

Abstract
Changes in hepatic vasculature accompany fibrogenesis, and targeting angiogenic molecules often attenuates fibrosis in animals. Aquaporin-1 (AQP1) is a water channel, overexpressed in cirrhosis, that promotes angiogenesis by enhancing endothelial invasion. The effect of AQP1 on fibrogenesis in vivo and the mechanisms driving AQP1 expression during cirrhosis remain unclear. The purpose of this study was to test the effect of AQP1 deletion in cirrhosis and explore mechanisms regulating AQP1. After bile duct ligation, wild-type mice overexpress AQP1 that colocalizes with vascular markers and sites of robust angiogenesis. AQP1 knockout mice demonstrated reduced angiogenesis compared with wild-type mice, as evidenced by immunostaining and endothelial invasion/proliferation in vitro. Fibrosis and portal hypertension were attenuated based on immunostaining, portal pressure, and spleen/body weight ratio. AQP1 protein, but not mRNA, was induced by hyperosmolality in vitro, suggesting post-transcriptional regulation. Endothelial cells from normal or cirrhotic mice were screened for microRNA (miR) expression using an array and a quantitative PCR. miR-666 and miR-708 targeted AQP1 mRNA and were decreased in cirrhosis and in cells exposed to hyperosmolality, suggesting that these miRs mediate osmolar changes via AQP1. Binding of the miRs to the untranslated region of AQP1 was assessed using luciferase assays. In conclusion, AQP1 promotes angiogenesis, fibrosis, and portal hypertension after bile duct ligation and is regulated by osmotically sensitive miRs.
Liver cirrhosis is the final common end point in a variety of toxic, metabolic, infectious, and autoimmune forms of chronic liver disease. Progression toward end-stage liver disease is characterized by an exaggerated wound healing response to long-term injury, culminating in regenerative nodules of hepatocytes surrounded by a dense scar of extracellular matrix.1 In concert with this progressive fibrogenesis, pathological changes in the hepatic angioarchitecture also occur and are thought to promote fibrosis, portal hypertension, and their clinical sequelae.2–4 Despite intensive investigations and significant insights into the basic mechanisms driving these processes, no effective anti-fibrotic therapies are yet available for use in patients with chronic liver diseases. Thus, further mechanistic insights into liver fibrogenesis and coinciding events, such as pathological angiogenesis, are needed to identify potential anti-fibrotic targets and translate those into advances in clinical care.
Aquaporins (AQPs) are a class of integral membrane channel proteins that facilitate the rapid, transmembrane flux of water that occurs passively and bidirectionally in response to local osmotic gradients. These proteins have well-characterized roles in epithelial secretion, absorption, and cell volume regulation.5–8 More recently, they have also been implicated in localized protrusions of plasma membranes, cell motility, and angiogenesis.9,10 Researchers11–13 have demonstrated robust overexpression of AQP1 in both human and rodent chronic liver disease. The increased expression during cirrhosis is localized to the pathological neovasculature and promotes dynamic membrane protrusions that facilitate invasion through the dense extracellular microenvironment associated with that disease. However, direct in vivo evidence of a contribution of AQP1 to liver fibrogenesis is lacking.
The molecular mechanisms driving the increased expression of AQP1 during cirrhosis are unknown; however, as our results will suggest, the mechanism may involve epigenetic responses to osmotic stress within the endothelium. microRNAs (miRs) are small noncoding nucleic acids 21 to 23 nucleotides long that have emerged as important post-transcriptional regulators of protein expression that affect a variety of developmental and pathobiological processes. miRs are initially transcribed by RNA polymerase II as monocistronic or polycistronic primary miRs and are further processed within and outside the nucleus into functionally active mature miRs that act by binding to target messenger RNAs and regulating stability or translational efficiency. The mechanisms regulating miR expression remain largely unclear, but there is precedent for the concept of osmotically sensitive miRs in human, zebra fish, and plant responses to osmotic stress.14–18 Furthermore, conceptual precedent for osmoregulation of AQP1 exists in recent articles19 showing increased AQP1 expression in response to osmolality in renal epithelial cells.
Therefore, the aims of the present study were to test the effect of genetic deletion of AQP1 on liver angiogenesis, fibrosis, and portal hypertension in an established murine model of cirrhosis and to examine the mechanisms by which AQP1 is overexpressed in cirrhotic endothelia. The experimental results demonstrate a prominent role for endothelial cell AQP1 in liver fibrogenesis after bile duct ligation (BDL) and propose a novel mechanism driving AQP1 expression involving osmotically regulated miRs.
Materials and Methods
Animal Models
Mice with global genetic knockout of AQP1 in a CD1 background were a gift from the laboratory of Dr. Alan Verkman (University of California San Francisco, San Francisco, CA).6 Fibrosis was induced at the age of 8 to 10 weeks by common BDL using a well-established protocol with appropriate Institutional Animal Care and Use Committee approval.20 Experiments were performed 4 weeks after BDL. Animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals by the National Academy of Sciences.
Cell Culture
Freshly isolated mouse hepatic sinusoidal endothelial cells (mHSEC) were obtained using an immunomagnetic bead isolation protocol and characterized using 3,3'-dioctadecylindocarbocyanine labeled low-density lipoprotein and staining for von Willebrand's factor (vWF), as previously described.11,21,22 The cells were cultured on collagen-coated plastic tissue culture dishes in endothelial cell media (ScienCell, Carlsbad, CA) containing 5% fetal bovine serum, 1% penicillin-streptomycin, and 1% ECGS supplement (ScienCell). Primary human hepatic sinusoidal endothelial cells (HHSECs; ScienCell), or transformed sinusoidal endothelial cells (TSECs), an SV40-immortalized mouse cell line derived from sinusoidal endothelial cells,23 were grown on uncoated plastic dishes in the same media. In some experiments, cells were incubated in an experimentally modified hypoxia chamber or in the presence of altered osmolality of the culture media. Osmolality was altered using variable concentrations of sodium chloride in the culture media and verified using an osmometer.
Immunoblotting
Western blot analyses were performed as previously described.5 Briefly, mouse liver, mouse sinusoidal endothelial cells, or TSECs were homogenized in lysis buffer and cleared by centrifugation. Each sample, 50 to 100 μg, was denatured, electrophoresed, transferred, blocked with milk, and incubated with antibodies to AQP1 (1:1000; α Diagnostic International, San Antonio, TX), β-actin (1:10,000; Sigma, St. Louis, MO), or total extracellular signal–regulated kinase (1:1000; BD Biosciences, Franklin Lakes, NJ) overnight at 4°C. Horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Piscataway, NJ) were used at 1:5000. Protein was detected using chemiluminescence (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and autoradiography.
Immunofluorescence
Immunofluorescence (IF) was performed as previously described.5 Liver was harvested, sliced, embedded, and flash frozen on dry ice. Sections were cut to 4 to 8 μm. Sections were fixed, quenched, blocked, and incubated with antibodies against AQP1 (1:500; α Diagnostics International), CD31 (1:250), vWF (1:250), vascular endothelial growth factor receptor 2 (VEGFR2; 1:250), and endothelial nitric oxide synthase (eNOS; 1:250). Fluorescently tagged secondary antibodies were used at 1:500. In some experiments, nuclear counterstaining was performed with TOTO-3 (Invitrogen, Carlsbad, CA). Slides were mounted with Vectashield (Vector, Burlingame, CA) and imaged by scanning laser confocal microscopy (Carl Zeiss MicroImaging, Berlin, Germany). Similar procedures were used to stain cultured cells using four-well chamber slides.
IHC Analyses
Whole liver was harvested, sliced, formalin fixed, and embedded in paraffin. Sections were cut to 4 to 8 μm and antigen unmasked with hot citrate buffer. After quenching of endogenous peroxidase, the sections were blocked and incubated with antibody against AQP1 (1:500; α Diagnostics International) overnight at 4°C. The remaining steps were performed using an immunoperoxidase detection kit (Vector Laboratories) and counterstaining with hematoxylin.
Cell Proliferation Assay
Cell proliferation rates of mHSEC, HHSECs, and TSECs were measured in 96-well plates using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI), which is a colorimetric method for determining the number of viable cells. The AQueous One Solution contains a tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; [MTS] and an electron coupling reagent (phenazine methosulfate; PES). The MTS tetrazolium compound is bioreduced by cells into a colored formazan product that is soluble in tissue culture medium. Assays were performed by adding the AQueous One Solution directly to culture wells, incubating for 1 to 4 hours, and measuring optical density at 490 nm with a plate reader at baseline and 48 hours to calculate the proliferation rate.
Portal Pressure Measurements
Portal pressure was directly measured using a digital blood pressure analyzer (Digi-Med, Louisville, KY) with a computer interface. Once the analyzer was calibrated, a 16-gauge catheter attached to the pressure transducer was inserted into the portal vein and sutured in place. The pressure was continuously monitored, and the average portal pressure was recorded. On sacrifice of the animal, the spleen was removed and weighed and the spleen/body weight ratio was calculated.
Serum Tests
Whole blood was obtained from each experimental animal via a heart puncture technique and transferred into 3.5-mL serum separating tubes. Specimens were processed and analyzed for serum transaminase and bilirubin levels by the Mayo Clinic Special Studies Laboratory (Rochester, MN), a clinically validated reference laboratory.
miR Microarray
Total RNA (including miRs) was isolated from endothelial cells derived from BDL or sham operated on mice using the MirVana miRNA Isolation Kit (Ambion, Inc., Austin, TX), according to the manufacturer's instructions. The samples were delivered to the Mayo Clinic Advanced Genomic Technology Center Microarray Shared Resource, where they were further analyzed. Briefly, the samples were hybridized to the GeneChip miRNA Array (Affymetrix, Santa Clara, CA), which interrogates miRs from >70 species, including the entire known murine transcriptome of miRs. Data were extracted, manually curated, and analyzed using QC Toolbox software (Affymetrix). Further rational processing of array data included assessment of relative expression levels in BDL versus sham endothelial cells. Down-regulated targets were analyzed using TargetScan (Whitehead Institute for Biomedical Research, Cambridge, MA) and Microcosm software (European Bioinformatics Institute, Cambridgeshire, UK) to identify down-regulated targets that had potential binding sites within the untranslated regions of AQP1 mRNA. TargetScan predicts biological targets of miRs by searching for the presence of conserved eight- and seven-base pair sites that match the seed region of each miR. Predictions are ranked based on the predicted efficacy of targeting, as calculated using the context scores of the sites. Microcosm uses an algorithm to identify potential binding sites for a given miR using dynamic alignment to identify highly complementary sequences. The algorithm uses a weighted scoring system and prioritizes complementarity at the 5′ end of the miR.
Quantitative RT-PCR
Total RNA (including miRs) was isolated as previously outlined. RNA was reverse transcribed using the miScript RT-PCR system (Qiagen), and cyber green–based real-time RT-PCR was performed using miScript Primer Assays (Qiagen) or AQP1-specific primers, according to the manufacturer's instructions.
Hydroxyproline Assays
Hydroxyproline content was quantified from whole liver tissue using a colorimetric assay, as described elsewhere.24 Briefly, frozen tissue was weighed and processed using hydrochloric acid hydrolysis and chloramine-T–dimethylaminobenzaldehyde incubation, and absorbance at 561 nm was recorded and compared with a standard prepared from commercial hydroxyproline (Sigma).
Luciferase Reporter Assays
Complementary oligonucleotides were designed to amplify a 300-bp portion of the untranslated region of mouse AQP1. The primers were synthesized to contain SpeI and HindIII restriction enzyme digestion sites and used to amplify the region of interest using RT-PCR. The amplified fragment was digested with SpeI and HindIII and ligated into the multiple cloning site of the pMIR-REPORT Luciferase vector (Ambion, Inc.). Chinese hamster ovary cells were transfected with the reporter construct and, in some experiments, cotransfected with miR precursors for miR-666, miR-708, or a control miR. This was followed by assessment of luciferase activity 24 hours after transfection. Luciferase activity was normalized to the expression of a control TK Renilla construct.
Endothelial Cell Biological PCR Array
RNA from TSECs overexpressing either AQP1 or a LacZ control gene was isolated using the QiaShredder and RNeasy kits (Qiagen), according to the manufacturer's instructions. RNA was used for reverse transcription using the RT2 kit (SA Biosciences, Frederick, MD). Cyber green–based real-time quantitative RT-PCR was performed with the Endothelial Cell Biology Array (SA Biosciences), according to the manufacturer's instructions. Array data were processed using the PCR Array Data Analysis Web Portal (SA Biosciences).
Statistical Analysis
Data are presented as the mean ± SEM. Bar graphs, blots, and micrographs represent typical experiments reproduced at least three times. Data analysis was performed using Graph Stat Prizm software (GraphPad Software, Inc., La Jolla, CA). Data were analyzed for normal gaussian distribution using the Kolmogorov-Smirnov normality test. For paired and normally distributed data, statistical analyses were performed using two-tailed Student's t-tests. For normally distributed multiple comparisons, statistical analyses were performed using one-way analysis of variance with a Tukey post-test. For all analyses, P < 0.05 was considered statistically significant.
Results
AQP1 Expression Is Increased after BDL
To directly test the role of AQP1 in chronic liver disease, we used mice from a CD1 background with global genetic deletion of AQP1 or age-matched, wild-type controls and induced cirrhosis and portal hypertension in these mice. We confirmed deletion of AQP1 in total liver lysates from knockout animals by using Western blot analysis (Figure 1A). We induced fibrosis in wild-type mice using BDL, a model of cholestatic liver injury, and confirmed that AQP1 protein levels were significantly increased after BDL using Western blot analysis (Figure 1B) and IF (Figure 1, C and D). Immunohistochemistry (IHC) for AQP1 in these animals demonstrated a similar increase in AQP1 protein levels and showed specific staining of vascular structures, including sinusoids, the portal vein, and the hepatic artery (Figure 1, E and F). IF costaining showed colocalization of AQP1 with several vascular markers (Figure 1, G–J; see also Supplemental Figure S1 at http://ajp.amjpathol.org), including CD31 (Figure 1G), vWF (Figure 1H), VEGFR2 (Figure 1I), and eNOS (Figure 1J).
Figure 1.
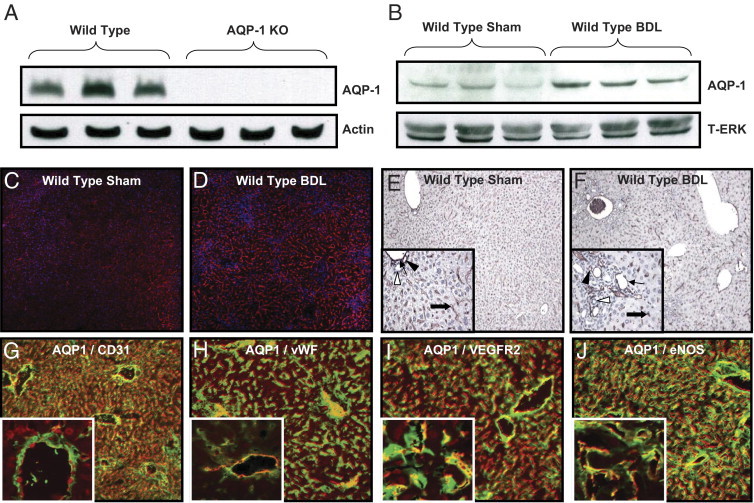
AQP1 is increased in liver endothelia after BDL. A: Representative immunoblots for AQP1 or actin (100 μg/lane) in wild-type or AQP1 knockout (KO) mouse liver tissue. B: Representative immunoblots for AQP1 or total extracellular signal–regulated kinase (T-ERK; 50 μg/lane) in wild-type sham operated on or bile duct–ligated mouse liver tissue. C and D: Representative images of sham operated on or bile duct–ligated mouse liver tissue stained with IF for AQP1 (red) and nuclear TOTO-3 (blue). Original magnification, ×7.5. E and F: Representative images of sham operated on or bile duct–ligated mouse liver tissue stained with IHC for AQP1 (brown) and counterstained with hematoxylin. Original magnification, ×4. Insets: High-power images. Original magnification, ×10. Arrows, portal vein; thick arrows, hepatic artery; black arrowheads, sinusoids; and white arrowheads, bile ducts. G–J: Representative images of bile duct–ligated mouse liver tissue costained with IF for AQP1 (red) and CD31, vWF, VEGFR2, and eNOS (green). Original magnification, ×10. Insets: High-power images. Original magnification, ×63. Individual color images are in Supplemental Figure S1 at http://ajp.amjpathol.org.
AQP1 Knockout Mice Have Reduced Pathological Angiogenesis after BDL
To test the effects of AQP1 deletion on the pathological angiogenesis that accompanies fibrosis, we measured endothelial cell invasion and proliferation, both required for angiogenesis. Liver endothelial cells were isolated from wild-type or AQP1 knockout mice after BDL or sham surgery and subjected to invasion and proliferation assays in vitro. Endothelial cell purity was validated using uptake of diI-labeled acetylated low-density lipoprotein and staining for vWF (see Supplemental Figure S2 at http://ajp.amjpathol.org). Cells isolated from wild-type animals showed markedly increased invasion after BDL, an effect that was largely absent in the knockout animals (Figure 2, A–E). Proliferation assays demonstrated a similar effect (Figure 2F). We also measured proliferation in two sinusoidal endothelial cell lines (TSECs and HHSECs) transduced with retroviral AQP1 or control gene and again found small, but statistically significant, increases in proliferation after overexpression of AQP1 (see Supplemental Figure S3A at http://ajp.amjpathol.org). We used IF staining for vWF in wild-type and knockout mice subjected to either BDL or sham surgery and quantified the fluorescence signals. We found significantly increased neovascularization after BDL in the wild-type animals, an effect that was partially abrogated in AQP1 knockout animals (Figure 2G). A similar effect was noted on staining for CD31 (see Supplemental Figure S3, B–F, at http://ajp.amjpathol.org).
Figure 2.
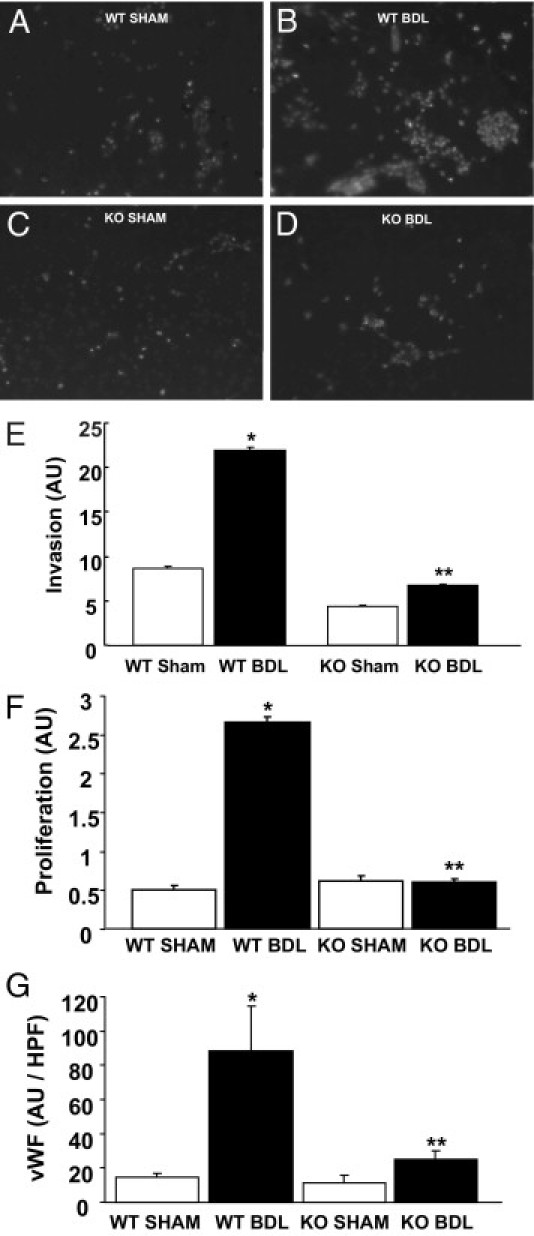
AQP1 knockout (KO) mice have reduced angiogenesis in response to BDL. A–D: Representative images of invaded cells isolated from wild-type (WT) and AQP1 KO mice, with and without BDL stained with IF for nuclear TOTO-3 (white). Original magnification, ×10. E: Quantification of the average total fluorescence signal per high-power field is shown (n = 10 wells per cell type, and n = 3 animals per group). Data are given as the mean ± SE. F: MTS proliferation assays were performed in endothelial cells isolated from WT and AQP1 KO mice, with and without BDL. Quantification of the average spectrophotometric signal per well is shown (n = 9 wells per animal, and n = 3 animals per group). Data are given as the mean ± SE. G: Tissue from WT and AQP1 KO mice, with and without BDL, was stained with IF for vWF. Quantification of the average total fluorescence signal per high-power field is shown (n = 10 fields per animal, and n = 3 animals per group). Data are given as the mean ± SE. *P < 0.05 versus WT sham; **P < 0.05 versus WT BDL. AU, arbitrary unit.
AQP1 Knockout Mice Have Reduced Fibrosis after BDL
To assess fibrogenesis, we immunostained for a standard panel of morphological markers of extracellular matrix deposition, stellate cell activation, and fibrosis, as well as complementary histochemical and biochemical approaches. After BDL, IF staining showed a prominent increase in the extracellular matrix components, collagen (Figure 3, A–E) and fibronectin (Figure 3F), that were partially inhibited in the AQP1 knockout animals. A well-accepted histochemical stain for fibrosis, Sirius red, showed similar effects (Figure 3G). Smooth muscle actin (a marker of stellate cell activation), H&E staining, and a biochemical correlate of cross-linked collagen, hydroxyproline content, further corroborated these findings (see Supplemental Figure S4 at http://ajp.amjpathol.org). Collectively, these results strongly support a role for endothelial cell AQP1 in fibrogenesis and its associated angiogenesis.
Figure 3.
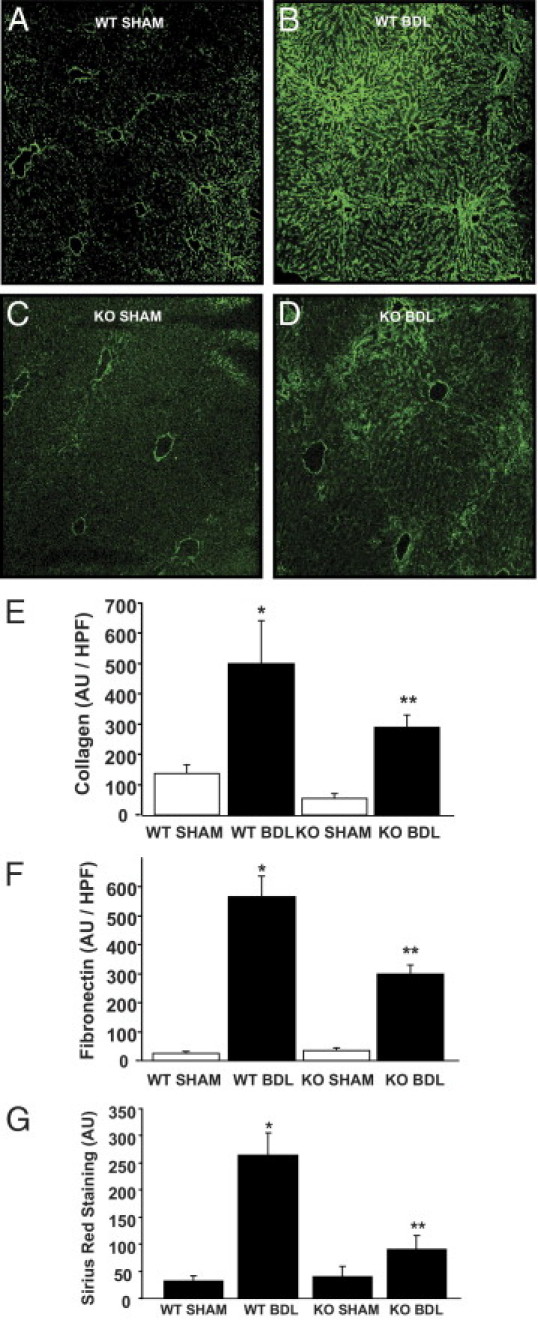
AQP1 knockout (KO) mice have reduced fibrosis in response to BDL. A–D: Representative images of wild-type (WT) and AQP1 knockout (KO) mice, with and without BDL, stained with IF for collagen (green). Original magnification, ×7.5. E: Quantification of the average total fluorescence signal per high-power field (HPF) is shown (n = 10 fields per animal, and n = 3 animals per group). Data are given as the mean ± SE. F: Tissue from AQP1 KO mice, with and without BDL, was stained with IF for fibronectin. Quantification of the average total fluorescence signal per HPF is shown (n = 10 fields per animal, and n = 3 animals per group). Data are given as the mean ± SE. G: Tissue from AQP1 KO mice, with and without BDL, was stained with Sirius red. Quantification of the average total signal per HPF is shown (n = 10 fields per animal, and n = 3 animals per group). Data are given as the mean ± SE. *P < 0.05 versus WT sham; **P < 0.05 versus WT BDL. See images in Supplemental Figure S4D at http://ajp.amjpathol.org. AU, arbitrary unit.
AQP1 Knockout Mice Have Less Portal Hypertension after BDL
We next assessed the level of portal hypertension in wild-type and AQP1 knockout mice after BDL using two complementary approaches. After direct cannulation of the portal vein and connection to a pressure transducer, we directly measured portal pressure. The BDL-induced increase in portal pressure seen in wild-type mice was reduced in the absence of AQP1 (Figure 4A). As a complementary approach, we also measured spleen/body weight ratio, an indirect assessment of portal hypertension. AQP1 knockout mice demonstrated splenomegaly at baseline (data not shown), possibly due to lack of AQP1 in red blood cells and impaired hematopoiesis.25 Nevertheless, after normalizing BDL values to the corresponding sham group, we again saw a significantly smaller increase in spleen size in AQP1 knockout animals compared with wild-type controls (Figure 4B). To broadly assess liver inflammation, which is closely associated with, and frequently parallels, angiogenesis, we also measured transaminase (aspartate aminotransferase and alanine aminotransferase) levels in the serum of these animals. There was a substantially smaller increase in both transaminases in AQP1 knockout animals after BDL (45% and 58% reduction for aspartate aminotransferase and alanine aminotransferase, respectively), albeit not statistically significant (Figure 4, C and D). This is consistent with the concept that the angiogenic neovasculature may be a source of inflammatory cytokines, driving chronic inflammation and progression of fibrosis. Total bilirubin levels were increased after BDL but unchanged in knockouts compared with wild-type controls (Figure 4E).
Figure 4.
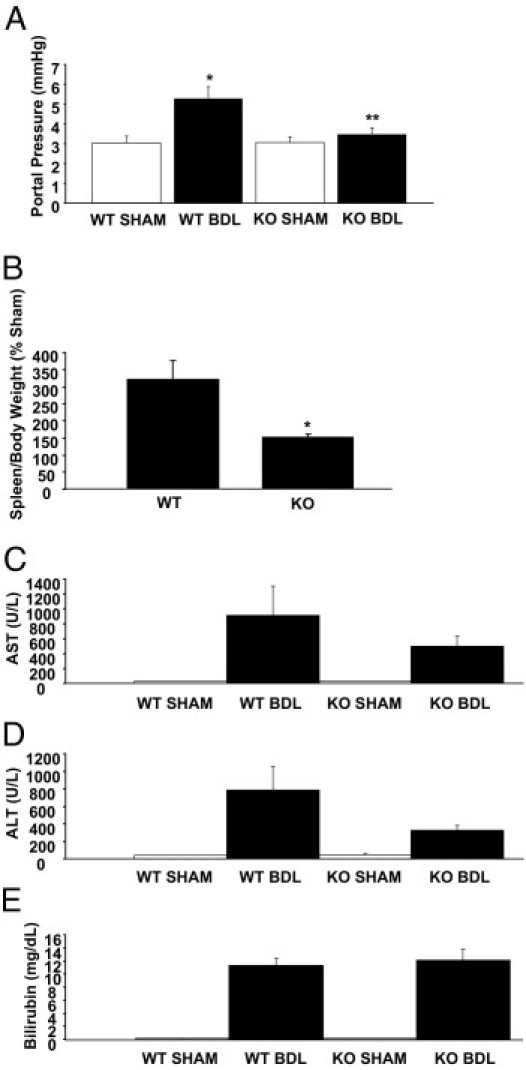
AQP1 knockout (KO) mice have reduced portal hypertension after BDL. A: Portal pressure was directly measured by portal vein cannulation in wild-type (WT) and AQP1 KO mice, with and without BDL. Quantification of the average portal pressure is shown (n = 6). Data are given as the mean ± SE. B: Spleen and body weight were measured in WT and AQP1 KO mice, with and without BDL. The average calculated spleen/body weight ratio of BDL animals normalized to the corresponding sham groups is shown (n = 6). Data are given as the mean ± SE. Serum was analyzed in a clinically verified reference laboratory to measure aspartate aminotransferase (AST) (C), alanine aminotransferase (ALT) (D), and total bilirubin (E) levels in WT and AQP1 KO mice, with and without BDL (n = 6). Data are given as the mean ± SE. *P < 0.05 versus WT; **P < 0.05 versus WT BDL.
AQP1 Protein Is Induced by Hypertonicity
Given the robust overexpression of AQP1 in human cirrhosis11–13 and in animal models and the functional consequences on liver angiogenesis, fibrosis, and portal pressure, we next sought to elucidate a molecular mechanism whereby AQP1 protein expression is increased during cirrhosis. We consistently saw that, although ECs isolated from cirrhotic mice expressed high levels of AQP1, this expression was rapidly lost in culture and that cells in long-term culture, such as the TSEC cell line, do not express AQP1 under normal culture conditions (see Supplemental Figure S5 at http://ajp.amjpathol.org). We anticipated that this could be the result of changes in the endothelial microenvironment in the context of cirrhosis. We explored two previously described AQP1 regulatory mechanisms, hypoxia and altered osmolality. Indeed, both have been linked to AQP expression19,26 and may be physiologically relevant in chronic liver disease.2,27,28 Therefore, we incubated TSECs, an immortalized cell line derived from sinusoidal endothelial cells,23 under normal conditions, under hypoxic conditions, and in the presence of varying external osmolalities (based on the sodium chloride content of the culture media). No significant induction of AQP1 was seen under hypoxic conditions (data not shown), but AQP1 protein levels were significantly induced by hyperosmolality (Figure 5A) and proportionally increased with increasing osmolality (Figure 5B). This finding was confirmed using IF staining, which showed an increasing number of cells staining positive for AQP1 as osmolality increased (Figure 5C). Higher-power images suggested that staining intensity within individual cells also increased in proportion to the osmolality (Figure 5C). We expected that the increase in AQP1 protein would be the result of transcriptional activation of the AQP1 gene. However, surprisingly, we could not detect any significant increase in AQP1 mRNA levels, as measured by quantitative RT-PCR (Figure 5D), suggesting a post-transcriptional regulatory mechanism, such as that conferred by miRs.
Figure 5.
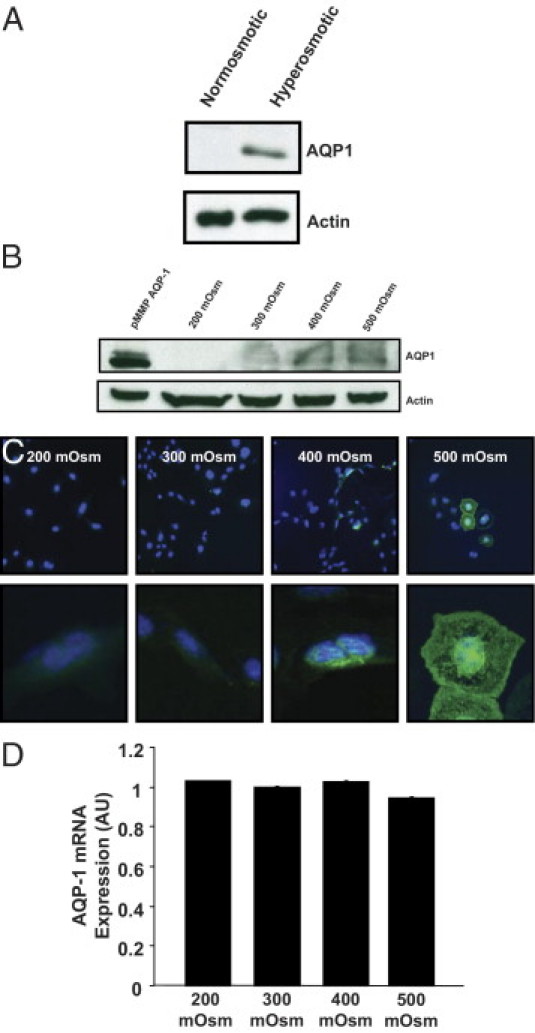
AQP1 protein is induced by hypertonicity. A: Representative immunoblots for AQP1 or actin (50 μg/lane) on lysates from TSECs exposed for 72 hours to normal osmolality (300 mosm) or hyperosmolality (600 mosm). B: Representative immunoblots for AQP1 or actin (100 μg/lane) on lysates from TSECs exposed for 72 hours to varying external osmolality (200 to 500 mosm). The far left lane is a positive control using the pMMP vector for retroviral overexpression of AQP1 in TSECs. C: Representative images of TSECs exposed for 72 hours to varying external osmolality (200 to 500 mosm) and stained using IF for AQP1 (green) and nuclear TOTO-3 (blue). Original magnification: ×20 (top); ×63 (bottom). D: Quantitative real-time RT-PCR was performed on RNA isolated from TSECs exposed for 72 hours to varying external osmolality (200 to 500 mosm). The average signal normalized to a housekeeping gene and relative to the control group is shown (n = 6). Data are given as the mean ± SE.
Osmotically Sensitive miRs Are Down-Regulated after BDL
To investigate whether post-transcriptional regulation of AQP1 by miRs may be occurring in the setting of cirrhosis, we initially used an miR array screening approach. We isolated total RNA (including miR) from endothelial cells derived from sham operated on or BDL mice and screened the relative expression levels of the entire mouse transcriptome of miRs, consisting of 610 distinct miRs. An in silico analysis using TargetScan and Microcosm software identified miRs with potential binding sites within the untranslated region of AQP1. These targets were then cross-referenced with the array data to identify potential regulators of AQP1 that were down-regulated in BDL compared with sham mice. This analysis yielded four miRs meeting these criteria (Table 1). We followed up these results using quantitative RT-PCR for three of the miRs to confirm the down-regulation (miR-574 could not be validated by this method because specific PCR primers for this miR are not available). We confirmed that the miRs were down-regulated in BDL compared with sham mice (miR-7a > miR-666 > miR-708; Figure 6A). Furthermore, in TSECs exposed to increasing osmolality, we found that two of the miRs (miR-666 > miR-708 but not miR-7a) were osmotically sensitive and down-regulated as osmolality increased (Figure 6B). Luciferase reporter assays assessed functional binding of miR-666 and miR-708 to the untranslated region of AQP1 (Figure 7); consistent with the PCR studies, miR-666 showed a larger and statistically significant effect size. In total, these data suggest a mechanism by which alterations in the local solute and water milieu may influence osmotically sensitive endothelial miR levels and, subsequently, AQP1 protein levels.
Table 1.
AQP1 Regulatory miRs Down-Regulated in BDL Endothelial Cells
| miR | Sham signal | BDL signal | Sequence |
|---|---|---|---|
| mmu-miR-574-5p | 6.565967 | 5.900687 | 5′-UGAGUGUGUGUGUGUGAGUGUGU-3′ |
| mmu-miR-7a | 4.270513 | 3.657709 | 5′-UGGAAGACUAGUGAUUUUGUUGU-3′ |
| mmu-miR-666-5p | 4.163507 | 3.991432 | 5′-AGCGGGCACAGCUGUGAGAGCC-3′ |
| mmu-miR-708 | 5.470477 | 5.236704 | 5′-AAGGAGCUUACAAUCUAGCUGGG-3′ |
Figure 6.
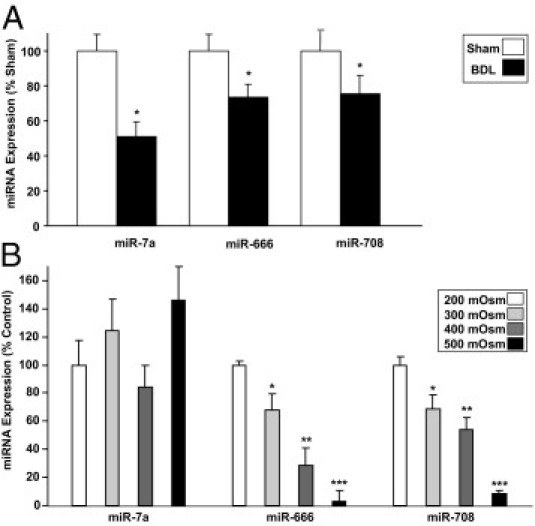
Osmotically sensitive miRs are down-regulated after BDL. A: Quantitative real-time RT-PCR was performed on total RNA (including miR) isolated from endothelial cells derived from wild-type sham or BDL mice. The signal normalized to a housekeeping RNA and relative to the control group is shown (n = 9). Data are given as the mean ± SE. B: Quantitative real-time RT-PCR was performed on total RNA (including miR) isolated from TSECs exposed for 72 hours to varying external osmolality (200 to 500 mosm). The average signal normalized to a housekeeping RNA and relative to the control group is shown (n = 9). Data are given as the mean ± SE. *P < 0.05 versus 200 mOsm. **P < 0.05 versus 300 mOsm. ***P < 0.05 versus 400 mOsm.
Figure 7.
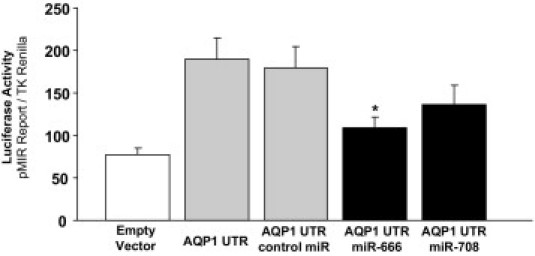
miR-666 and miR-708 functionally bind the UTR of AQP1. A reporter construct containing the potential binding sites for miR-666 and miR-708 in the UTR of AQP1 was generated. Chinese hamster ovary cells were transiently cotransfected for 24 hours with the reporter construct and mimics of miR-666 and/or miR-708. Luciferase activities were measured and normalized to the control TK Renilla luciferase level. Bars represent the mean ± SE from three independent experiments. *P < 0.05 versus cells transfected with the reporter construct only.
Discussion
Extensive dynamic changes in the hepatic vasculature occur in the setting of chronic liver disease and progression toward liver cirrhosis.29,30 The mechanisms by which pathological angiogenesis may promote fibrosis are not entirely clear, but it is tempting to speculate that targeting early vascular changes might afford an opportunity to intervene on chronic liver disease at a stage before irreversible fibrosis. Indeed, most strategies targeting angiogenic molecules have shown benefit in preclinical animal models of liver disease.11,31–34 In this context, our study extends the current knowledge of an emerging anti-angiogenic target, AQP1, by providing direct in vivo evidence that AQP1 regulates the angiogenesis, fibrosis, and portal hypertension that occurs after BDL; and defining a novel, molecular, fine-tuning mechanism involving osmotically sensitive miRs that may contribute to the pathological overexpression of AQP1 during cirrhosis.
We previously demonstrated that AQP1 is overexpressed in the angiogenic neovasculature within fibrotic septa in human cirrhosis and in CCl4-induced liver injury in C57 black mice.11 We also showed in vitro that AQP1 promotes angiogenic invasion and dynamic membrane protrusions. To more directly test the role of AQP1 in angiogenesis and fibrosis in vivo, we used mice from a CD1 background with global genetic deletion of AQP1. Concordant with the prior findings, we describe herein significant overexpression of AQP1 in liver after BDL. The signal was prominently localized to endothelia and, more important, there was complete absence of AQP1 expression seen in biliary epithelia in this strain of mice (in contrast to C57 black mice). This excludes potentially confounding effects of biliary AQP1 in a fibrosis model with prominent biliary proliferation, such as BDL.
Vascular remodeling was significantly inhibited in the AQP1 knockout animals, with a much smaller increase in the number of vWF-positive neovessels. This is consistent with reduced angiogenesis seen in these animals in a tumor implantation model,9 but it also extends this concept to a pathophysiologically relevant context in the liver. We also noted a prominent increase in endothelial cell invasion and proliferation after BDL that was absent in the knockout animals. A smaller, but statistically significant, effect of AQP1 overexpression on proliferation was also noted in two sinusoidal endothelial cell lines. The smaller effect size may reflect a high baseline in vitro proliferation rate in these cell lines.
Conflicting reports9,35–37 exist in the literature regarding the effect of AQPs on cell proliferation, possibly reflecting cell type–specific phenomena. Conceptually, however, a role for AQPs can be envisioned in the cell volume regulation required for mitosis38–40 and in the dramatic cell shape changes accompanying cytokinesis. Indeed, we see prominent localization of AQP1 to the cleavage furrow between daughter cells during cytokinesis (data not shown). A recently proposed poroelastic biophysical model of the cytoplasm allows rapid localized water transport to affect cellular shape changes on time scales relevant to these processes.41 Regardless, the considerable effect of AQP1 knockout in our model suggests a prominent role for this protein in the pathological angiogenesis that occurs during chronic liver disease.
Fibrosis, portal hypertension, and liver inflammation were also reduced after BDL in the knockout animals, suggesting that AQP1 may be a promising potential anti-fibrotic target. Although compounds, such as mercuric chloride, can inhibit AQP function in vitro,42 no clinically useful inhibitors of AQPs are known. There are, however, significant pharmacological efforts underway to develop such compounds.43–46 Understanding the mechanisms by which angiogenesis can promote progression of liver fibrosis is an area of ongoing research, but the field is moving toward a concept of an activated endothelial phenotype that occurs during chronic liver injury. This pathological phenotype is characterized by a transition from a normal, fenestrated, sinusoidal endothelium that lacks a basement membrane to a more capillarized and less compliant vascular system, with endothelial cells that are angiogenic, invasive, and proliferative and may serve as paracrine sources of inflammatory cytokines/chemokines. Indeed, we see alterations in the expression of inflammatory cytokines/chemokines in endothelial cells overexpressing AQP1, which could potentially serve as a paracrine source of stellate cell activation (see Supplemental Figure S6 at http://ajp.amjpathol.org). AQP1 seems to be a prominent marker of this type of activated endothelium in the liver and seems to contribute to several angiogenic features, including invasion and proliferation. Increased bulk angiogenesis may also provide a conduit for delivery of a variety of circulating inflammatory and fibrogenic cell types.
We have identified a mechanism by which increasing osmolality can induce the expression of AQP1 protein in liver endothelial cells. These findings are particularly interesting in light of the fact that patients with cirrhosis have avid sodium retention and, thus, have long-term sodium overload.28 However, ensuing water retention generally results in cirrhotic patients with low serum osmolality. The local solute concentration in the cirrhotic microenvironment is not known, but it seems clear that osmoregulation is generally an important variable to be considered in this group of patients. A similar phenomenon recently described in hyperosmotic regions of the kidney implicates transcriptional regulation of the AQP1 gene by the transcription factor TonEBP.19 However, surprisingly, in liver endothelial cells, we could not detect increased AQP1 mRNA after a hyperosmotic stress, which led us to consider post-transcriptional regulation via miRs, an emerging regulatory mechanism in liver.47–50 We show a subset of endothelial miRs that are down-regulated during cirrhosis and have potential to repress translation of AQP1 mRNA. Furthermore, a subset of those miRs appears to be regulated by changes in external osmolality. These results suggest a mechanism by which liver endothelial cells can sense and respond to local osmotic shifts during disease. Interestingly, several matrix molecules and inflammatory cytokines/chemokines are also potentially regulated by these miRs and could theoretically provide an additional and parallel mechanism for regulation of hepatic stellate cell activation and fibrosis. Furthermore, miR-666 exists within a region of the genome containing an unusually large cluster of miRs, suggesting that many other miRs could also potentially be induced by osmolality if driven by the same promoter elements/transcription factors. Of course, these studies do not exclude other parallel mechanisms of AQP1 regulation, such as altered rates of protein degradation.
In summary, the current study shows a dramatic in vivo effect of AQP1 knockout on the angiogenesis, fibrosis, and portal hypertension that follows BDL in mice, further validating this protein as a logical treatment target in chronic liver disease. Furthermore, our experiments on osmotically regulated miR expression suggest a novel mechanism contributing to the intense overexpression of AQP1 in cirrhotic endothelial cells, which may ultimately provide pathophysiological insight and additional points of therapeutic intervention at the molecular level.
Acknowledgments
We acknowledge Theresa Johnson for secretarial support.
Footnotes
Supported by the Loan Repayment Program (R.C.H.) and grants (DK24031 and P30DK084567 to N.F.L.; DK59615-06 and HL086990 to V.H.S.) from the NIH; and by the Mayo Foundation.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.045.
Contributor Information
Robert C. Huebert, Email: huebert.robert@mayo.edu.
Vijay H. Shah, Email: shah.vijay@mayo.edu.
Supplementary data
AQP1 colocalizes with vascular markers. Representative images of bile duct–ligated mouse liver tissue costained with IF for AQP1 (red; top) and CD31, vWF, VEGFR2, or eNOS (green; middle). Original magnification, ×10. Bottom: Merged images (yellow). High-power images are in Figure 1.
Characterization of isolated endothelial cells. Top: Representative images of isolated endothelial cells characterized using diI-labeled acetylated low-density lipoprotein (acLDL; red) or IF staining for vWF (green). Original magnification, ×10. Bottom: Corresponding bright-field images.
AQP1 increases endothelial cell angiogenesis in vitro. A: MTS proliferation assays were performed in TSECs or HHSECs after overexpressing LacZ, YFP, or AQP1 using a retroviral transduction system. Quantification of the average spectrophotometric signal per well is shown (n = 9 wells per condition). Data are given as the mean ± SE. *P < 0.05 versus control. B–E: Representative images of wild-type (WT) or AQP1 knockout (KO) mouse tissue, with or without BDL, stained for CD31 (green). Original magnification, ×10. F: Quantification of the number of CD31-positive lumens per high-power field (HPF) (n = 3 to 8 fields per slide, and n = 3 animals per group). Data are given as the mean ± SE. *P < 0.05 versus WT sham; **P < 0.05 versus WT BDL. AU, arbitrary unit.
AQP1 knockout (KO) mice have reduced fibrosis in response to BDL. A: Representative images of wild-type (WT) and AQP1 KO mice, with and without BDL, stained with IF for smooth muscle actin (SMA; green). Original magnification, ×7.5. Quantification of the average total fluorescence signal per high-power field (HPF) is shown (n = 10 fields per animal, and n = 3 animals per group). Data are given as the mean ± SE. B: Representative images of WT and AQP1 KO mice, with and without BDL, stained with H&E. Original magnification, ×10. C: Hydroxyproline assays were performed on liver tissue derived from WT and AQP1 KO mice, with and without BDL. The average spectrophotometric signal normalized to the control group is shown (n = 5). Data are given as the mean ± SE. D: Representative images of WT and AQP1 KO mice, with and without BDL, stained with Sirius red. The graphical result is in Figure 3. Original magnification, ×10. *P < 0.05 versus WT sham; **P < 0.05 versus WT BDL.
Endothelial AQP1 protein expression is lost in culture. Representative immunoblots for AQP1 or actin (50 μg/lane) on lysates from freshly isolated or cultured ECs.
Cells expressing AQP1 have increased cytokine expression. Gene expression patterns were compared with TSECs overexpressing AQP1 or a control gene (LacZ). mRNA expression profiles were assessed by real-time quantitative RT-PCR using the Endothelial Cell Biology Pathway Specific Quantitative PCR Array (SA Biosciences). A: Heat mapping represents the fold change in expression in TSEC-AQP1, compared with TSEC-LacZ. B: The scatterplot shows the number of genes with similar expression levels (within black lines), up-regulated (red) or down-regulated (green) in TSEC-AQP1.
References
- 1.Schuppan D., Afdhal N.H. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paternostro C., David E., Novo E., Parola M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J Gastroenterol. 2010;16:281–288. doi: 10.3748/wjg.v16.i3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwakiri Y., Grisham M., Shah V. Vascular biology and pathobiology of the liver: report of a single-topic symposium. Hepatology. 2008;47:1754–1763. doi: 10.1002/hep.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huebert R.C., Shah V. Hepatic Sinusoidal Endothelial Cells. In: Dufour J.F., Clavien P.A., editors. Springer-Verlag; Berlin, Heidelberg: 2009. pp. 79–91. [Google Scholar]
- 5.Huebert R.C., Splinter P.L., Garcia F., Marinelli R.A., LaRusso N.F. Expression and localization of aquaporin water channels in rat hepatocytes: evidence for a role in canalicular bile secretion. J Biol Chem. 2002;277:22710–22717. doi: 10.1074/jbc.M202394200. [DOI] [PubMed] [Google Scholar]
- 6.Ma T., Yang B., Gillespie A., Carlson E.J., Epstein C.J., Verkman A.S. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J Biol Chem. 1998;273:4296–4299. doi: 10.1074/jbc.273.8.4296. [DOI] [PubMed] [Google Scholar]
- 7.Liu X., Bandyopadhyay B.C., Nakamoto T., Singh B., Liedtke W., Melvin J.E., Ambudkar I. A role for AQP5 in activation of TRPV4 by hypotonicity: concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery. J Biol Chem. 2006;281:15485–15495. doi: 10.1074/jbc.M600549200. [DOI] [PubMed] [Google Scholar]
- 8.Li Y.H., Eto K., Horikawa S., Uchida S., Sasaki S., Li X.J., Noda Y. Aquaporin-2 regulates cell volume recovery via tropomyosin. Int J Biochem Cell Biol. 2009;41:2466–2476. doi: 10.1016/j.biocel.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Saadoun S., Papadopoulos M.C., Hara-Chikuma M., Verkman A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005;434:786–792. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]
- 10.La Porta C. AQP1 is not only a water channel: it contributes to cell migration through Lin7/beta-catenin. Cell Adh Migr. 2010;4:204–206. doi: 10.4161/cam.4.2.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huebert R.C., Vasdev M.M., Shergill U., Das A., Huang B.Q., Charlton M.R., LaRusso N.F., Shah V.H. Aquaporin-1 facilitates angiogenic invasion in the pathologic neovasculature that accompanies cirrhosis. Hepatology. 2010;52:238–248. doi: 10.1002/hep.23628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokomori H., Oda M., Yoshimura K., Watanabe S., Hibi T. Aberrant expressions of aquaporin-1 in association with capillarized sinusoidal endothelial cells in cirrhotic rat liver. Med Mol Morphol. 2010;43:6–12. doi: 10.1007/s00795-009-0475-6. [DOI] [PubMed] [Google Scholar]
- 13.Xian Z.H., Cong W.M., Wang Y.H., Wang B., Wu M.C. Expression and localization of aquaporin-1 in human cirrhotic liver. Pathol Res Pract. 2009;205:774–780. doi: 10.1016/j.prp.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Lee H.J., Palkovits M., Young W.S., 3rd miR-7b, a microRNA up-regulated in the hypothalamus after chronic hyperosmolar stimulation, inhibits Fos translation. Proc Natl Acad Sci U S A. 2006;103:15669–15674. doi: 10.1073/pnas.0605781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uney J.B., Lightman S.L. MicroRNAs and osmotic regulation. Proc Natl Acad Sci U S A. 2006;103:15278–15279. doi: 10.1073/pnas.0607655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang W., Liu H., Wang T., Zhang T., Kuang J., Luo Y., Chung S.S., Yuan L., Yang J.Y. Tonicity-responsive microRNAs contribute to the maximal induction of osmoregulatory transcription factor OREBP in response to high-NaCl hypertonicity. Nucleic Acids Res. 2011;39:475–485. doi: 10.1093/nar/gkq818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flynt A.S., Thatcher E.J., Burkewitz K., Li N., Liu Y., Patton J.G. miR-8 microRNAs regulate the response to osmotic stress in zebrafish embryos. J Cell Biol. 2009;185:115–127. doi: 10.1083/jcb.200807026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao B., Ge L., Liang R., Li W., Ruan K., Lin H., Jin Y. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol Biol. 2009;10:29. doi: 10.1186/1471-2199-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanaspa M.A., Andres-Hernando A., Li N., Rivard C.J., Cicerchi C., Roncal-Jimenez C., Schrier R.W., Berl T. The expression of aquaporin-1 in the medulla of the kidney is dependent on the transcription factor associated with hypertonicity, TonEBP. J Biol Chem. 2010;285:31694–31703. doi: 10.1074/jbc.M109.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semela D., Das A., Langer D.A., Kang N., Leof E., Shah V.H. Platelet-derived growth factor signaling through ephrin-B2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671–679. doi: 10.1053/j.gastro.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeCouter J., Moritz D.R., Li B., Phillips G.L., Liang X.H., Gerber H.P., Hillan K.J., Ferrara N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 22.Follenzi A., Benten D., Novikoff P., Faulkner L., Raut S., Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118:935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huebert R.C., Jagavelu K., Liebl A.F., Huang B.Q., Splinter P.L., LaRusso N.F., Urrutia R.A., Shah V.H. Immortalized liver endothelial cells: a cell culture model for studies of motility and angiogenesis. Lab Invest. 2010;90:1770–1781. doi: 10.1038/labinvest.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L., Chan C.C., Kwon O.S., Liu S., McGhee J., Stimpson S.A., Chen L.Z., Harrington W.W., Symonds W.T., Rockey D.C. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–G911. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 25.Zeidel M.L., Ambudkar S.V., Smith B.L., Agre P. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry. 1992;31:7436–7440. doi: 10.1021/bi00148a002. [DOI] [PubMed] [Google Scholar]
- 26.Wu Q.Q., Chen Y.C., Jiang X.F., Zeng Z. AQP1 expression in vascular endothelial cells in vitro under hypoxia conditions. Sichuan Da Xue Xue Bao Yi Xue Ban. 2008;39:916–920. [in Chinese] [PubMed] [Google Scholar]
- 27.Rosmorduc O., Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin Liver Dis. 2010;30:258–270. doi: 10.1055/s-0030-1255355. [DOI] [PubMed] [Google Scholar]
- 28.Oliver J.A., Verna E.C. Afferent mechanisms of sodium retention in cirrhosis and hepatorenal syndrome. Kidney Int. 2010;77:669–680. doi: 10.1038/ki.2010.4. [DOI] [PubMed] [Google Scholar]
- 29.Iwakiri Y., Groszmann R.J. Vascular endothelial dysfunction in cirrhosis. J Hepatol. 2007;46:927–934. doi: 10.1016/j.jhep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Hennenberg M., Trebicka J., Kohistani A.Z., Heller J., Sauerbruch T. Vascular hyporesponsiveness to angiotensin II in rats with CCl(4)-induced liver cirrhosis. Eur J Clin Invest. 2009;39:906–913. doi: 10.1111/j.1365-2362.2009.02181.x. [DOI] [PubMed] [Google Scholar]
- 31.Tugues S., Fernandez-Varo G., Munoz-Luque J., Ros J., Arroyo V., Rodes J., Friedman S.L., Carmeliet P., Jimenez W., Morales-Ruiz M. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46:1919–1926. doi: 10.1002/hep.21921. [DOI] [PubMed] [Google Scholar]
- 32.Jagavelu K., Routray C., Shergill U., O'Hara S.P., Faubion W., Shah V.H. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology. 2010;52:590–601. doi: 10.1002/hep.23739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spirli C., Okolicsanyi S., Fiorotto R., Fabris L., Cadamuro M., Lecchi S., Tian X., Somlo S., Strazzabosco M. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology. 2010;138 doi: 10.1053/j.gastro.2009.09.005. 360.e7-371.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patsenker E., Popov Y., Stickel F., Schneider V., Ledermann M., Sägesser H., Niedobitek G., Goodman S.L., Schuppan D. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50:1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shankardas J., Patil R.V., Vishwanatha J.K. Effect of down-regulation of aquaporins in human corneal endothelial and epithelial cell lines. Mol Vis. 2010;16:1538–1548. [PMC free article] [PubMed] [Google Scholar]
- 36.Kang S.K., Chae Y.K., Woo J., Kim M.S., Park J.C., Lee J., Soria J.C., Jang S.J., Sidransky D., Moon C. Role of human aquaporin 5 in colorectal carcinogenesis. Am J Pathol. 2008;173:518–525. doi: 10.2353/ajpath.2008.071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoque M.O., Soria J.C., Woo J., Lee T., Lee J., Jang S.J., Upadhyay S., Trink B., Monitto C., Desmaze C., Mao L., Sidransky D., Moon C. Aquaporin 1 is overexpressed in lung cancer and stimulates NIH-3T3 cell proliferation and anchorage-independent growth. Am J Pathol. 2006;168:1345–1353. doi: 10.2353/ajpath.2006.050596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boucrot E., Kirchhausen T. Mammalian cells change volume during mitosis. PLoS One. 2008;3:e1477. doi: 10.1371/journal.pone.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Habela C.W., Sontheimer H. Cytoplasmic volume condensation is an integral part of mitosis. Cell Cycle. 2007;6:1613–1620. doi: 10.4161/cc.6.13.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Habela C.W., Ernest N.J., Swindall A.F., Sontheimer H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J Neurophysiol. 2009;101:750–757. doi: 10.1152/jn.90840.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchison T.J., Charras G.T., Mahadevan L. Implications of a poroelastic cytoplasm for the dynamics of animal cell shape. Semin Cell Dev Biol. 2008;19:215–223. doi: 10.1016/j.semcdb.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castle N.A. Aquaporins as targets for drug discovery. Drug Discov Today. 2005;10:485–493. doi: 10.1016/S1359-6446(05)03390-8. [DOI] [PubMed] [Google Scholar]
- 43.Haddoub R., Rützler M., Robin A., Flitsch S.L. Design, synthesis and assaying of potential aquaporin inhibitors. Handb Exp Pharmacol. 2009;(190):385–402. doi: 10.1007/978-3-540-79885-9_19. [DOI] [PubMed] [Google Scholar]
- 44.Mola M.G., Nicchia G.P., Svelto M., Spray D.C., Frigeri A. Automated cell-based assay for screening of aquaporin inhibitors. Anal Chem. 2009;81:8219–8229. doi: 10.1021/ac901526k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yool A.J. Functional domains of aquaporin-1: keys to physiology, and targets for drug discovery. Curr Pharm Des. 2007;13:3212–3221. doi: 10.2174/138161207782341349. [DOI] [PubMed] [Google Scholar]
- 46.Monzani E., Shtil A.A., La Porta C.A. The water channels, new druggable targets to combat cancer cell survival, invasiveness and metastasis. Curr Drug Targets. 2007;8:1132–1137. doi: 10.2174/138945007782151342. [DOI] [PubMed] [Google Scholar]
- 47.Bala S., Marcos M., Szabo G. Emerging role of microRNAs in liver diseases. World J Gastroenterol. 2009;15:5633–5640. doi: 10.3748/wjg.15.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang X., Tsitsiou E., Herrick S.E., Lindsay M.A. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–2021. doi: 10.1111/j.1742-4658.2010.07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kren B.T., Wong P.Y., Shiota A., Zhang X., Zeng Y., Steer C.J. Polysome trafficking of transcripts and microRNAs in regenerating liver after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1181–G1192. doi: 10.1152/ajpgi.90636.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Venugopal S.K., Jiang J., Kim T.H., Li Y., Wang S.S., Torok N.J., Wu J., Zern M.A. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol. 2010;298:G101–G106. doi: 10.1152/ajpgi.00220.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AQP1 colocalizes with vascular markers. Representative images of bile duct–ligated mouse liver tissue costained with IF for AQP1 (red; top) and CD31, vWF, VEGFR2, or eNOS (green; middle). Original magnification, ×10. Bottom: Merged images (yellow). High-power images are in Figure 1.
Characterization of isolated endothelial cells. Top: Representative images of isolated endothelial cells characterized using diI-labeled acetylated low-density lipoprotein (acLDL; red) or IF staining for vWF (green). Original magnification, ×10. Bottom: Corresponding bright-field images.
AQP1 increases endothelial cell angiogenesis in vitro. A: MTS proliferation assays were performed in TSECs or HHSECs after overexpressing LacZ, YFP, or AQP1 using a retroviral transduction system. Quantification of the average spectrophotometric signal per well is shown (n = 9 wells per condition). Data are given as the mean ± SE. *P < 0.05 versus control. B–E: Representative images of wild-type (WT) or AQP1 knockout (KO) mouse tissue, with or without BDL, stained for CD31 (green). Original magnification, ×10. F: Quantification of the number of CD31-positive lumens per high-power field (HPF) (n = 3 to 8 fields per slide, and n = 3 animals per group). Data are given as the mean ± SE. *P < 0.05 versus WT sham; **P < 0.05 versus WT BDL. AU, arbitrary unit.
AQP1 knockout (KO) mice have reduced fibrosis in response to BDL. A: Representative images of wild-type (WT) and AQP1 KO mice, with and without BDL, stained with IF for smooth muscle actin (SMA; green). Original magnification, ×7.5. Quantification of the average total fluorescence signal per high-power field (HPF) is shown (n = 10 fields per animal, and n = 3 animals per group). Data are given as the mean ± SE. B: Representative images of WT and AQP1 KO mice, with and without BDL, stained with H&E. Original magnification, ×10. C: Hydroxyproline assays were performed on liver tissue derived from WT and AQP1 KO mice, with and without BDL. The average spectrophotometric signal normalized to the control group is shown (n = 5). Data are given as the mean ± SE. D: Representative images of WT and AQP1 KO mice, with and without BDL, stained with Sirius red. The graphical result is in Figure 3. Original magnification, ×10. *P < 0.05 versus WT sham; **P < 0.05 versus WT BDL.
Endothelial AQP1 protein expression is lost in culture. Representative immunoblots for AQP1 or actin (50 μg/lane) on lysates from freshly isolated or cultured ECs.
Cells expressing AQP1 have increased cytokine expression. Gene expression patterns were compared with TSECs overexpressing AQP1 or a control gene (LacZ). mRNA expression profiles were assessed by real-time quantitative RT-PCR using the Endothelial Cell Biology Pathway Specific Quantitative PCR Array (SA Biosciences). A: Heat mapping represents the fold change in expression in TSEC-AQP1, compared with TSEC-LacZ. B: The scatterplot shows the number of genes with similar expression levels (within black lines), up-regulated (red) or down-regulated (green) in TSEC-AQP1.