevance of targeting HDAC6 in selected subtypes of lymphoma. On the other hand, it is important to confirm whether some of the primary cases truly lack HDAC6 expression, or simply have low level of HDAC6 that was below detection by immunohistochemical methods. This issue can be clarified by performing correlative biomarker studies on tissue specimens obtained from lymphoma patients treated with pan HDAC inhibitors, as tumours with low HDAC6 levels are expected to demonstrate an increase in tubulin acetylation in response to pan HDAC inhibitors.
Even if targeting HDAC6 is not clinically relevant in DLBCL and HL, it may be an important target in other types of lymphoma and non-lymphoid tumours. Moreover, the possible lack of HDAC6 expression does not necessarily mean that class I inhibitors should be preferentially used in these lymphoma subtypes, as pan HDAC inhibitors can inhibit other class II enzymes that may be involved in the lymphomagenesis process. In fact, our data, demonstrating a wide range of expression for HDACs 5, 6, and 10, suggest that class II HDACs may have such a role in lymphoma, especially that class-II enzymes usually demonstrate tissue specificity and are primarily expressed in non-lymphoid organs. Knockdown experiments of these individual HDACs in lymphoid cell lines will provide valuable information about the potential therapeutic value of targeting class II HDACs for lymphoma therapy.
This study is the first to report on the pattern of class IV HDAC11 expression in lymphoma. HDAC11 was found to be expressed in all lymphoid cell lines (Fig 1). Interestingly, HDAC11 was expressed in primary NHL cases but not in HL cases. HDAC11 is primarily expressed in heart, smooth muscle, kidney, and brain tissues. There are currently no data on the phenotype of HDAC11-deficient mice, and the role of HDAC11 in the carcinogenetic process remains unknown. However, in a recent elegant study, Villagra et al (2008) reported that overexpression of HDAC11 inhibited interleukin-10 expression and induced inflammatory antigen-presenting cells that were able to prime naive T cells and restore the responsiveness of tolerant CD4(+) T cells.(Villagra, et al 2008) Conversely, disruption of HDAC11 in antigen-presenting cells led to upregulation of expression of the gene encoding IL-10 and impairment of antigen-specific T cell responses. Whether the lack of HDAC11 expression in HL contributes to the immune tolerance of HRS cells is currently unknown, but deserves future investigation.
Finally, it is important to note that several HDACs were expressed by not only the malignant HRS cells, but also by the surrounding reactive cells in the microenvironment. This observation suggest that some of the HDAC inhibitors may induce clinical responses in vivo in HL patients by a dual effect; a direct antiproliferative effect on the malignant cells and indirect immune modulatory effect on the reactive surrounding cells.(Buglio, et al 2008)
In summary, our data suggest that class I HDACs are ubiquitously expressed across different types of lymphomas suggesting that their expression cannot be used as a predictive marker for treatment response. Furthermore, the lack of HDAC6 expression suggests that global or pan HDAC inhibitors may not be required for optimal therapeutic efficacy.
Acknowledgments
Supported in part by NCI grant 1 R21 CA117070-01 (AY), Clay Chiles Lymphoma Fund, and Jack L. Stotsky Memorial Fund and by a grant from the Ministerodella Salute, Rome, contract no. F/07/01B (A.C.).* The Authors thank Elisa Bonomi and Barbara Vergani for their technical assistance in performing the immunohistochemical studies, and Dawn Chalaire for the editorial assistance.
Footnotes
Within the framework of the ProgettoIntegratoOncologia-Advanced Molecular Diagnostics: Multidimentional Characterization of Tumours.
Authorship
A.G. performed the experiments, evaluated the results, and helped in writing the manuscript
D.B. performed the experiments, evaluated the results, and helped in writing the manuscript
N.K. performed the experiments, and evaluated the results
G.G. performed the experiments, and evaluated the results
R.Z.O. provided important reagents for the studies
S.N. provided important reagents for the studies
A.C. Design the experiments, evaluated the results, and helped in writing the manuscript
A.Y. Designed the experiments, evaluated the results, and wrote the manuscript
References
- Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104:19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin HM, McDonnell TJ, Medeiros LJ, Rassidakis GZ, Leventaki V, O’Connor SL, Keating MJ, Lai R. Characterization of 4 mantle cell lymphoma cell lines. Arch Pathol Lab Med. 2003;127:424–431. doi: 10.5858/2003-127-0424-COMCLC. [DOI] [PubMed] [Google Scholar]
- Buglio D, Georgiakis GV, Hanabuchi S, Arima K, Khaskhely NM, Liu YJ, Younes A. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood. 2008;112:1424–1433. doi: 10.1182/blood-2008-01-133769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus R, Lubbert M. Epigenetic targets in hematopoietic malignancies. Oncogene. 2003;22:6489–6496. doi: 10.1038/sj.onc.1206814. [DOI] [PubMed] [Google Scholar]
- Drexler HG. Recent results on the biology of Hodgkin and Reed-Sternberg cells. II. Continuous cell lines. Leuk Lymphoma. 1993;9:1–25. doi: 10.3109/10428199309148499. [DOI] [PubMed] [Google Scholar]
- Duvic M, Talpur R, Zhang C, Goy A, Richon V, Frankel SR. Phase II trial of oral suberoylanilide hydroxamic acid (SAHA) for cutaneous T-cell lymphoma (CTCL) unresponsive to conventional therapy. Journal of Clinical Oncology. 2005;23:577S–577S. [Google Scholar]
- Fiskus W, Rao R, Fernandez P, Herger B, Yang Y, Chen J, Kolhe R, Mandawat A, Wang Y, Joshi R, Eaton K, Lee P, Atadja P, Peiper S, Bhalla K. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood. 2008;112:2896–2905. doi: 10.1182/blood-2007-10-116319. [DOI] [PubMed] [Google Scholar]
- Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, Gillespie J, Wang JJ, Ste-Croix H, Rahil J, Lefebvre S, Moradei O, Delorme D, Macleod AR, Besterman JM, Li Z. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- Georgakis GV, Li Y, Rassidakis GZ, Martinez-Valdez H, Medeiros LJ, Younes A. Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxy-geldanamycin in Hodgkin’s lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death. Clin Cancer Res. 2006;12:584–590. doi: 10.1158/1078-0432.CCR-05-1194. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- Gogusev J, Telvi L, Nezelof C. Molecular cytogenetic aberrations in CD30+ anaplastic large cell lymphoma cell lines. Cancer Genet Cytogenet. 2002;138:95–101. doi: 10.1016/s0165-4608(02)00589-7. [DOI] [PubMed] [Google Scholar]
- Goy A, Gilles F, Remache Y, Filippa D, Portlock CS, Jhanwar SC, Zelenetz AD. Establishment of a human cell line (SKI-DLCL-1) with a t(1;14)(q21;q32) translocation from the ascites of a patient with diffuse large cell lymphoma. Leuk Lymphoma. 2001;40:419–423. doi: 10.3109/10428190109057942. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2008;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102:8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Ishihama K, Yamakawa M, Semba S, Takeda H, Kawata S, Kimura S, Kimura W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol. 2007;60:1205–1210. doi: 10.1136/jcp.2005.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Suzuki T, Miyata N. Isoform-selective histone deacetylase inhibitors. Curr Pharm Des. 2008;14:529–544. doi: 10.2174/138161208783885335. [DOI] [PubMed] [Google Scholar]
- Kaler P, Sasazuki T, Shirasawa S, Augenlicht L, Klampfer L. HDAC2 deficiency sensitizes colon cancer cells to TNFalpha-induced apoptosis through inhibition of NF-kappaB activity. Exp Cell Res. 2008;314:1507–1518. doi: 10.1016/j.yexcr.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. Drug Insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat Clin Pract Oncol. 2008;5:714–726. doi: 10.1038/ncponc1238. [DOI] [PubMed] [Google Scholar]
- Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68:7561–7569. doi: 10.1158/0008-5472.CAN-08-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquard L, Gjerdrum LM, Christensen IJ, Jensen PB, Sehested M, Ralfkiaer E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology. 2008;53:267–277. doi: 10.1111/j.1365-2559.2008.03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Drexler HG, Harashima A, Okochi A, Hasegawa A, Kojima K, Orita K. Induction of CD28 on the new myeloma cell line MOLP-8 with t(11;14)(q13;q32) expressing delta/lambda type immunoglobulin. Leuk Res. 2004;28:869–877. doi: 10.1016/j.leukres.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Morgan R, Smith SD, Hecht BK, Christy V, Mellentin JD, Warnke R, Cleary ML. Lack of involvement of the c-fms and N-myc genes by chromosomal translocation t(2;5)(p23;q35) common to malignancies with features of so-called malignant histiocytosis. Blood. 1989;73:2155–2164. [PubMed] [Google Scholar]
- Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, Zelenetz AD, Frankel S, Richon V, Marks P, Kelly WK. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24:166–173. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- Park BL, Kim YJ, Cheong HS, Lee SO, Han CS, Yoon JH, Park JH, Chang HS, Park CS, Lee HS, Shin HD. HDAC10 promoter polymorphism associated with development of HCC among chronic HBV patients. Biochem Biophys Res Commun. 2007;363:776–781. doi: 10.1016/j.bbrc.2007.09.026. [DOI] [PubMed] [Google Scholar]
- Prince HM, George D, Patnaik A, Mita M, Dugan M, Butterfoss D, Masson E, Culver KW, Burris HA, III, Beck J. Phase I study of oral LBH589, a novel deacetylase (DAC) inhibitor in advanced solid tumors and non-hodgkin’s lymphoma. J Clin Oncol (Meeting Abstracts) 2007;25:3500. [Google Scholar]
- Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe H, Shibahara T, Tanzawa H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol. 2006;29:117–124. [PubMed] [Google Scholar]
- Spencer A, DeAngelo DJ, Prince HM, Bhalla KN, Fischer T, Liu A, Parker K, Jalaluddin M, Scott JW, Ottmann OG. Oral Panobinostat (LBH589), a novel deacetylase inhibitor (DACi) demonstrates clinical activity in relapsed/refractory Hodgkin lymphoma (HL) Ann Oncol. 2008;19:136. (Abstract) [Google Scholar]
- Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–147. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- Su H, Altucci L, You Q. Competitive or noncompetitive, that’s the question: research toward histone deacetylase inhibitors. Mol Cancer Ther. 2008;7:1007–1012. doi: 10.1158/1535-7163.MCT-07-2289. [DOI] [PubMed] [Google Scholar]
- Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2008 doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltregny D, De Leval L, Glenisson W, Ly Tran S, North BJ, Bellahcene A, Weidle U, Verdin E, Castronovo V. Expression of histone deacetylase 8, a class I histone deacetylase, is restricted to cells showing smooth muscle differentiation in normal human tissues. Am J Pathol. 2004;165:553–564. doi: 10.1016/S0002-9440(10)63320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2008;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745–1755. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- Younes A, Pro B, Fanale M, McLaughlin P, Neelapu S, Fayad L, Wedgwood A, Buglio D, Patterson T, Dubay M, Li Z, Martell RE, Ward MR, Bociek RG. Isotype-Selective HDAC Inhibitor MGCD0103 Decreases Serum TARC Concentrations and Produces Clinical Responses in Heavily Pretreated Patients with Relapsed Classical Hodgkin Lymphoma (HL) Blood (ASH Annual Meeting Abstracts) 2007a;110:2566. [Google Scholar]
- Younes A, Wedgwood A, McLaughlin P, Andreadis C, Assouline SE, Li Z, Martell RE, Dubay M, Patterson TA, Ward MR, Crump M. Treatment of Relapsed or Refractory Lymphoma with the Oral Isotype-Selective Histone Deacetylase Inhibitor MGCD0103: Interim Results from a Phase II Study. Blood (ASH Annual Meeting Abstracts) 2007b;110:2571. [Google Scholar]
- Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi S, Iwase H. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10:6962–6968. doi: 10.1158/1078-0432.CCR-04-0455. [DOI] [PubMed] [Google Scholar]
- Zheng B, Fiumara P, Li YV, Georgakis G, Snell V, Younes M, Vauthey JN, Carbone A, Younes A. MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood. 2003;102:1019–1027. doi: 10.1182/blood-2002-11-3507. [DOI] [PubMed] [Google Scholar]
- Zhou N, Moradei O, Raeppel S, Leit S, Frechette S, Gaudette F, Paquin I, Bernstein N, Bouchain G, Vaisburg A, Jin Z, Gillespie J, Wang J, Fournel M, Yan PT, Trachy-Bourget MC, Kalita A, Lu A, Rahil J, MacLeod AR, Li Z, Besterman JM, Delorme D. Discovery of N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide (MGCD0103), an orally active histone deacetylase inhibitor. J Med Chem. 2008;51:4072–4075. doi: 10.1021/jm800251w. [DOI] [PubMed] [Google Scholar]