

Abstract
Neurofibrillary tangles (NFT), intracellular inclusions of abnormal fibrillar forms of microtubule associated protein tau, accumulate in Alzheimer's disease (AD) and other tauopathies and are believed to cause neuronal dysfunction, but the mechanism of tau-mediated toxicity are uncertain. Tau overexpression in cell culture impairs localization and trafficking of organelles. Here we tested the hypothesis that, in the intact brain, changes in mitochondrial distribution occur secondary to pathological changes in tau. Array tomography, a high-resolution imaging technique, was used to examine mitochondria in the reversible transgenic (rTg)4510, a regulatable transgenic, mouse model and AD brain tissue. Mitochondrial distribution is progressively disrupted with age in rTg4510 brain, particularly in somata and neurites containing Alz50-positive tau aggregates. Suppression of soluble tau expression with doxycycline resulted in complete recovery of mitochondrial distribution, despite the continued presence of aggregated tau. The effect on mitochondrial distribution occurs without concomitant alterations in neuropil mitochondrial size, as assessed by both array tomography and electron microscopy. Similar mitochondrial localization alterations were also observed in human AD tissue in Alz50+ neurons, confirming the relevance of tau to mitochondrial trafficking observed in this animal model. Because abnormalities reverted to normal if soluble tau was suppressed in rTg4510 mice, even in the continued presence of fibrillar tau inclusions, we suggest that soluble tau plays an important role in mitochondrial abnormalities, which likely contribute to neuronal dysfunction in AD.
Tau, a microtubule associated protein, is the major constituent of neurofibrillary tangles (NFT). These deposits are the most common neuronal inclusions in Alzheimer's disease (AD).1,2 As a result of their correlation with synapse and neuronal loss and severity and duration of dementia in AD,3–6 NFT have been thought to indicate neural dysfunction and impending neuronal death. Recent studies oppose this view and suggest that other pathological changes in tau, such as mislocalization to the somatodendritic compartment, hyperphosphorylation, and conformational changes, may be more detrimental to neurons than tangles. Animal models have demonstrated dissociations between NFT, neuronal dysfunction,7 and neuronal loss.8–12 Meanwhile, tau overexpression, which results in abnormally high levels of soluble tau throughout the cell in the absence of aggregates, alters fast axonal transport, particularly in the anterograde or kinesin-dependent direction.13–18 This transport deficit is thought to lead to depletion of necessary materials, deterioration of the synapse, and a “dying back” of the neuron.19–21 We previously found in cultured neurons that alterations in axonal transport due to soluble tau over-expression resulted in perinuclear clumping of mitochondria.18 Disruptions in mitochondrial distribution, morphology and function have been linked to several diseases and implicated as early pathogenic steps in neurodegenerative processes.22–28
Studies of axonal transport and mitochondrial distribution have largely been limited to cell culture models, in which the effects of soluble tau species versus NFT have not been determined. In the current study, we tested the hypothesis that mitochondrial trafficking defects would occur in vivo as a consequence of tau over-expression and/or its mislocalization and aggregation, and also asked whether defects in mitochondrial trafficking could be detected in human AD. Evidence of an imbalance of mitochondrial fission and fusion processes has been observed in Alzheimer's brain,26–31 which could contribute to mitochondrial dysfunction and changes in mitochondrial distribution. We investigated somatic and neuritic mitochondrial distribution and mitochondrial volumes in neurons and neuropil of rTg4510 mice, which over-express a human mutant form of tau (P301L) known to lead to dementia. The rTg4510 mice demonstrate age-related cognitive impairment, accumulation of NFT and neuronal loss but also harbor a doxycycline-regulatory domain that can be used as an “off-switch” for this mutant tau over-expression. Doxycycline treatment results in stabilization of neuronal number and recovery of cognitive function even in the face of continued accumulation of NFT.9,10
Since mitochondria are quite small, on the order of the limits of light microscopy, we used a recently developed high-resolution microscopy technique, array tomography, for precise localization of mitochondria as well as accompanying mitochondrial volume quantification.32,33 Array tomography overcomes the approximate 1 μm z-resolution limitation of conventional confocal, and multiphoton microscopy, which is larger than the width of a single mitochondrion, by ultrathin sectioning of samples into ribbons of 70 nm sections followed by immunofluorescence imaging and three-dimensional reconstructions of structures of interest. This allows precise quantification of the number, volume, and protein labeling of small structures including mitochondria in a high-throughput automated fashion with thousands of mitochondria imaged per case. Electron micrographs were used to confirm mitochondrial volume data derived from the array tomography method, which validates the use of this automated technique for volume measurements. We also applied array tomography to human brain tissue to observe mitochondrial distribution changes in AD, highlighting the efficacy of this technique for human pathological analyses.
Our data demonstrate that early in the course of disease, mitochondrial distribution is altered, particularly in those cells or neurites bearing aggregates of tau. These patterns persist and become more severe with age. Doxycycline treatment of a subset of rTg4510 (regulatable transgenic) mice remarkably restored mitochondrial distribution to near normal, even in the continued presence of aggregated misfolded tau. Interestingly, these distribution changes were not accompanied by alterations in mitochondrial volume in the neuropil. Mitochondrial distribution in human AD brain demonstrated patterns that mirrored those seen in rTg4510 mice. Taken together, our findings indicate that mitochondrial distribution changes occur in vivo as a consequence of tau over-expression, and may be predominantly due to soluble tau species.
Materials and Methods
Animals
For this study we used a well-characterized, regulatable mouse model of tauopathy that over-expresses human mutant (P301L) tau that can be suppressed with doxycycline (dox) treatment. Mice were generated as previously described.9,10 The responder transgene contains cDNA of human four-repeat tau with the P301L mutation downstream of a tetracycline-operon-responsive element (TRE). The activator transgene consists of a tet-off open reading frame downstream of Ca2+ calmodulin kinase II promoter elements. This bigenic system results in over-expression of human mutant tau in forebrain structures when both promoter and activator are present. Littermate animals with only the activator transgene, which don't over-express tau were used as controls. Two age groups, 5.5 and 8.5 month old, of tau over-expressing (rTg4510) and control animals (nonTg) were used (n = 4 per group). Doxycycline was administered at 200ppm in the food for 6 weeks between 7 and 8.5 months of age in a subset of rTg4510 and nonTg animals (n = 4 per group), leading to suppression of tau as previously described.9 Animals were housed and treated in accordance with institutional guidelines and those of the National Institutes of Health.
Human Brain Tissue
Tissue from the superior temporal gyrus of subjects with either an AD diagnosis or no cognitive impairment was obtained from the Alzheimer Disease Research Center at Massachusetts General Hospital. All human tissue was handled in agreement with local and national IRB guidelines. Ten AD cases (61–90 years of age) and four cognitively normal controls (62–75 years of age) were included in this study (Table 1).
Table 1.
AD Diagnoses in 10 Patients
| Case | Age | Sex | Diagnosis |
|---|---|---|---|
| 1 | 75 | M | No cognitive impairment (prior stroke in contralateral hemisphere) |
| 2 | 66 | M | No cognitive impairment |
| 3 | 69 | M | No cognitive impairment |
| 4 | 62 | M | No cognitive impairment |
| 5 | 89 | M | AD; Braak stage VI/VI |
| 6 | 61 | M | AD; Braak stage V/VI |
| 7 | 77 | M | AD; Braak stage V/VI |
| 8 | 74 | F | AD; Braak stage VI/VI |
| 9 | 90 | M | AD; Braak stage IV/VI |
| 10 | 80 | F | AD; Braak stage VI/VI |
| 11 | 83 | F | AD; Braak stage VI/VI |
| 12 | 84 | M | AD; Braak stage V/VI |
| 13 | 81 | M | AD; Braak stage VI/VI |
| 14 | 84 | F | AD; Braak stage VI/VI |
Braak staging defines pathological progression of AD in six stages (I-VI), with VI being the most severe.
AD, Alzheimer's disease; F, female; M, male.
Sample Preparation
Tissue was prepared for array tomography as previously described.32–34 Brains were removed from mice immediately after euthanasia by CO2 inhalation. Human tissue samples were collected within 24 hours of autopsy. Small blocks (∼1 mm3) of primary somatosensory cortex from mice or temporal cortex from human cases were fixed by immersion in 4% paraformaldehyde and 2.5% sucrose in phosphate-buffered saline solution (PBS) for 3 hours at room temperature. Tissue was then dehydrated through a graded series of ethanols, and into LRWhite resin (Electron Microscopy Sciences, Hatfield PA), embedded in gelatin capsules with LRWhite and polymerized at 53C for 24 hours. Blocks were then removed from gelatin capsules and cut into ribbons of 7 to 150 ultra-thin 70 nm sections with a Jumbo Histo Diamond Knife (Diatome, Hatfield PA) and mounted on gel-subbed coverslips (Fisher Scientific, Pittsburgh PA; 12-544-E; No. 1.5; 0.16- to 0.19-μm thick).
Immunohistochemistry and Microscopy
Immunostaining for analysis of mitochondrial localization was performed as follows for both mouse and human cases. Ribbons were washed in 50 mmol/L glycine in Tris-buffered saline (TBS) and blocked in 0.05% Tween and 0.1% BSA in TBS. Primary antibodies mouse IgM Alz50 (a generous gift of Peter Davies, Albert Einstein College of Medicine), mouse IgG anti-tubulin (Sigma, St. Louis MO), and rabbit anti-VDAC/porin (Abcam, Cambridge MA) were diluted 1:50 in block buffer and applied to ribbons for 2 hours then rinsed off with TBS. Fluorescent secondary antibodies, donkey anti-mouse IgM Cy3, donkey anti-mouse IgG Alexa-Fluor 488, and donkey anti-rabbit Cy5 (Jackson ImmunoResearch, Westgrove PA) were diluted 1:100 in block buffer and incubated on ribbons for 30 minutes. 1024 × 1024 pixel images of regions of interest (cell bodies or neurites) within a single 70 nm section were acquired with a Leica DMRE confocal microscope (Wetzlar, Germany) and a 63 × 1.4 numerical aperture Plan Apochromatic oil objective.
For analysis of mitochondrial volume, ribbons of at least 10 ultrathin sections were stained as described above with rabbit anti-VDAC/porin (Abcam) at 1:200 followed by fluorescent secondary donkey anti-rabbit Cy3 at 1:100. Areas of interest, containing cell bodies (used as fiduciary markers) were imaged on 10 to 12 serial sections. Images of 1024 × 1024 pixels were collected with a Zeiss Axioplan LSM510 confocal/multiphoton microscope (Ziess, Thornwood, NY) with a 63 × 1.2 numerical aperture Plan Apochromatic water objective.
To confirm that astrocytic processes were not included in analyses, we performed two array immunostains of rTg4510 mouse tissue as described above but using the following antibodies. The first included mouse IgM Alz50 (1:100), mouse IgG anti-tubulin (1:300), rabbit anti-glial fibrillary acidic protein (1:300; Sigma), and secondaries goat anti-rabbit Cy3, goat anti-mouse IgG Alexa-Fluor 488, goat anti-mouse IgM Cy5 (1:100) and DAPI (Invitrogen, Eugene OR). The second included mouse IgM Alz50 (1:100), rat anti-tubulin (1:100; Abcam), mouse anti-glutamine synthetase (1:50; Millipore, Billerica MA) and secondaries goat anti-mouse IgG Alexa-Fluor 488 (1:50), goat anti-rat Cy3 and goat anti-mouse IgM Cy5 (1:100) and DAPI. Single section images of 1024 × 1024 pixels were collected with a Zeiss Axioplan LSM510 confocal/multiphoton microscope with a 63 × 1.4 numerical aperture Plan Apochromatic oil DIC objective.
For whole cell reconstruction by array tomography a ribbon of 150 80 nm sections was stained as described previously with rabbit anti-VDAC/porin (1:300), mouse IgG anti-tubulin (1:300), and mouse IgM Alz50 (1:100) followed by goat anti-rabbit Alexa Fluor 488 (1:100), chicken anti-mouse IgG Alexa-Fluor 647 (1:300), goat anti-mouse IgM Cy3 (1:100) and DAPI. A region of interest (1024 × 1024 pixels, zoom 3) containing the entirety of an Alz50+ and neighboring Alz50− cell was serially collected from each of 109 sections with a Zeiss Axioplan LSM510 confocal/multiphoton microscope and a 63 × 1.4 numerical aperture Plan Apochromatic oil DIC objective.
Electron Microscopy
Electron micrographs of two 8.5-month-old rTg4510 and one age-matched nonTg were acquired as previously described.35 In short, mice were anesthetized with an intraperitoneal injection of sodium pentobarbital and transcardially perfused with fixative solution containing 1% paraformaldehyde and 1.25% glutaraldehyde in 0.1M cacodylate buffer (pH 7.2–7.4) at 37°C. Following perfusion, the heads of the mice, with calvaria removed, were submerged in a solution of 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1M cacodylate buffer and kept at 4°C for 12 hours. One-millimeter thick coronal slices of the dorsal premotor cortex were obtained and divided into smaller pieces for embedding. Tissue was rinsed in 0.1M sodium cacodylate buffer, osmicated with 1% osmium tetroxide in cacodylate buffer, dehydrated through ascending ethyl alcohol concentrations, rinsed with propylene oxide and immersed in 1:1 propylene oxide and Araldite 502 plastic (Ernest F. Fullam, Inc, Redding CA) overnight. Cortical pieces were placed in pure Araldite, rotated for 6 hours, transferred to Beem capsules and hardened at 60°C. An RMC MT6000-XL ultramicrotome (Boeckeler, Tuscon AZ) was used to cut thin sections, which were mounted on copper grids then stained with uranyl acetate and lead citrate and photographed using a JEOL 100S electron microscope (JEOL, USA, Peabody MA). Negatives were scanned at 800 dpi with an Epson Perfection V700 photo scanner.
Image Analysis
Images were viewed and analyzed with Image J (National Institutes of Health open software; http://rsbweb.nih.gov/ij). Analysis of mitochondrial localization was performed as previously described.18 Approximately 50 cells and 40 neurites were imaged for each animal or human case. The mitochondrial channel of each image was opened and re-named for blinded thresholding. Once thresholds were determined, the tubulin channel was used to identify and circle the somatic or neuritic cytoplasm (excluding the nucleus). This region of interest (ROI) was then applied to the thresholded mitochondrial image and the ‘Analyze Particles’ feature of Image J applied to determine percentage of the ROI occupied by mitochondria. For each cell or neurite analyzed, presence or absence of Alz50 staining was also determined. This percent area occupied by mitochondria in the soma and neurites is referred to as mitochondrial distribution and is used as a readout for the ability of mitochondria to be trafficked to all parts of the cell body and neurites as in our previous study, which showed changes in mitochondrial distribution in the soma and axon were associated with reduced anterograde trafficking of mitochondria and fewer mitochondria reaching the periphery of the cell body (resulting in perinuclear clumping) and fewer reaching the axon.18
In the whole cell reconstruction, the tubulin images were opened sequentially and converted to a stack. An ROI for an Alz50+ and an Alz50− cell was defined in each image. These ROIs were then applied to the corresponding, individually thresholded mitochondrial channel image and the ‘Analyze Particles’ feature of Image J applied to determine percentage of the ROI occupied by mitochondria. The area of each ROI was also measured. These outputs were used to calculate a percent volume fraction occupied by mitochondria in an Alz50+ and an Alz50− cell (sum of total area occupied by mitochondria in each section multiplied by 0.08 divided by the sum of total area of the ROIs multiplied by 0.08).
For analysis of mitochondrial volume,32 images from each ribbon were opened sequentially, converted to a stack and aligned with the MultiStackReg and StackReg plugins (courtesy of B. Busse and36). Crop boxes (10.01 μm2) of neuropil were selected so as to exclude neuronal cell bodies or other obscuring features, realigned and re-cropped to exclude empty space created by realignment. Crops were thresholded and run through an automated, threshold based detection program that counts puncta appearing in more than one consecutive section and reports dimensions of each (WaterShed program provided by B. Busse, S. Smith, and K. Micheva, Stanford University). Two cortical blocks from the somatosensory cortex from each 8.5-month-old rTg4510 and control mouse (n = 4 per group) were imaged, yielding 45 crop boxes.
To confirm the accuracy of the threshold based detection program, mitochondrial volumes were confirmed by electron microscopy. All mitochondria in two images for each of the three animals, accounting for greater than 600 mitochondria, were measured at their widest and longest points with the line-measuring tool of Image J after setting the scale (157.48 pix/μm for 5,000x magnification and 125.98 pix/μm for 4,000x magnification). Estimates of the ellipsoidal volume of the mitochondria were calculated with the following formula 4/3π x r1 × r2 × r3, with r1 and r2 each representing ½ the length and width, respectively, and r3 estimated as ½ the shorter of the two measurements.
Also in these electron micrographs, the cytoplasm of each cell was defined as an ROI and its area measured. The sum of the areas of each of the mitochondria measured within this ROI was used to calculate an approximation of the mitochondrial percent area fraction in NFT+ and NFT− cells in control and rTg4510 mice (n = 2 NFT+, 4 NFT−).
Statistics
For mitochondrial distribution analysis, area fraction of cytoplasm occupied by mitochondria was recorded for each soma or neurite and grouped by age, genotype, treatment and whether or not aggregated tau was present (Alz50+/−). Automated mitochondrial volume output were recorded and grouped by genotype. Mitochondrial volume distribution was then compared to the measurements acquired by electron microscopy. Normality of data was assessed with a Shapiro-Wilks test. Mann-Whitney U-tests were applied to non-normal data (mitochondrial distribution) and student's t-test to normally distributed data (mitochondrial volumes). Significance was determined as P < 0.05. Non-normally distributed data are presented as box plots, which display the median value (line inside the box), upper quartile (top of the box), lower quartile (bottom of the box), 90th percentile (top whisker), 10th percentile (bottom whisker), with all values below the 10th and above the 90th percentile (potential outliers) shown as dots. Normal data are shown as bar graphs of the mean with SD.
Results
Age-Dependent Disruption of Mitochondrial Distribution with Aggregated Tau
We used array tomography32–34 on the rTg4510 mouse brain9,10 to determine somatic and neuritic mitochondrial distribution. This can be used as an indirect assessment of neuronal transport18 in the presence of over-expressed human mutant tau. Array tomography uses ultrathin sections to provide a physical z-axis resolution that is >10 times greater than that obtained with standard or even confocal light microscopy techniques. Reconstructions of whole cells (Figure 1; see also Supplemental Video S1 at http://ajp.amjpathol.org) show mitochondrial distribution throughout the somatic cytoplasm of neurons without pathological tau accumulation (Alz50−) which appears reduced in neurons with tau pathology (Alz50+). The 3D reconstructions using array tomography on over 100 serial sections require extensive sectioning and imaging time, thus we compared the volume fraction of mitochondria in whole cell 3D volumes with the area fraction of mitochondria in single 70 nm array sections and found them to be almost identical. The percent volume fraction calculated for both the Alz50+ and Alz50− cells were within 2% of those generated by single section analyses. This confirms the use of single section array tomography as a valuable technique for quickly acquiring large quantities of precise data on mitochondrial distribution.
Figure 1.
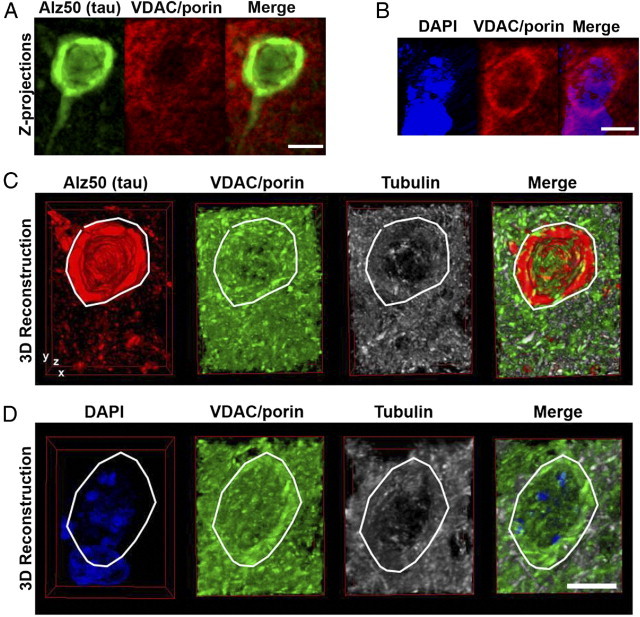
Three-dimensional analysis of mitochondrial content of neurons using array tomography. Z-stack projections of 99 sequential 80-nm array tomography images of an Alz50+ cell (A) and a nearby Alz50− cell (B) from rTg4510 brain exhibits the perinuclear localization of mitochondria (VDAC/porin) in Alz50− cells which appears reduced in Alz50+ cells. Panels C and D show partial 3-dimensional reconstructions through the same cells, the cytoplasm of which are encircled by the white lines. Scale bars = 5 μm.
Using single sections from array tomograms, neurons of the somatosensory cortex of 5.5 month-old rTg4510 mice were investigated as a model of early disease, as at this age, this region has not suffered significant neuronal loss although some NFT are present.10 Percent area fraction of the cytoplasm of soma and neurites occupied by mitochondria was compared across neurons with pathological tau accumulation (Alz50+ cell bodies and neuropil threads) and those without tau pathology (Alz50−) and cell bodies and neurites in wild type littermate controls. A decrease in the percentage of the cytoplasm occupied by mitochondria was interpreted as a neuronal transport deficit resulting in perinuclear clustering of mitochondria and reduction in mitochondrial content in neurites.18 As shown in Figure 2A and C, Alz50+ cell bodies demonstrate significant (*P < 0.05) reduction in cytoplasmic area-fraction occupied by mitochondria in comparison to neurons of nonTg animals. A trend, which was statistically not significant, toward a reduction in Alz50+ cells in comparison to Alz50− neurons within rTg4510 cortex was also seen at this age (P = 0.09). These data suggest that pathological accumulation of tau influences mitochondrial distribution in the cell body. These findings support the suggestion that mitochondrial distribution alterations are an early pathogenic step in neurodegenerative processes.24,28
Figure 2.
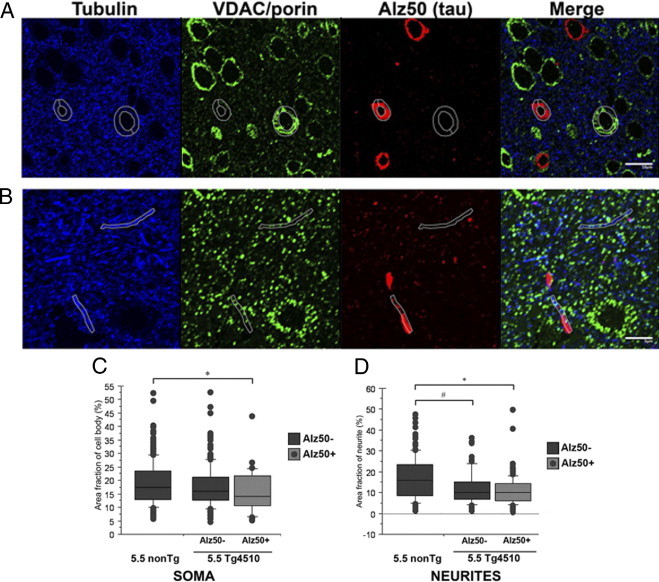
Mitochondrial distribution is disrupted in 5.5-month-old rTg4510 neurons. Array tomography images of single 70-nm sections from layer II/III of somatosensory cortex in rTg4510 were used to analyze percent area occupied by mitochondria (stained with VDAC/porin – green) within the soma (A) and neurites (B) (identified with tubulin staining – white outlines). Scale bars: 10 μm (A); 5 μm (B). Quantification reveals that soma with Alz50+ tau accumulations (red) have depleted mitochondrial fraction (C) and neurites both with and without pathological tau accumulations have altered mitochondrial distribution (D). *P < 0.0001, **P < 0.05.
Analysis of mitochondrial distribution in neurites shows that regardless of whether or not aggregated tau was present, neurites in rTg4510 cortex were significantly depleted of mitochondria (Figure 2, B and D). Alz50− neurites exhibited significantly reduced neurite area fraction occupied by mitochondria when compared to neurites of nonTg animals (P < 0.05). Neuropil thread containing (Alz50+) neurites also displayed a significant decrease in mitochondrial content (P < 0.0001). Within the 5.5 month-old rTg4510 neurons, the Alz50− and Alz50+ neurites did not significantly differ from one another in terms of mitochondrial distribution. Since mitochondrial mislocalization is evident even in the absence of aggregated tau, these data indicate that though aggregated tau may influence mitochondrial localization, hyperphosphorylated or other pathological forms of tau also contribute to such deficits.
Analysis of mitochondrial distribution within Alz50+ and Alz50− cell bodies of rTg4510 mice at 8.5 months of age (Figure 3A), when pathology is more advanced, shows that Alz50+ neurons have a worsened phenotype of mitochondrial distribution deficits than present at 5.5 months (P < 0.05), with a greatly reduced cytoplasmic area fraction occupied by mitochondria when compared to nonTg neurons (P < 0.0001). Meanwhile, Alz50− cell bodies continued to maintain mitochondrial distribution similar to that of control animals.
Figure 3.
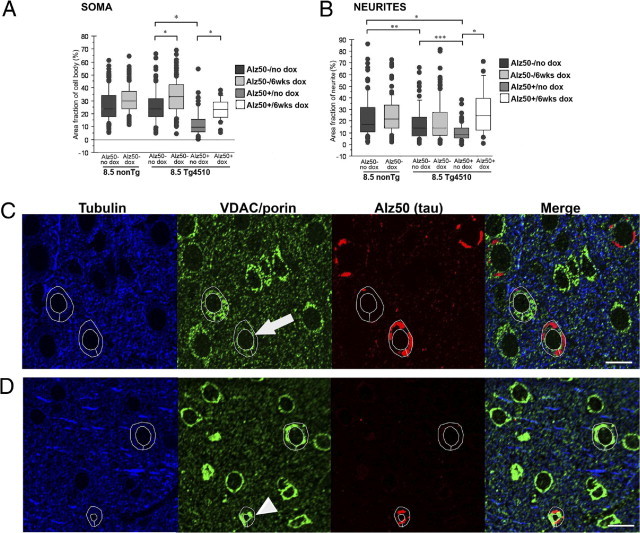
rTg4510 neurons at 8.5 months of age have disrupted mitochondrial distribution, which can be ameliorated with transgene suppression. Box and whisker plots demonstrate percent cytoplasm occupied by mitochondria in soma (A) and neurites (B) of both untreated and doxycycline-treated 8.5-month-old rTg4510 and nonTg mice. As with the younger age group, neurites in the rTg4510, regardless of whether or not tau aggregates are present, are depleted of mitochondrial content whereas somatic mitochondrial distribution is only impacted with the presence of aggregated tau. In addition, 6 weeks of doxycycline treatment allows for recovery of mitochondrial distribution to near that of nonTg levels in both the somata and neurites. Representative array tomography images of mitochondrial distribution in single 70-nm sections of untreated 8.5-month-old rTg4510 (C) and doxycycline-treated 8.5-month-old rTg4510 (D) exhibit depletion of somatic mitochondrial content in Alz50+ cells, indicated by the arrow in C, whereas the arrowhead in D indicates recovery to near control levels following 6 weeks of doxycycline treatment. *P < 0.0001, **P < 0.005, and ***P < 0.0001. Scale bars = 10 μm.
In accord with the data from the 5.5 month-old age group, mitochondria were significantly reduced in neurites, with or without the concomitant presence of aggregated tau (Figure 3B). Alz50+ neurites were the most severely affected, demonstrating significantly depleted mitochondrial content in comparison to both nonTg neurites (P < 0.0001), and Alz50− neurites (P < 0.05). Alz50−, tubulin-stained neurites of rTg4510 also exhibited aberrant mitochondrial localization with decreased area fraction occupied by mitochondria when compared to neurites of nonTg animals (P < 0.05); this result must be interpreted in the context that it is not known if some of these Alz50− neurites in rTg4510 mice would be contiguous with a section of neurite that would be Alz50+ outside of the block examined. Nonetheless, the more severe effect evident in Alz50+ neurites and soma indicates that either tau aggregates are present in cells with very high levels of soluble tau, which impairs mitochondrial distribution, or that aggregates themselves block transport. To distinguish between these possibilities, we treated animals with doxycycline to reduce soluble tau levels by suppressing its production. Aggregated tau persists with several weeks of transgene suppression, allowing the examination of mitochondrial distribution in structures with aggregated tau in the context of greatly reduced soluble tau levels.
To ensure that neurite analyses excluded astrocytic processes, we performed two additional array analyses for astrocytes (see Supplemental Figure S1 at http://ajp.amjpathol.org). The pattern of staining with tubulin, which was used to outline neurites and neuronal cell bodies for mitochondrial analysis, was completely different from glial fibrillary acidic protein or glutamine synthetase, both of which are astrocyte markers. Since the glial stains and tubulin never overlap, we are confident that the somata and neurites analyzed are neuronal.
Suppression of Soluble Mutant Tau Overexpression Ameliorates Aberrant Mitochondrial Distribution Patterns
The bigenic ‘tet-off’ rTg4510 mouse model allows for suppression of soluble tau over-expression by treatment with doxycycline (dox). Previous studies have shown that dox treatment for six weeks can ameliorate cognitive deficits and halt neuronal loss and reduce tau expression by approximately 80% without removing existing aggregates.9 We treated a subset of rTg4510 (n = 4) and nonTg (n = 4) animals with dox between 7 and 8.5 months of age, then analyzed somatic and neuritic mitochondrial distribution as previously described. As shown in Figure 3, dox treatment resulted in redistribution of mitochondrial content throughout somatic and neuritic cytoplasm, even with the continued presence of Alz50+ aggregates and neuropil threads. Percent cytoplasmic area fraction occupied by mitochondria within the soma of Alz50+ cells increased by 139% (P < 0.0001), to a level not significantly different from cells of nonTg. Non-tangle bearing neurons of the rTg4510 also exhibited a dramatic increase of ∼40% in cytoplasmic mitochondrial content following dox treatment, thereby significantly surpassing mitochondrial distribution levels seen even in nonTg cells (P < 0.0001). Dox treatment also increased area fraction of cytoplasm occupied by mitochondria within nonTg cells by 24% (P < 0.0001), suggesting that doxycycline may have additional effects on mitochondrial distribution independent of tau suppression. Since the dox-related increase evident in nonTg cells was substantially smaller than the effect seen in rTg4510 animals and similar dox effects were not seen in other experiments to be discussed below, we interpret our results as demonstrating significant redistribution of mitochondrial content within the cell body due to tau suppression rather than as non-specific effects of dox treatment. These data suggest that, although presence of Alz50+ aggregates may coincide with mitochondrial distribution deficits, reduction of the soluble tau species improves this condition, indicating soluble tau as more influential than aggregated tau on patterns of mitochondrial localization.
Mitochondrial distribution within neurites of the rTg4510 was similarly affected by dox treatment. Even with continued presence of neuropil threads, the cytoplasmic area occupied by mitochondria increased from 8.4% to 25.45%, in Alz50+ neurites, representing a dramatic increase of 303% (P < 0.0001). This striking increase in mitochondrial content results in Alz50+ neurites no longer being significantly different from neurites of nonTg wild type mice. In contrast, both non-neuropil thread containing neurites (Alz50−) in the rTg4510 and neurites in the wild type mice demonstrated a nonsignificant change in mitochondrial content following doxycycline, suggesting that non-specific dox effects do not play a critical role in the dramatic mitochondrial localization changes evident in soma and neurites of Alz50+ cells. These data implicate both soluble tau over-expression and aggregated tau as influential in mitochondrial localization. Furthermore, these data suggest that the presence of aggregated tau may not exert its effects on mitochondrial distribution by existing as a space occupying lesion, since mitochondria are able to redistribute subsequent to suppression of soluble tau, even in the continued presence of aggregated tau deposits.
Mitochondrial Volume Is Maintained Although Distribution Alterations Occur
In addition to being indicative of neuronal transport deficits, changes in mitochondrial distribution have been linked to disruptions in fission and fusion dynamics thereby influencing mitochondrial size and number.26–29,37 To assess whether global changes in fission/fusion dynamics accompany the evident transport deficits, we measured mitochondrial volumes in the neuropil in the 8.5 month-old rTg4510 and nonTg mice by array tomography. Nearly 4000 mitochondria were measured and demonstrated no significant difference in volume between rTg4510 and nonTg mice (Figure 4). To further evaluate this observation, obtained using array tomography-derived reconstructions of mitochondria, we compared the output from array tomography to estimations of volumes from measurements of mitochondrial profiles in electron microscopy. The volumes reported from the array tomography method were consistent with those measured in electron microscopy (Figure 4). In addition, mitochondria measured within the cytoplasm of the cells present in the electron micrographs (total of 6 cells) were summed to generate an approximation of a percent area fraction for Alz50+ and Alz50− cells of rTg4510 and nonTg mice. Though absolute values of these measurements were lower than those obtained by array tomography (presumably due to overlapping fluorescence of adjacent, clumping mitochondria in the cell body), the patterns still persisted, with cells of nonTg having the highest mitochondrial percent area fraction, and Alz50+ cells of rTg4510 having the lowest.
Figure 4.
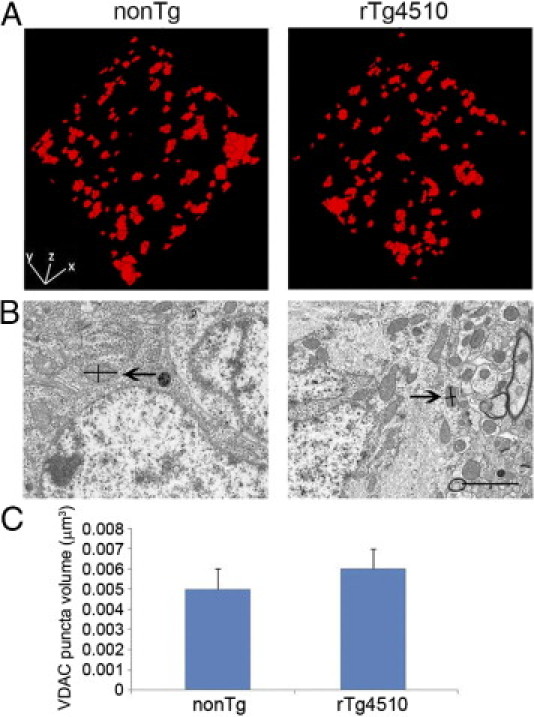
Mitochondrial volume is maintained even in the presence of distribution deficits. Three-dimensional volume reconstructions (A) of 8 × 70 nm sections immunofluorescently stained for VDAC in 8.5-month-old nonTg (left) and Tg4510 (right) mice were used to measure volumes of individual mitochondria in the neuropil. These volume measurements were verified by comparison with volumes estimated from cross-sectional measurements of mitochondria on electron microscopy images (B). Each mitochondrion was measured at its widest and longest points as demonstrated with lines within the mitochondria beside the arrows. An ellipsoidal volume approximation was calculated from these diameters. Mitochondrial volumes approximated from electron micrographs confirmed the mitochondrial volumes obtained with the automated output of the array tomography method. Quantification of array volumes of n = 3922 mitochondria demonstrated no significant differences in volumes of individual mitochondria of the neuropil (C) between 8.5-month-old Tg4510 and nonTg mice (depicted as medians with SD). Scale bar = 2 μm.
Alterations in Mitochondrial Distribution Are Evident in Human AD Brain
The rTg4510 mouse model exhibits a 13-fold over-expression of a human Frontotemporal dementia associated mutant tau9 and does not present with any amyloid plaque pathology. To determine whether our findings of mitochondrial localization in rTg4510 mice could be extrapolated to human AD, we performed array tomographic studies in human AD tissue. As shown in Figure 5, percent cytoplasmic area occupied by mitochondria in the soma of Alz50+ and Alz50− cells were significantly decreased from that of healthy control neurons (P < 0.0001). Tangle bearing neurons of human AD brain were more severely affected than Alz50− neurons (P < 0.0001), in accord with data obtained from the mouse model. In contrast to the mouse data, is the finding that Alz50− neurons are also significantly depleted of their mitochondria in AD brain, a result not seen in either 5.5- or 8.5-month-old mice. This may be consequent to human AD cases representing a more severe stage of disease progression than the 8.5-month-old mice or a consequence of the additional pathological changes seen in AD brain, including Aβ plaque accumulation, soluble oligomeric Aβ burden or progressive synaptic degeneration and glial activation.
Figure 5.
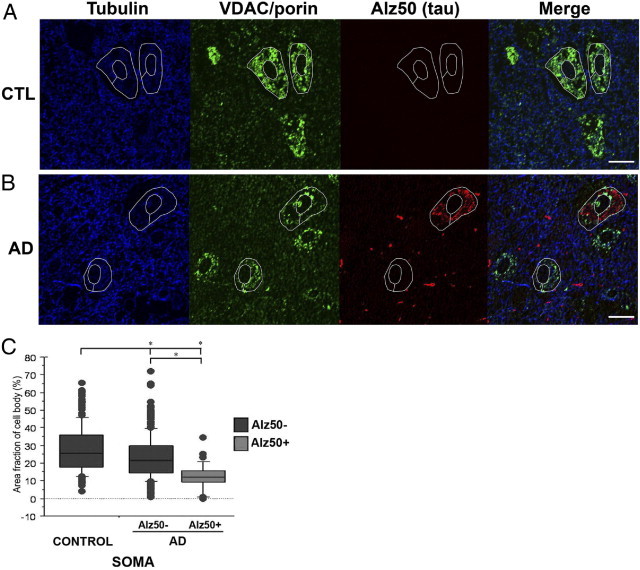
Somatic mitochondrial distribution is disrupted in neurons in human AD brain. Array tomography images of single 70-nm sections from human control (CTL) (A) and human AD brain (B) reveal mitochondrial distribution is altered in both Alz50+ and Alz50− neurons compared with control brain (quantified in C). *P < 0.0001. Scale bars = 10 μm.
Discussion
Cell culture data in both neurons and non-neuronal cells link tau over-expression to mitochondrial trafficking deficits,18,38,39 which have been associated with negative consequences to mitochondrial distribution and function. Regulation of mitochondrial distribution and function is essential in neurons, which have higher energy demands and extended processes, requiring regulated transport.29,40 Dysregulation of these processes has been shown to result in impaired synaptic function and synapse loss30 and is thus proposed as an early pathogenic step in AD.22,24,28,41 Here, we tested the hypothesis that tau mislocalization to the somatodendritic compartment, and subsequent aggregation, in the intact brain of both a mouse model of tauopathy and human AD would affect mitochondrial distribution. Further, we tested the hypothesis that soluble, perhaps in addition to, fibrillar tau would be important for these changes. As a model of tauopathy, we used rTg4510 animals, which over-express a human mutant form of tau leading to neurofibrillary pathology and neuronal loss.9,10 Our results indicate that aberrant mitochondrial distribution is evident in neurites of rTg4510 mice, even at an early stage, regardless of whether or not aggregated tau is present in the form of neuropil threads. Meanwhile, the somatic distribution is less affected with only cells with coincident somatic accumulation of misfolded tau showing an indication of perinuclear mitochondrial clumping at 5.5 months. In the aged rTg4510 mice (8.5 months), a worsening of this mitochondrial distribution phenotype is observed with further decreased mitochondrial area fractions in both neurites and in cell bodies with Alz50+ tau accumulations. Mitochondrial distribution in somas of neurons in human AD brain closely reflects the findings in the mouse model, with dramatic depletion in mitochondrial cytoplasmic area fraction in Alz50+ cells.
Interestingly, in the regulated mouse line the suppression of soluble tau expression, which does not remove the Alz50+ aggregates, allows full recovery of mitochondrial distribution, thereby supporting an important role for soluble tau in this process. These data also indicate that mitochondrial distribution deficits are not the result of exclusion by space occupying aggregates of tau, but that some other mechanism is involved. The presence of NFT within the neuronal cell body may represent the result of reaching a cellular threshold for soluble, mislocalized tau accumulation, which our data suggest more directly interferes with mitochondrial trafficking. Our data demonstrate an early-onset and pervasive mislocalization of mitochondria, which can be alleviated by removing soluble tau species. In addition, our findings in the rTg4510 mouse model are mirrored in human AD brain, and thereby implicate soluble tau, alongside aggregated forms, as detrimental to normal neuronal function in human tauopathies such as AD.
The mechanism by which tau over-expression influences mitochondrial distribution is yet to be precisely determined. However, our results from the intact rTg4510 mouse brain are consistent with findings from previous in vitro studies implicating soluble tau over-expression as detrimental to axonal transport and mitochondrial distribution and function. The fact that alterations in mitochondrial distribution are evident at early stages, in the 5.5 month rTg4510, supports the notion that transport deficits and accompanying mitochondrial changes in distribution are early events in neurodegenerative cascades.19,23,24,42,43 Several potential mechanisms for tau overexpression contributing to transport deficits have previously been explored. (1) Studies of tau over-expression in cells and in vitro assays have demonstrated negative consequences to anterograde cellular transport, by directly inhibiting progress of kinesin-1, by forming a “road block” on the microtubule where excessive tau is bound.14,15,44 (2) Tau-induced trafficking impairments could also result from destabilization of microtubules due to hyperphosphorylated tau losing its affinity for the microtubule.45,46 Microtubule stabilizing compounds such as taxol have been shown to alleviate tau-induced mitochondrial depletion in vitro.47 However, it should be noted that several studies have demonstrated that microtubule networks remain intact and stable even with a loss or complete knock-out of tau,48 suggesting that other mechanisms may be involved. (3) Other studies indicate that tau may interfere with signaling pathways, or molecules such as GSK3β, thereby exerting more indirect control over transport processes.20,49 Our data support tau-induced anterograde transport deficits as the general mechanism by which mitochondria become improperly distributed. The re-distribution of mitochondria following doxycycline treatment in the rTg4510, even in the continued presence of neuropil threads or NFT, suggests that the presence of aggregates of tau may not, in fact, serve as roadblocks, but rather that soluble tau species interfere with mitochondrial trafficking by some other means. Another mechanism by which tau over-expression may influence mitochondrial distribution is by interfering with fission and fusion dynamics. Changes to this equilibrium can result in mitochondrial and neuronal dysfunction and synaptic loss or neuronal cell death.26–29 Manipulation of the primary mammalian fission (fragmentation) GTPase, dynamin-related protein-1 (Drp-1) has been shown to result in mitochondrial distribution deficits with perinuclear clumping.26,28,30,31,37 It has been posited that abnormal proteins present in neurodegenerative diseases, such as tau, may interact directly with mitochondria and/or Drp-1 and initiate the cascade of mitochondrial morphological and functional changes and eventual cellular dysfunction and death.28,37 Interestingly, it has been demonstrated recently that tau does directly interact with mitochondria and in this way may influence their distribution and function.50,51 Furthermore, tau over-expression in vitro has been shown to result in increased mitochondrial fragmentation, alterations in mitochondrial membrane potential, impaired calcium buffering and oxidative stress responses, and diminished respiratory chain function.19,43,52 For these reasons, we assessed mitochondrial volumes in the 8.5 month-old rTg4510 and nonTg mice to determine whether concomitant disequilibrium in fission and fusion dynamics is evident alongside mitochondrial distribution deficits. We did not find significant differences in mitochondrial size in the neuropil, which is consistent with a proteomic study of P301L mutant tau over-expressing mice.52 In addition, we confirmed that the mitochondrial volumes generated by the array tomography automated analysis program were similar to those estimated from electron microscopy measurements, for both rTg4510 and nonTg mice. These findings suggest that mitochondrial mislocalization is indicative of tau-induced anterograde transport deficits rather than interference with mitochondrial fission and fusion dynamics.
Though our data suggest little change, if any, in mitochondrial fission and fusion machinery, the mitochondrial mislocalization that is evident likely has significant and deleterious effects on mitochondrial and neuronal function and survival. Failure to properly distribute mitochondria throughout the cell leaves synapses deprived of their energy requirements, susceptible to reactive oxygen species, and with inefficient calcium handling and can result in synaptic dysfunction and subsequent dying back of the neuron.15,19,20,53 When improperly distributed, mitochondria are thought to have impaired function25,54 and this can lead to increased neuronal susceptibility to excitotoxicity or neurodegenerative stressors such as amyloid-β.2 Changes to mitochondrial distribution have been linked to failure to maintain mitochondrial membrane potential, resulting in release and activation of pro-apoptotic molecules such as caspase. Tau is a caspase-3 substrate and its cleavage is thought to potentiate oligomerization and enhance tau-induced mitochondrial alterations and other negative consequences to the neuron.1,43,52
Our data also demonstrate several methodological advances for the field of pathological analyses of mitochondria. First, we confirmed that single section analysis of cytoplasmic mitochondrial area fraction is comparable to volume fractions from whole cell reconstructions. This allows for much faster, yet still accurate data collection with single slice array tomography. We also confirmed that the automated volume calculations from the immunofluorescent labeled mitochondria in array tomograms are valid by comparing them to volumes estimated from measuring mitochondrial profiles on electron micrographs. This is important since array tomography generates data from thousands of mitochondria per case, allowing analysis of a large sample size, which is prohibitively time consuming by serial reconstructions in electron microscopy. Lastly, we applied array tomography to human brain tissue collected at autopsy and fixed according to the array tomography protocols. This technique was developed for synapse analysis in mice32–34 and has subsequently been adapted for use in zebrafish,55 songbirds,56 and on one case of collagen imaging in a human optic nerve.57 We demonstrate that in human brain, array tomography can be used for analysis of mitochondria and other structures at or near the limit of z-resolution of conventional microscopy, adding an important tool in the arsenal of neuropathological analyses.
Taken together, our data support the hypothesis that tau mislocalization to the somatodendritic compartment and subsequent aggregation is associated with mitochondrial distribution deficits. Furthermore, by suppressing soluble tau species recovery of mitochondrial distribution to near control levels occurs, suggesting that soluble tau species play a more significant role in mitochondrial distribution than aggregated forms. These findings are in agreement with several studies that propose that oligomeric species are more toxic than aggregates.1,2,8,13,58–60 Meanwhile, tau-induced deficiencies in mitochondrial distribution are likely a consequence of axonal transport deficits, and may result in disrupted mitochondrial and neuronal function and subsequent synaptic failure and/or cell death. The resemblance of mitochondrial distribution patterns in neurons in human AD brain to those in rTg4510 suggests that soluble tau species play a role in mitochondrial localization in human neurodegenerative disease and may serve as a useful therapeutic target.
Acknowledgments
We thank Karlotta Fitch and the Massachusetts Alzheimer's Disease Research Center.
Footnotes
Supported by National Institute of Health Grants AG08487, T32AG000277, AG026249, K99AG33670, P50AG05134 and the Alzheimer's Association Zenith Award.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.004.
Supplementary data
3-dimensional array tomography reconstruction of a tangle from rTg4510 mouse brain stained with conformational tau antibody Alz50. Images from 100 sequential 80nm sections were used for reconstruction.
Tubulin and astrocyte staining patterns do not coincide. Array tomography images of single 70-nm sections from layer II/III of somatosensory cortex of nonTg (A and C) and rTg4510 (B and D) demonstrate that tubulin staining and GFAP or glutamine synthetase staining do not coincide. Neurites, indicated by arrows, exemplify those that would be selected for analysis. Scale bars = 10 μm. DAPI, 4′6-diamidino-2-phenylindole•2HCl staining; GFAP, glial fibrillary acidic protein staining.
References
- 1.Spires-Jones T.L., Stoothoff W.H., de Calignon A., Jones P.B., Hyman B.T. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32:150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Gendron T.F., Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 4.Gomez-Isla T., Hollister R., West H., Mui S., Growdon J.H., Petersen R.C., Parisi J.E., Hyman B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 5.Giannakopoulos P., Herrmann F.R., Bussiere T., Bouras C., Kovari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 6.Braak H., Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 7.Rocher A.B., Crimins J.L., Amatrudo J.M., Kinson M.S., Todd-Brown M.A., Lewis J., Luebke J.I. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2010;223:385–393. doi: 10.1016/j.expneurol.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lasagna-Reeves C.A., Castillo-Carranza D.L., Guerrero-Muoz M.J., Jackson G.R., Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- 9.Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K.H. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spires T.L., Orne J.D., SantaCruz K., Pitstick R., Carlson G.A., Ashe K.H., Hyman B.T. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wittmann C.W., Wszolek M.F., Shulman J.M., Salvaterra P.M., Lewis J., Hutton M., Feany M.B. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 12.de Calignon A., Fox L.M., Pitstick R., Carlson G.A., Bacskai B.J., Spires-Jones T.L., Hyman B.T. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger Z., Roder H., Hanna A., Carlson A., Rangachari V., Yue M., Wszolek Z., Ashe K., Knight J., Dickson D., Andorfer C., Rosenberry T.L., Lewis J., Hutton M., Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixit R., Ross J.L., Goldman Y.E., Holzbaur E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubey M., Chaudhury P., Kabiru H., Shea T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: neurofilaments attenuate tau-mediated neurite instability. Cell Motil Cytoskeleton. 2008;65:89–99. doi: 10.1002/cm.20243. [DOI] [PubMed] [Google Scholar]
- 16.LaPointe N.E., Morfini G., Pigino G., Gaisina I.N., Kozikowski A.P., Binder L.I., Brady S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thies E., Mandelkow E.M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoothoff W., Jones P.B., Spires-Jones T.L., Joyner D., Chhabra E., Bercury K., Fan Z., Xie H., Bacskai B., Edd J., Irimia D., Hyman B.T. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J Neurochem. 2009;111:417–427. doi: 10.1111/j.1471-4159.2009.06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckert A., Schulz K.L., Rhein V., Gotz J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107–114. doi: 10.1007/s12035-010-8109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morfini G.A., Burns M., Binder L.I., Kanaan N.M., LaPointe N., Bosco D.A., Brown R.H., Jr., Brown H., Tiwari A., Hayward L., Edgar J., Nave K.A., Garberrn J., Atagi Y., Song Y., Pigino G., Brady S.T. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muresan V., Muresan Z. Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer's disease? Future Neurol. 2009;4:761–773. doi: 10.2217/fnl.09.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 23.Querfurth H.W., LaFerla F.M. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 24.Swerdlow R.H., Burns J.M., Khan S.M. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20(Suppl 2):S265–S279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X., Schwarz T.L. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X., Su B., Lee H.G., Li X., Perry G., Smith M.A., Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X., Su B., Zheng L., Perry G., Smith M.A., Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;109(Suppl 1):153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 29.Du H., Guo L., Yan S., Sosunov A.A., McKhann G.M., Yan S.S. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Verstreken P., Ly C.V., Venken K.J., Koh T.W., Zhou Y., Bellen H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 32.Koffie R.M., Meyer-Luehmann M., Hashimoto T., Adams K.W., Mielke M.L., Garcia-Alloza M., Micheva K.D., Smith S.J., Kim M.L., Lee V.M., Hyman B.T., Spires-Jones T.L. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Micheva K.D., Smith S.J. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Micheva K.D., Busse B., Weiler N.C., O'Rourke N., Smith S.J. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ludvigson A.E., Luebke J.I., Lewis J., Peters A. Structural abnormalities in the cortex of the rTg4510 mouse model of tauopathy: a light and electron microscopy study. Brain Struct Funct. 2010;216:31–42. doi: 10.1007/s00429-010-0295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thevenaz P., Ruttimann U.E., Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- 37.Reddy P.H., Reddy T.P., Manczak M., Calkins M.J., Shirendeb U., Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev. 2011;67:103–118. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebneth A., Godemann R., Stamer K., Illenberger S., Trinczek B., Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamer K., Vogel R., Thies E., Mandelkow E., Mandelkow E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hollenbeck P.J., Saxton W.M. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreira P.I., Carvalho C., Zhu X., Smith M.A., Perry G. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 42.Decker H., Lo K.Y., Unger S.M., Ferreira S.T., Silverman M.A. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintanilla R.A., Matthews-Roberson T.A., Dolan P.J., Johnson G.V. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284:18754–18766. doi: 10.1074/jbc.M808908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baas P.W., Qiang L. Neuronal microtubules: when the MAP is the roadblock. Trends Cell Biol. 2005;15:183–187. doi: 10.1016/j.tcb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Ballatore C., Lee V.M., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 46.Bretteville A., Planel E. Tau aggregates: toxic, inert, or protective species. J Alzheimers Dis. 2008;14:431–436. doi: 10.3233/jad-2008-14411. [DOI] [PubMed] [Google Scholar]
- 47.Zempel H., Thies E., Mandelkow E., Mandelkow E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites: Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris M., Maeda S., Vossel K., Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tackenberg C., Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009;29:14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amadoro G., Corsetti V., Stringaro A., Colone M., D'Aguanno S., Meli G., Ciotti M., Sancesario G., Cattaneo A., Bussani R., Mercanti D., Calissano P. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis. 2010;21:445–470. doi: 10.3233/JAD-2010-100120. [DOI] [PubMed] [Google Scholar]
- 51.Bobba A., Petragallo V.A., Marra E., Atlante A. Alzheimer's proteins, oxidative stress, and mitochondrial dysfunction interplay in a neuronal model of Alzheimer's disease. Int J Alzheimers Dis. 2010 doi: 10.4061/2010/621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.David D.C., Hauptmann S., Scherping I., Schuessel K., Keil U., Rizzu P., Ravid R., Drose S., Brandt U., Muller W.E., Eckert A., Gotz J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. 2005;280:23802–23814. doi: 10.1074/jbc.M500356200. [DOI] [PubMed] [Google Scholar]
- 53.Darios F., Muriel M.P., Khondiker M.E., Brice A., Ruberg M. Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. J Neurosci. 2005;25:4159–4168. doi: 10.1523/JNEUROSCI.0060-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H., Chan D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robles E., Smith S.J., Baier H. Characterization of genetically targeted neuron types in the zebrafish optic tectum. Front Neural Circuits. 2011;5:1. doi: 10.3389/fncir.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oberti D., Kirschmann M.A., Hahnloser R.H. Correlative microscopy of densely labeled projection neurons using neural tracers. Front Neuroanat. 2010;4:24. doi: 10.3389/fnana.2010.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winkler M., Jester B., Nien-Shy C., Massei S., Minckler D.S., Jester J.V., Brown D.J. High resolution three-dimensional reconstruction of the collagenous matrix of the human optic nerve head. Brain Res Bull. 2010;81:339–348. doi: 10.1016/j.brainresbull.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.L., Ashe K.H., Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kimura T., Fukuda T., Sahara N., Yamashita S., Murayama M., Mizoroki T., Yoshiike Y., Lee B., Sotiropoulos I., Maeda S., Takashima A. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem. 2010;285:38692–38699. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lasagna-Reeves C.A., Castillo-Carranza D.L., Sengupta U., Clos A.L., Jackson G.R., Kayed R. Tau Oligomers Impair Memory and Induce Synaptic and Mitochondrial Dysfunction in Wild-type Mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3-dimensional array tomography reconstruction of a tangle from rTg4510 mouse brain stained with conformational tau antibody Alz50. Images from 100 sequential 80nm sections were used for reconstruction.
Tubulin and astrocyte staining patterns do not coincide. Array tomography images of single 70-nm sections from layer II/III of somatosensory cortex of nonTg (A and C) and rTg4510 (B and D) demonstrate that tubulin staining and GFAP or glutamine synthetase staining do not coincide. Neurites, indicated by arrows, exemplify those that would be selected for analysis. Scale bars = 10 μm. DAPI, 4′6-diamidino-2-phenylindole•2HCl staining; GFAP, glial fibrillary acidic protein staining.